
Abstract
Pericytes, the mural cells that constitute the capillaries along with endothelial cells, have been associated with the pathobiology of diabetic retinopathy; however, therapeutic implications of this association remain largely unexplored. Pericytes appear to be highly susceptible to the metabolic challenges associated with a diabetic environment, and there is substantial evidence that their loss may contribute to microvascular instability leading to the formation of microaneurysms, microhemorrhages, acellular capillaries, and capillary nonperfusion. Since pericytes are strategically located at the interface between the vascular and neural components of the retina, they offer extraordinary opportunities for therapeutic interventions in diabetic retinopathy. Moreover, the availability of novel imaging methodologies now allows for the in vivo visualization of pericytes, enabling a new generation of clinical trials that use pericyte tracking as clinical endpoints. The recognition of multiple signaling mechanisms involved in pericyte development and survival should allow for a renewed interest in pericytes as a therapeutic target for diabetic retinopathy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pericytes, the vascular cells that together with the endothelial cells make up the capillaries, have long been associated with the pathobiology of diabetic retinopathy (DR). However, translational applications of this link remain largely unexplored. Here, we review cell signaling mechanisms of pericytes in models of diabetes as well as new imaging technologies that open the door for novel therapeutic interventions and the possibility of assessing pericytes as clinical end points in clinical trials.
Microvascular Degeneration and Neurovascular Unit Dysfunction in Diabetic Retinopathy
The retinal requirements for oxygen and nutrients rank as the highest among all the other tissues in the human body, exceeding even that of the brain [1–4]. Thus, it is not surprising that two independent blood supplies operate to meet the demand. The superficial layers of the retina, including the ganglion cell layers and the inner nuclear layer, are nourished through a network of capillaries deriving from the central retinal artery. This vascular network has been the focus of intense research in the field of DR, as it is the target of overt degenerative changes that are easily detected by clinicians using standard fundoscopy examination or retinal photographs [5]. In fact, DR is classified according to the nature of changes observed in the retinal microvasculature. Early-stage DR, also known as nonproliferative DR, is characterized by the presence of microaneurysms, microhemorrhages, cotton-wool spots (an ischemic lesion of the nerve fiber layer), venous caliber changes, and intraretinal microvascular abnormalities; late-stage DR, also known as proliferative DR, is defined by the presence of aberrant neovascular outgrowths originating from the retinal vessels [6–9]. The outer retina, including the photoreceptors, receives a majority of its oxygen and nutrients through diffusion from a rich plexus of fenestrated capillaries located in the choroid. This vascular network, called the choriocapillaris, is separated from the retina by Bruch’s membrane and a monolayer of retinal pigment epithelium (RPE).
Multiple lines of evidence coming from studies in cell culture, animal models, and human studies strongly indicate that all the components of the retina, not only its vessels but also the neural elements, the choroid, and the RPE, are impacted by the metabolic and signaling challenges imposed by diabetes [10–14]. Neuronal apoptosis, astrocyte dysfunction, microglial activation, Müller cell dysfunction, and loss of RPE barrier function have all been observed in diabetic retinopathy [5, 15–18]. Moreover, studies of human specimens and animal models of diabetes have revealed a key role for inflammatory cells and cytokines (reviewed in [19] and [20]; also see [21–23]).
Recognizing the complexity of DR, however, should not detract from efforts to pursue therapeutic approaches that focus on specific cellular targets and signaling pathways; such targeted approaches may be clinically effective and involve fewer side effects. Based on this premise, this review focuses on the critical role of pericytes, a cell that, along with endothelial cells, comprises the retinal capillaries and is an early target of diabetes in both humans and experimental animals.
Cogan and colleagues first reported the loss of pericytes in DR in 1961 [24]. Termed as pericyte dropout, a loss of retinal pericytes was the earliest morphological change observed in diabetic retinopathy, with the endothelial cell-to-pericyte ratio dropping from 1:1 in normal retinal tissue to 4:1 in diabetic retinas [25]. Studies of the time course of diabetic complications in humans revealed that pericyte dropout in DR is linked to the development of microangiopathies such as microaneurysms, acellular capillaries, vessel tortuosity, hyperpermeability, and capillary nonperfusion [26, 24, 27–31]. This correlation was corroborated by evidence from animal models of diabetes consistently showing that pericyte loss preceded the development of microangiopathies (STZ, db/db, and galactosemia models) (summarized in [32]). Pericytes, however, are not the only cell type damaged in the course of diabetes. Rather, there is overwhelming evidence that pericytes are an integral component of what is known as the neurovascular unit, a functional and architectural arrangement of cells comprising vascular and neural components that support visual perception by maintaining an appropriate blood supply to retinal tissues [33, 34].
Pericytes and endothelial cells both synthesize and share a common basement membrane [35]. Discontinuities in the basement membrane allow intercellular contact and, thus, communication between pericytes and endothelial cells [36, 37]. These junctions consist of membrane invaginations that are rich in adherence and gap junctions [36–39]. Heterotypic cell-cell interactions also take place on the other side of the neurovascular unit with glial cells extending end-feet processes that surround vessels and regulate their function through diffusible molecules. In fact, there is evidence that the formation and maintenance of a functional blood-barrier is highly dependent on factors derived from glial cells [40]. In the superficial vascular plexus, astrocytes play a major role in regulating vascular integrity whereas Müller cells contribute to vascular integrity in the inner nuclear layer [41]. Accordingly, ablation of Müller cells has been shown to trigger photoreceptor apoptosis, vascular telangiectasias, and breakdown of the blood-retina barrier [41]. Similarly, genetic manipulations affecting astrocyte development have been shown to be associated with vascular abnormalities [42–44]. In consideration of the strong associations among the different cellular components of the neurovascular unit, pericyte loss should be understood both as cause and consequence of the dysfunction associated with diabetes.
Heterotypic Cell Signaling in the Neurovascular Unit
As described in detail above, pericytes and endothelial cells communicate through junctions that extend through discontinuities in a shared basement membrane. These direct cell-cell interactions, as well as the close proximity of the two cell types in vivo, are thought to facilitate cell signaling through key signaling pathways including PDGF-B/platelet-derived growth factor receptor beta (PDGFRβ), TGFβ, and angiopoietin-1/Tie2. Cell loss as a result of high glucose, inflammation, and/or the thickening and rarefaction of the basement membrane, all hallmarks of DR, may disrupt or abrogate cell signaling [45, 46]. In fact, not only a significant thickening of the capillary basement membrane in DR but also a change in the composition of diabetic basement membranes, with increased production of both collagen type IV and laminin, is found [47, 48]. The summary of cell signaling pathways linked to DR, the model system in which these observations were made, and the conclusions of the study are shown in Table 1. PDGF-B/PDGFRβ, TGFβ, and angiopoietin-1/Tie2 are discussed in detail because they are potentially amenable to therapeutic intervention.
PDGF-B/PDGFRβ
Pericyte recruitment is coordinated by the interplay of the PDGF-B acting through PDGFRβ. Proliferating endothelial cells secrete PDGF-B, whereas pericytes and their precursors express PDGFRβ, the receptor for PDGF-B. In development, PDGF-B is secreted from the endothelium of angiogenic sprouts and newly formed vessels where it serves as an attractant for PDGFRβ-expressing co-migrating pericytes or pericyte precursors [49–51]. Consistent with this function, transgenic mouse models clearly indicate a role for PDGF-B and PDGFRβ in the development of a mature neurovascular unit. Pdgfb and Pdgfrb-deficient mice display vascular abnormalities, including microaneurysms and increased microvascular permeability, associated with the absence of pericytes and/or abnormal endothelial cell ultrastructure [49, 52–54]. As a systemic knockout of the Pdgfb and/or Pdgfrb genes is embryonically lethal, mice heterozygous for PDGF-B+/− and mice with an endothelial cell-specific conditional knockout of PDGF-B were created to study the loss of PDGF-B on pericytes [30, 51, 55, 56]. Although pericyte loss occurs in these models, the extent of impact is variable. The pericyte population in PDGF-B+/− mice is reportedly reduced by approximately 30 %, and a slight increase in acellular capillaries is observed [30]. Mice with an endothelial cell-specific conditional knockout of PDGF-B begin to display microaneurysms and increased microvascular regression when pericyte coverage is less than 50 % [55, 56]. Additionally, studies have shown that hyperglycemia leads to persistent activation of protein kinase C delta (PKCδ) and p38α MAPK, which results in increased expression of Src homology region 2 domain-containing phosphatase 1 (SHP-1) and PDGFβ dephosphorylation [57]. Unlike diabetic control mice, diabetic null for the PKCδ gene showed a reduced number of acellular capillaries compared to controls. Thus, these studies indicate that pericytes and PDGF-B play an important role in the development of the neurovascular unit and have generally been interpreted as further evidence of the key role played by pericytes in vascular integrity in diabetes.
However, whether PDGF signaling plays a role in pericyte survival and maintenance in adult tissues requires further analyses involving conditional knockouts. This question is highly relevant now that PDGF signaling inhibitors (Fovista™ (E10030), Ophthotech) are entering clinical trials for neovascular conditions in the eye including neovascular age-related macular degeneration (AMD) [58].
In wet AMD, neovascular outgrowths, which originate from the choriocapillaris and grow into the subretinal space, are treated with anti-VEGF therapies [59–64]. This therapy is generally effective at reducing vascular permeability and inducing vascular regression. However, it has been observed that pericyte investment of vascular outgrowths correlates with poor response to anti-VEGF therapy. This is not surprising as the association of the pericyte with the new vessels leads to increased vessel stability. The addition of anti-PDGF therapy [65–67] to anti-VEGF is based on the premise that pericyte association with nascent vessels makes those vessels relatively (but not totally) insensitive to vascular regression by VEGF neutralization; however, considering the precedent of pericyte dropout in DR, it is essential to evaluate long-term safety of this treatment in both diabetic and nondiabetic eyes.
TGFβ/TGFβ Receptors
Both pericytes and endothelial cells express TGFβ as well as TGFβ receptors, and the interactions between these two cell types are important in TGFβ signaling. Studies using co-culture of pericytes and endothelial cells demonstrated that physical contact between the cells is necessary for activation of latent TGFβ1 [68, 69]. Furthermore, in vivo studies targeting endothelial cell-specific deletion of activin receptor-like kinase 5 (Alk-5), a TGFβ type I receptor, showed a reduction in endothelial cell secretion of TGFβ1 [70]. This, in turn, resulted in reduced signaling of TGFβ/Alk-5 in pericytes [70]. Taken together, these data suggest an important role for TGFβ signaling between pericytes and endothelial cells.
Both TGFβ and angiopoietin-1 (Ang-1)/Tie2 (discussed in detail below) signaling between pericytes and endothelial cells appears to play a significant role in vessel stability. The majority of TGFβ/TGFβ receptor knockout models (Tgfb1, Alk5, Eng, and Smad5) are embryonically lethal due to major abnormalities in the vasculature [71–75]. Furthermore, mice deficient of either Alk-5, a TGFβ type I receptor, or endoglin, a TGFβ binding protein, lack smooth muscle cell investment and formation, respectively [72, 75]. Consistent with this biology, misregulation of TGFβ signaling has been associated with DR in humans and animal models, including thickening of the basal lamina [76] and cicatricial contraction of proliferative fibrous membranes [77]. Moreover, there is evidence indicating that TGFβ is also an indirect target of drugs that prevents experimental DR. Concordant treatment of diabetic rats with the aldose reductase inhibitor sorbinil and aspirin reduced the diabetes-induced upregulation of genes in the TGFβ pathway [78], and inhibition of ROCK, a key downstream mediator of TGFβ, dramatically suppressed PDR/PVR-induced collagen gel contraction [77], albeit clinical trials of sorbinil in humans did not show clinical efficacy [79]. Notwithstanding this set back, a recent systemic meta-analysis of vitreous biomarkers associated with DR identified blockade of TGFβ using cell therapy as a viable therapeutic candidate for DR therapy [80].
Ang-1/Tie2
Many studies have indicated the importance of pericyte and endothelial cell interactions in angiopoietin-1/Tie2 signaling, a system known to participate in vascular development. Ang-1 is produced by the pericytes, whereas endothelial cells express Tie2 [39, 81–83]. Studies have shown that Ang-1 signaling via Tie2 is important for capillary sprouting, endothelial cell survival, and vascular remodeling [81, 83–85]. Ang-1 has also been implicated in the stabilization of vessels by pericytes and smooth muscle cells to the vessel wall [86]. Ang-2, another member of the angiopoietin family, can act as a competitive inhibitor of Ang-1 signaling through Tie2 [87, 88]. Overexpression of Ang-2 in transgenic mice disrupts blood vessel formation during embryo development [87] and, over the course of several months, causes the pericyte dropout and the formation of acellular capillaries, similar to that observed in early-stage DR [86, 89, 90]. Similar to TGFβ/TGFβ receptor knockout models, Ang-1 and Tie2-deficient mice lack pericytes and are embryonically lethal due cardiovascular failure at mid-gestation [84, 91]. In addition, intravitreal injection of Ang-1 has been shown to rescue high-order architecture of the developing vasculature in an anti-PDGFRβ antibody model of pericyte deficiency [92]. The Ang-1/Tie2 signaling system has also been shown to play a major role in the pericyte loss observed in DR by modulating pericyte migration [89] and influencing the activation state and recruitment of pericytes [93]. Additionally, Ang-1 may be useful for reducing microvascular leakage, as vessels in transgenic mice overexpressing Ang-1 were not only nonleaky but also resistant to leaks caused by inflammatory agents [94]. Targeting of the Ang-1/Tie2 signaling system for treatment of diabetic macular edema (DME) in humans has shown to be of promise, as a phase 1 trial using a subcutaneously administered agonist of this signaling system has been completed and a phase 2 trial is currently ongoing in patients with DME (ClinicalTrials.gov identifier: NCT02050828).
While extensive research has shown that the PDGF-B/PDGFRβ, TGFβ, and Ang-1/Tie2 signaling systems all play an important role in the pathogenesis of DR, a number of other pathways also have been identified to be associated with pericyte loss. These include the complement system, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), forkhead box protein O1 (FOXO1), glucose metabolism (polyol/sorbitol), poly (ADP-ribose) polymerase (PARP), NF-κB, and vitamins thiamine and benfotiamine (see Table 1).
Tracking Pericytes in the Neurovascular Unit
Measurements of pericyte loss have been used extensively as an indication of diabetic retinopathy in animal models [57, 95–97] and in postmortem studies in human tissues [79, 98]. Pericytes can be identified in whole-mount retinal preparations using a combination of their location and one or more of a number of markers of differentiation, such as alpha-smooth muscle actin (α-SMA), desmin, and regulator of G protein signaling 5 (RGS-5) [39, 51, 99–105]. Cell surface molecule expressed by pericytes include neuron-glial antigen 2 (NG2), PDGFRβ, and vascular cell adherence molecule 1 (VCAM-1) [49, 51, 99, 106–108]. To date, no specific molecular marker has been identified that will reliably label and differentiate pericytes from other cell types found in the retina. Instead, the markers described above must be used in combination and with contextual information such as species, vessel type, developmental or angiogenic stage, tissue specificity, and/or pathology in order to confirm pericyte identity.
Quantification of pericyte loss has historically been conducted using a method referred to as “trypsin digest” in which the neural components of the retina are digested away, leaving the microvasculature intact [109]. Subsequent optimization of the trypsin digest protocol [110, 111] revealed that elastase, not trypsin, was the active component of the trypsin solution and that, in the fixed retina, elastase preserves the microvasculature structure. Elastase digests can be stained using standard immunohistochemical methods. Furthermore, at least in humans, pericytes and endothelial cells are further distinguishable by their nuclear shape. Pericytes have small, dark-staining round or slightly oval nuclei; although they are completely enveloped by basement membrane, they protrude from the abluminal wall of the capillary [25, 109, 112]. Endothelial cells have larger oval or ellipsoid nuclei that lie in the axis of blood flow [25, 109, 112].
However, distinguishing between pericytes and endothelial cells is not as straightforward in mice, which are widely used to study the role of specific genes in diabetic retinopathy. Moreover, in an elastase digest, the analysis of the microvasculature is performed out of the context as other components of the neurovascular unit including glial cells and neurons are removed to allow visualization. Of great promise are methods like CLARITY, which allows for the transformation of intact biological tissue into an optically transparent and chemically permeable hydrogel-based nanoporous structure that retains structural integrity and relevant molecules such as native antigens, neurotransmitters, soluble and cell membrane proteins, and mRNAs [113]. CLARITY methodology removes lipid bilayers and replaces them with hydrogel monomers that are covalently linked to remaining biomolecules. The resultant tissue preparation can be further visualized and analyzed, enabling intact tissue in situ hybridization, immunohistochemistry, and antibody labeling of the intact tissue or organ. CLARITY has not yet been applied to the eye, but its successful use on tissue of the mouse brain [113, 114] is promising. More recently, another method for the optical clearing of tissues, named CUBIC, has been developed. As for the CLARITY methodology, the CUBIC protocol creates optically transparent tissue while retaining subcellular structures; however, CUBIC has the advantages of utilizing nontoxic water-soluble chemicals and not requiring expensive clearing reagents or specialized devices [115]. Application of these methods, which retain the full tissue architecture, to the eye should help to increase our understanding of how pericyte loss influences the neurovascular unit and how changes in other components of the unit directly affect pericyte homeostasis and function.
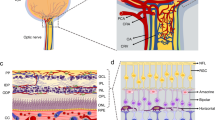
In addition to these new advances in tissue processing, a next generation of live imaging is also now being developed. These new imaging technologies can be used to noninvasively visualize retinal pericytes in the living eye. Using a two-channel, adaptive optics scanning laser ophthalmoscope (AOSLO) [116], retinas of transgenic mice expressing fluorescent pericytes (NG2, DsRed) were imaged in vivo (Fig. 1). The first channel imaged vascular perfusion with new infrared light, while the second channel simultaneously imaged fluorescent retinal pericytes [117]. Pericyte morphology and topography observed from in vivo imaging were confirmed using flat mounts with conventional fluorescent microscopy. This is the first demonstration of high-resolution imaging of retinal pericytes in situ, and this promises to provide the basis to track and quantify pericyte topography, morphology, and function. Because of its noninvasive nature, visualization can be accomplished in the living retina over time, allowing for the progressive monitoring of microvascular disease, like DR. Intravital imaging technology using AOSLO is also being used in the human retinas, allowing for the imaging of vascular wall structures, including nuclei, and for the clear visualization of a variety of retinal vascular and nonvascular changes in subjects with mild to moderate nonproliferative DR [98, 96]. Thus, this new imaging innovation has the potential to revolutionize our understanding of the disease itself.

Simultaneous in vivo imaging of vascular perfusion and fluorescent retinal pericytes using a two-channel, adaptive optics scanning laser ophthalmoscope (AOSLO). a Wide-field HRA Spectralis image shows approximately 30° field of the mouse retina. Superimposed on the fundus image are motion-contrast AOSLO fields demonstrating capillary perfusion with micron-level resolution. b Two-channel imaging using AOSLO in vivo. Channel 1 collects NIR motion contrast demonstrating capillary perfusion (magenta, moving blood cells), while channel 2 simultaneously images DsRed fluorescently labeled pericytes (green). c AOSLO field (5°) demonstrates the association of pericytes (green) with perfused capillaries (magenta). Montage is from a single capillary stratification (reprinted from [117])
Conclusion
In summary, the loss of retinal pericytes is likely an early factor contributing to the onset and progression of clinically relevant vascular pathology in DR. Renewed focus on pericyte protection or replacement as a therapeutic goal for the treatment of patients with diabetic retinopathy may one day result in the prevention or delay of vision loss. Translational efforts that combine a focus on cell signaling regulation in conjunction with in vivo imaging studies of pericytes may ultimately lead to the development of innovative approaches to treat diabetic retinopathy.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Anderson Jr B, Saltzman HA. Retinal oxygen utilization measured by hyperbaric blackout. Arch Ophthalmol. 1964;72:792–5.
Ames 3rd A, Li YY, Heher EC, Kimble CR. Energy metabolism of rabbit retina as related to function: high cost of Na+ transport. J Neurosci. 1992;12(3):840–53.
Yu DY, Cringle SJ. Oxygen distribution and consumption within the retina in vascularised and avascular retinas and in animal models of retinal disease. Prog Retin Eye Res. 2001;20(2):175–208.
Wong-Riley MT. Energy metabolism of the visual system. Eye Brain. 2010;2:99–116. doi:10.2147/EB.S9078.
Barber AJ, Gardner TW, Abcouwer SF. The significance of vascular and neural apoptosis to the pathology of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2011;52(2):1156–63. doi:10.1167/iovs.10-6293.
Cogan DG. Diabetic retinopathy. N Engl J Med. 1964;270:787–8. doi:10.1056/NEJM196404092701508.
Antonetti DA, Klein R, Gardner TW. Diabetic retinopathy. N Engl J Med. 2012;366(13):1227–39. doi:10.1056/NEJMra1005073. This recent review article highlights the principles underlying metabolic control and anti-VEGF therapies in the treatment of diabetic retinopathy. The molecular interactions of neuronal, glial, and vascular cells in the retina as the basis of the neurovascular unit and the effect of diabetes on the function of the neurovascular unit in order to highlight new therapeutic approaches are discussed.
Taylor E, Dobree JH. Proliferative diabetic retinopathy. Site and size of initial lesions. Br J Ophthalmol. 1970;54(1):11–8.
Sapieha P, Hamel D, Shao Z, Rivera JC, Zaniolo K, Joyal JS, et al. Proliferative retinopathies: angiogenesis that blinds. Int J Biochem Cell Biol. 2010;42(1):5–12. doi:10.1016/j.biocel.2009.10.006.
MacGregor LC, Rosecan LR, Laties AM, Matschinsky FM. Altered retinal metabolism in diabetes. I. Microanalysis of lipid, glucose, sorbitol, and myo-inositol in the choroid and in the individual layers of the rabbit retina. J Biol Chem. 1986;261(9):4046–51.
Bradbury MW, Lightman SL. The blood-brain interface. Eye (Lond). 1990;4(Pt 2):249–54. doi:10.1038/eye.1990.36.
Alder VA, Su EN, Yu DY, Cringle S, Yu P. Overview of studies on metabolic and vascular regulatory changes in early diabetic retinopathy. Aust N Z J Ophthalmol. 1998;26(2):141–8.
Barot M, Gokulgandhi MR, Patel S, Mitra AK. Microvascular complications and diabetic retinopathy: recent advances and future implications. Future Med Chem. 2013;5(3):301–14. doi:10.4155/fmc.12.206.
Lutty GA. Effects of diabetes on the eye. Invest Ophthalmol Vis Sci. 2013;54(14):ORSF81-7. doi:10.1167/iovs.13-12979.
Hammes HP, Federoff HJ, Brownlee M. Nerve growth factor prevents both neuroretinal programmed cell death and capillary pathology in experimental diabetes. Mol Med. 1995;1(5):527–34.
Kerrigan LA, Zack DJ, Quigley HA, Smith SD, Pease ME. TUNEL-positive ganglion cells in human primary open-angle glaucoma. Arch Ophthalmol. 1997;115(8):1031–5.
Barber AJ, Antonetti DA, Kern TS, Reiter CE, Soans RS, Krady JK, et al. The Ins2Akita mouse as a model of early retinal complications in diabetes. Invest Ophthalmol Vis Sci. 2005;46(6):2210–8. doi:10.1167/iovs.04-1340.
Krady JK, Basu A, Allen CM, Xu Y, LaNoue KF, Gardner TW, et al. Minocycline reduces proinflammatory cytokine expression, microglial activation, and caspase-3 activation in a rodent model of diabetic retinopathy. Diabetes. 2005;54(5):1559–65.
Antonetti DA, Barber AJ, Bronson SK, Freeman WM, Gardner TW, Jefferson LS, et al. Diabetic retinopathy: seeing beyond glucose-induced microvascular disease. Diabetes. 2006;55(9):2401–11. doi:10.2337/db05-1635.
Gologorsky D, Thanos A, Vavvas D. Therapeutic interventions against inflammatory and angiogenic mediators in proliferative diabetic retinopathy. Mediat Inflamm. 2012;2012:629452. doi:10.1155/2012/629452.
Joussen AM, Poulaki V, Le ML, Koizumi K, Esser C, Janicki H, et al. A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J. 2004;18(12):1450–2. doi:10.1096/fj.03-1476fje.
Avery RL. Regression of retinal and iris neovascularization after intravitreal bevacizumab (Avastin) treatment. Retina. 2006;26(3):352–4.
Mason 3rd JO, Nixon PA, White MF. Intravitreal injection of bevacizumab (Avastin) as adjunctive treatment of proliferative diabetic retinopathy. Am J Ophthalmol. 2006;142(4):685–8. doi:10.1016/j.ajo.2006.04.058.
Cogan DG, Toussaint D, Kuwabara T. Retinal vascular patterns. IV. Diabetic retinopathy. Arch Ophthalmol. 1961;66:366–78.
Speiser P, Gittelsohn AM, Patz A. Studies on diabetic retinopathy. 3. Influence of diabetes on intramural pericytes. Arch Ophthalmol. 1968;80(3):332–7.
Cogan DG, Kuwabara T. The mural cell in perspective. Arch Ophthalmol. 1967;78(2):133–9.
Engerman RL. Pathogenesis of diabetic retinopathy. Diabetes. 1989;38(10):1203–6.
Klein R, Klein BE, Moss SE, Davis MD, DeMets DL. The Wisconsin epidemiologic study of diabetic retinopathy. III. Prevalence and risk of diabetic retinopathy when age at diagnosis is 30 or more years. Arch Ophthalmol. 1984;102(4):527–32.
Klein R, Klein BE, Moss SE, Davis MD, DeMets DL. The Wisconsin epidemiologic study of diabetic retinopathy. II. Prevalence and risk of diabetic retinopathy when age at diagnosis is less than 30 years. Arch Ophthalmol. 1984;102(4):520–6.
Hammes HP, Lin J, Renner O, Shani M, Lundqvist A, Betsholtz C, et al. Pericytes and the pathogenesis of diabetic retinopathy. Diabetes. 2002;51(10):3107–12.
Sasongko MB, Wong TY, Nguyen TT, Cheung CY, Shaw JE, Wang JJ. Retinal vascular tortuosity in persons with diabetes and diabetic retinopathy. Diabetologia. 2011;54(9):2409–16. doi:10.1007/s00125-011-2200-y.
Robinson R, Barathi VA, Chaurasia SS, Wong TY, Kern TS. Update on animal models of diabetic retinopathy: from molecular approaches to mice and higher mammals. Dis Model Mech. 2012;5(4):444–56. doi:10.1242/dmm.009597.
Abcouwer SF, Gardner TW. Diabetic retinopathy: loss of neuroretinal adaptation to the diabetic metabolic environment. Ann N Y Acad Sci. 2014;1311:174–90. doi:10.1111/nyas.12412.
Fisher M. Pericyte signaling in the neurovascular unit. Stroke. 2009;40(3 Suppl):S13–5. doi:10.1161/STROKEAHA.108.533117.
Mandarino LJ, Sundarraj N, Finlayson J, Hassell HR. Regulation of fibronectin and laminin synthesis by retinal capillary endothelial cells and pericytes in vitro. Exp Eye Res. 1993;57(5):609–21.
Cuevas P, Gutierrez-Diaz JA, Reimers D, Dujovny M, Diaz FG, Ausman JI. Pericyte endothelial gap junctions in human cerebral capillaries. Anat Embryol (Berl). 1984;170(2):155–9.
Carlson EC. Fenestrated subendothelial basement membranes in human retinal capillaries. Invest Ophthalmol Vis Sci. 1989;30(9):1923–32.
Tilton RG, Kilo C, Williamson JR. Pericyte-endothelial relationships in cardiac and skeletal muscle capillaries. Microvasc Res. 1979;18(3):325–35.
Armulik A, Abramsson A, Betsholtz C. Endothelial/pericyte interactions. Circ Res. 2005;97(6):512–23. doi:10.1161/01.RES.0000182903.16652.d7.
Kim JH, Yu YS, Kim DH, Kim KW. Recruitment of pericytes and astrocytes is closely related to the formation of tight junction in developing retinal vessels. J Neurosci Res. 2009;87(3):653–9. doi:10.1002/jnr.21884.
Shen W, Fruttiger M, Zhu L, Chung SH, Barnett NL, Kirk JK, et al. Conditional Mullercell ablation causes independent neuronal and vascular pathologies in a novel transgenic model. J Neurosci. 2012;32(45):15715–27. doi:10.1523/JNEUROSCI.2841-12.2012.
Gnanaguru G, Bachay G, Biswas S, Pinzon-Duarte G, Hunter DD, Brunken WJ. Laminins containing the beta2 and gamma3 chains regulate astrocyte migration and angiogenesis in the retina. Development. 2013;140(9):2050–60. doi:10.1242/dev.087817.
Stone J, Dreher Z. Relationship between astrocytes, ganglion cells and vasculature of the retina. J Comp Neurol. 1987;255(1):35–49. doi:10.1002/cne.902550104.
Fruttiger M, Calver AR, Kruger WH, Mudhar HS, Michalovich D, Takakura N, et al. PDGF mediates a neuron-astrocyte interaction in the developing retina. Neuron. 1996;17(6):1117–31.
Ashton N. Vascular basement membrane changes in diabetic retinopathy. Montgomery lecture, 1973. Br J Ophthalmol. 1974;58(4):344–66.
Garner A. Histopathology of diabetic retinopathy in man. Eye (Lond). 1993;7(Pt 2):250–3. doi:10.1038/eye.1993.58.
Ross MH, Reith EJ, Romrell LJ. Histology: a text and atlas. Baltimore: Williams and Williams; 1989.
Abrahamson DR. Recent studies on the structure and pathology of basement membranes. J Pathol. 1986;149(4):257–78. doi:10.1002/path.1711490402.
Lindahl P, Johansson BR, Leveen P, Betsholtz C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 1997;277(5323):242–5.
Gerhardt H, Betsholtz C. Endothelial-pericyte interactions in angiogenesis. Cell Tissue Res. 2003;314(1):15–23. doi:10.1007/s00441-003-0745-x.
Armulik A, Genove G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21(2):193–215. doi:10.1016/j.devcel.2011.07.001.
Leveen P, Pekny M, Gebre-Medhin S, Swolin B, Larsson E, Betsholtz C. Mice deficient for PDGF B show renal, cardiovascular, and hematological abnormalities. Genes Dev. 1994;8(16):1875–87.
Soriano P. Abnormal kidney development and hematological disorders in PDGF beta-receptor mutant mice. Genes Dev. 1994;8(16):1888–96.
Hellstrom M, Gerhardt H, Kalen M, Li X, Eriksson U, Wolburg H, et al. Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. J Cell Biol. 2001;153(3):543–53.
Enge M, Bjarnegard M, Gerhardt H, Gustafsson E, Kalen M, Asker N, et al. Endothelium-specific platelet-derived growth factor-B ablation mimics diabetic retinopathy. EMBO J. 2002;21(16):4307–16.
Bjarnegard M, Enge M, Norlin J, Gustafsdottir S, Fredriksson S, Abramsson A, et al. Endothelium-specific ablation of PDGFB leads to pericyte loss and glomerular, cardiac and placental abnormalities. Development. 2004;131(8):1847–57. doi:10.1242/dev.01080.
Geraldes P, Hiraoka-Yamamoto J, Matsumoto M, Clermont A, Leitges M, Marette A, et al. Activation of PKC-delta and SHP-1 by hyperglycemia causes vascular cell apoptosis and diabetic retinopathy. Nat Med. 2009;15(11):1298–306. doi:10.1038/nm.2052.
Boyer DS. The Ophthotech anti-PDGF in AMD study group. Combined inhibition of platelet derived (PDGF) and vascular endothelial (VEGF) growth factors for the treatment of neovascular age-related macular degeneration (NV-AMD)—results of a phase I study. Invest Ophthalmol Vis Sci. 2009;50 e-Abstract 1260.
Brown DM, Kaiser PK, Michels M, Soubrane G, Heier JS, Kim RY, et al. Ranibizumab versus verteporfin for neovascular age-related macular degeneration. N Engl J Med. 2006;355(14):1432–44. doi:10.1056/NEJMoa062655.
Chang TS, Bressler NM, Fine JT, Dolan CM, Ward J, Klesert TR. Improved vision-related function after ranibizumab treatment of neovascular age-related macular degeneration: results of a randomized clinical trial. Arch Ophthalmol. 2007;125(11):1460–9. doi:10.1001/archopht.125.11.1460.
Brown DM, Regillo CD. Anti-VEGF agents in the treatment of neovascular age-related macular degeneration: applying clinical trial results to the treatment of everyday patients. Am J Ophthalmol. 2007;144(4):627–37. doi:10.1016/j.ajo.2007.06.039.
Regillo CD, Brown DM, Abraham P, Yue H, Ianchulev T, Schneider S, et al. Randomized, double-masked, sham-controlled trial of ranibizumab for neovascular age-related macular degeneration: PIER study year 1. Am J Ophthalmol. 2008;145(2):239–48. doi:10.1016/j.ajo.2007.10.004.
Abraham P, Yue H, Wilson L. Randomized, double-masked, sham-controlled trial of ranibizumab for neovascular age-related macular degeneration: PIER study year 2. Am J Ophthalmol. 2010;150(3):315–24 e1. doi:10.1016/j.ajo.2010.04.011.
Mousa SA, Mousa SS. Current status of vascular endothelial growth factor inhibition in age-related macular degeneration. BioDrugs. 2010;24(3):183–94. doi:10.2165/11318550-000000000-00000.
Jo N, Mailhos C, Ju M, Cheung E, Bradley J, Nishijima K, et al. Inhibition of platelet-derived growth factor B signaling enhances the efficacy of anti-vascular endothelial growth factor therapy in multiple models of ocular neovascularization. Am J Pathol. 2006;168(6):2036–53. doi:10.2353/ajpath.2006.050588.
Spaide RF. Rationale for combination therapy in age-related macular degeneration. Retina. 2009;29(6 Suppl):S5–7. doi:10.1097/IAE.0b013e3181ad237a.
Chaoran Z, Zhirong L, Gezhi X. Combination of vascular endothelial growth factor receptor/platelet-derived growth factor receptor inhibition markedly improves the antiangiogenic efficacy for advanced stage mouse corneal neovascularization. Graefes Arch Clin Exp Ophthalmol. 2011;249(10):1493–501. doi:10.1007/s00417-011-1709-6.
Sato Y, Rifkin DB. Inhibition of endothelial cell movement by pericytes and smooth muscle cells: activation of a latent transforming growth factor-beta 1-like molecule by plasmin during co-culture. J Cell Biol. 1989;109(1):309–15.
Antonelli-Orlidge A, Saunders KB, Smith SR, D’Amore PA. An activated form of transforming growth factor beta is produced by cocultures of endothelial cells and pericytes. Proc Natl Acad Sci U S A. 1989;86(12):4544–8.
Carvalho RL, Jonker L, Goumans MJ, Larsson J, Bouwman P, Karlsson S, et al. Defective paracrine signalling by TGFbeta in yolk sac vasculature of endoglin mutant mice: a paradigm for hereditary haemorrhagic telangiectasia. Development. 2004;131(24):6237–47. doi:10.1242/dev.01529.
Dickson MC, Martin JS, Cousins FM, Kulkarni AB, Karlsson S, Akhurst RJ. Defective haematopoiesis and vasculogenesis in transforming growth factor-beta 1 knock out mice. Development. 1995;121(6):1845–54.
Larsson J, Goumans MJ, Sjostrand LJ, van Rooijen MA, Ward D, Leveen P, et al. Abnormal angiogenesis but intact hematopoietic potential in TGF-beta type I receptor-deficient mice. EMBO J. 2001;20(7):1663–73. doi:10.1093/emboj/20.7.1663.
Chang H, Huylebroeck D, Verschueren K, Guo Q, Matzuk MM, Zwijsen A. Smad5 knockout mice die at mid-gestation due to multiple embryonic and extraembryonic defects. Development. 1999;126(8):1631–42.
Yang X, Castilla LH, Xu X, Li C, Gotay J, Weinstein M, et al. Angiogenesis defects and mesenchymal apoptosis in mice lacking SMAD5. Development. 1999;126(8):1571–80.
Li DY, Sorensen LK, Brooke BS, Urness LD, Davis EC, Taylor DG, et al. Defective angiogenesis in mice lacking endoglin. Science. 1999;284(5419):1534–7.
Van Geest RJ, Klaassen I, Vogels IM, Van Noorden CJ, Schlingemann RO. Differential TGF-{beta} signaling in retinal vascular cells: a role in diabetic retinopathy? Invest Ophthalmol Vis Sci. 2010;51(4):1857–65. doi:10.1167/iovs.09-4181.
Kita T, Hata Y, Arita R, Kawahara S, Miura M, Nakao S, et al. Role of TGF-beta in proliferative vitreoretinal diseases and ROCK as a therapeutic target. Proc Natl Acad Sci U S A. 2008;105(45):17504–9. doi:10.1073/pnas.0804054105.
Gerhardinger C, Dagher Z, Sebastiani P, Park YS, Lorenzi M. The transforming growth factor-beta pathway is a common target of drugs that prevent experimental diabetic retinopathy. Diabetes. 2009;58(7):1659–67. doi:10.2337/db08-1008.
A randomized trial of sorbinil, an aldose reductase inhibitor, in diabetic retinopathy. Sorbinil Retinopathy Trial Research Group. Arch Ophthalmol. 1990;108(9):1234–44.
McAuley AK, Sanfilippo PG, Hewitt AW, Liang H, Lamoureux E, Wang JJ, et al. Vitreous biomarkers in diabetic retinopathy: a systematic review and meta-analysis. J Diabetes Complicat. 2014;28(3):419–25. doi:10.1016/j.jdiacomp.2013.09.010.
Davis S, Aldrich TH, Jones PF, Acheson A, Compton DL, Jain V, et al. Isolation of angiopoietin-1, a ligand for the TIE2 receptor, by secretion-trap expression cloning. Cell. 1996;87(7):1161–9.
Sundberg C, Kowanetz M, Brown LF, Detmar M, Dvorak HF. Stable expression of angiopoietin-1 and other markers by cultured pericytes: phenotypic similarities to a subpopulation of cells in maturing vessels during later stages of angiogenesis in vivo. Lab Investig. 2002;82(4):387–401.
Sato TN, Tozawa Y, Deutsch U, Wolburg-Buchholz K, Fujiwara Y, Gendron-Maguire M, et al. Distinct roles of the receptor tyrosine kinases Tie-1 and Tie-2 in blood vessel formation. Nature. 1995;376(6535):70–4. doi:10.1038/376070a0.
Suri C, Jones PF, Patan S, Bartunkova S, Maisonpierre PC, Davis S, et al. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell. 1996;87(7):1171–80.
Koblizek TI, Weiss C, Yancopoulos GD, Deutsch U, Risau W. Angiopoietin-1 induces sprouting angiogenesis in vitro. Curr Biol. 1998;8(9):529–32.
Hammes HP, Lin J, Wagner P, Feng Y, Vom Hagen F, Krzizok T, et al. Angiopoietin-2 causes pericyte dropout in the normal retina: evidence for involvement in diabetic retinopathy. Diabetes. 2004;53(4):1104–10.
Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science. 1997;277(5322):55–60.
Moss A. The angiopoietin:Tie 2 interaction: a potential target for future therapies in human vascular disease. Cytokine Growth Factor Rev. 2013;24(6):579–92. doi:10.1016/j.cytogfr.2013.05.009.
Pfister F, Feng Y, vom Hagen F, Hoffmann S, Molema G, Hillebrands JL, et al. Pericyte migration: a novel mechanism of pericyte loss in experimental diabetic retinopathy. Diabetes. 2008;57(9):2495–502. doi:10.2337/db08-0325.
Feng Y, vom Hagen F, Pfister F, Djokic S, Hoffmann S, Back W, et al. Impaired pericyte recruitment and abnormal retinal angiogenesis as a result of angiopoietin-2 overexpression. Thromb Haemost. 2007;97(1):99–108.
Patan S. TIE1 and TIE2 receptor tyrosine kinases inversely regulate embryonic angiogenesis by the mechanism of intussusceptive microvascular growth. Microvasc Res. 1998;56(1):1–21. doi:10.1006/mvre.1998.2081.
Uemura A, Ogawa M, Hirashima M, Fujiwara T, Koyama S, Takagi H, et al. Recombinant angiopoietin-1 restores higher-order architecture of growing blood vessels in mice in the absence of mural cells. J Clin Invest. 2002;110(11):1619–28. doi:10.1172/JCI15621.
Cai J, Kehoe O, Smith GM, Hykin P, Boulton ME. The angiopoietin/Tie-2 system regulates pericyte survival and recruitment in diabetic retinopathy. Invest Ophthalmol Vis Sci. 2008;49(5):2163–71. doi:10.1167/iovs.07-1206.
Thurston G, Suri C, Smith K, McClain J, Sato TN, Yancopoulos GD, et al. Leakage-resistant blood vessels in mice transgenically overexpressing angiopoietin-1. Science. 1999;286(5449):2511–4.
Kern TS, Engerman RL. Galactose-induced retinal microangiopathy in rats. Invest Ophthalmol Vis Sci. 1995;36(2):490–6.
Burns SA, Elsner AE, Chui TY, Vannasdale Jr DA, Clark CA, Gast TJ, et al. In vivo adaptive optics microvascular imaging in diabetic patients without clinically severe diabetic retinopathy. Biomed Opt Express. 2014;5(3):961–74. doi:10.1364/BOE.5.000961.
Steinle JJ, Kern TS, Thomas SA, McFadyen-Ketchum LS, Smith CP. Increased basement membrane thickness, pericyte ghosts, and loss of retinal thickness and cells in dopamine beta hydroxylase knockout mice. Exp Eye Res. 2009;88(6):1014–9. doi:10.1016/j.exer.2008.12.015.
Chui TY, Gast TJ, Burns SA. Imaging of vascular wall fine structure in the human retina using adaptive optics scanning laser ophthalmoscopy. Invest Ophthalmol Vis Sci. 2013;54(10):7115–24. doi:10.1167/iovs.13-13027.
Diaz-Flores L, Gutierrez R, Madrid JF, Varela H, Valladares F, Acosta E, et al. Pericytes. Morphofunction, interactions and pathology in a quiescent and activated mesenchymal cell niche. Histol Histopathol. 2009;24(7):909–69.
Herman IM, D’Amore PA. Microvascular pericytes contain muscle and nonmuscle actins. J Cell Biol. 1985;101(1):43–52.
DeNofrio D, Hoock TC, Herman IM. Functional sorting of actin isoforms in microvascular pericytes. J Cell Biol. 1989;109(1):191–202.
Fujimoto T, Singer SJ. Immunocytochemical studies of desmin and vimentin in pericapillary cells of chicken. J Histochem Cytochem. 1987;35(10):1105–15.
Nehls V, Denzer K, Drenckhahn D. Pericyte involvement in capillary sprouting during angiogenesis in situ. Cell Tissue Res. 1992;270(3):469–74.
Bondjers C, Kalen M, Hellstrom M, Scheidl SJ, Abramsson A, Renner O, et al. Transcription profiling of platelet-derived growth factor-B-deficient mouse embryos identifies RGS5 as a novel marker for pericytes and vascular smooth muscle cells. Am J Pathol. 2003;162(3):721–9. doi:10.1016/S0002-9440(10)63868-0.
Cho H, Kozasa T, Bondjers C, Betsholtz C, Kehrl JH. Pericyte-specific expression of Rgs5: implications for PDGF and EDG receptor signaling during vascular maturation. FASEB J. 2003;17(3):440–2. doi:10.1096/fj.02-0340fje.
Schlingemann RO, Rietveld FJ, de Waal RM, Ferrone S, Ruiter DJ. Expression of the high molecular weight melanoma-associated antigen by pericytes during angiogenesis in tumors and in healing wounds. Am J Pathol. 1990;136(6):1393–405.
Ozerdem U, Grako KA, Dahlin-Huppe K, Monosov E, Stallcup WB. NG2 proteoglycan is expressed exclusively by mural cells during vascular morphogenesis. Dev Dyn. 2001;222(2):218–27. doi:10.1002/dvdy.1200.
Carter RA, Wicks IP. Vascular cell adhesion molecule 1 (CD106): a multifaceted regulator of joint inflammation. Arthritis Rheum. 2001;44(5):985–94. doi:10.1002/1529-0131(200105)44:5<985::AID-ANR176>3.0.CO;2-P.
Kuwabara T, Cogan DG. Studies of retinal vascular patterns. I. Normal architecture. Arch Ophthalmol. 1960;64:904–11.
Laver NM, Robison Jr WG, Pfeffer BA. Novel procedures for isolating intact retinal vascular beds from diabetic humans and animal models. Invest Ophthalmol Vis Sci. 1993;34(6):2097–104.
Zhang Q, Guy K, Pagadala J, Jiang Y, Walker RJ, Liu L, et al. Compound 49b prevents diabetes-induced apoptosis through increased IGFBP-3 levels. Invest Ophthalmol Vis Sci. 2012;53(6):3004–13. doi:10.1167/iovs.11-8779.
Kuwabara T, Cogan DG. Retinal vascular patterns. VI. Mural cells of the retinal capillaries. Arch Ophthalmol. 1963;69:492–502.
Chung K, Wallace J, Kim SY, Kalyanasundaram S, Andalman AS, Davidson TJ, et al. Structural and molecular interrogation of intact biological systems. Nature. 2013;497(7449):332–7. doi:10.1038/nature12107. The CLARITY technique creates optically transparent tissue while retaining subcellular structures. The resultant tissue preparation can be further visualized and analyzed, enabling intact tissue in situ hybridization, immunohistochemistry, and antibody labeling of the intact tissue or organ.
Poguzhelskaya E, Artamonov D, Bolshakova A, Vlasova O, Bezprozvanny I. Simplified method to perform CLARITY imaging. Mol Neurodegener. 2014;9(1):19. doi:10.1186/1750-1326-9-19.
Susaki EA, Tainaka K, Perrin D, Kishino F, Tawara T, Watanabe TM, et al. Whole-brain imaging with single-cell resolution using chemical cocktails and computational analysis. Cell. 2014;157(3):726–39. doi:10.1016/j.cell.2014.03.042.
Geng Y, Dubra A, Yin L, Merigan WH, Sharma R, Libby RT, et al. Adaptive optics retinal imaging in the living mouse eye. Biomed Opt Express. 2012;3(4):715–34. doi:10.1364/BOE.3.000715.
Schallek J, Geng Y, Nguyen H, Williams DR. Morphology and topography of retinal pericytes in the living mouse retina using in vivo adaptive optics imaging and ex vivo characterization. Invest Ophthalmol Vis Sci. 2013;54(13):8237–50. doi:10.1167/iovs.13-12581. These new imaging technologies can be used to noninvasively visualize retinal pericytes in the living eye.
Li Y, Smith D, Li Q, Sheibani N, Huang S, Kern T, et al. Antibody-mediated retinal pericyte injury: implications for diabetic retinopathy. Invest Ophthalmol Vis Sci. 2012;53(9):5520–6. doi:10.1167/iovs.12-10010.
Madsen-Bouterse S, Mohammad G, Kowluru RA. Glyceraldehyde-3-phosphate dehydrogenase in retinal microvasculature: implications for the development and progression of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2010;51(3):1765–72. doi:10.1167/iovs.09-4171.
Alikhani M, Roy S, Graves DT. FOXO1 plays an essential role in apoptosis of retinal pericytes. Mol Vis. 2010;16:408–15.
Jadeja S, Mort RL, Keighren M, Hart AW, Joynson R, Wells S, et al. A CNS-specific hypomorphic Pdgfr-beta mutant model of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2013;54(5):3569–78. doi:10.1167/iovs.12-11125.
Dagher Z, Park YS, Asnaghi V, Hoehn T, Gerhardinger C, Lorenzi M. Studies of rat and human retinas predict a role for the polyol pathway in human diabetic retinopathy. Diabetes. 2004;53(9):2404–11.
Zheng L, Szabo C, Kern TS. Poly(ADP-ribose) polymerase is involved in the development of diabetic retinopathy via regulation of nuclear factor-kappaB. Diabetes. 2004;53(11):2960–7.
Beltramo E, Nizheradze K, Berrone E, Tarallo S, Porta M. Thiamine and benfotiamine prevent apoptosis induced by high glucose-conditioned extracellular matrix in human retinal pericytes. Diabetes Metab Res Rev. 2009;25(7):647–56. doi:10.1002/dmrr.1008.
Acknowledgments
This study received NIH grants EY005318 (P.A.D.) and EY021624 (J.A.-V.), American Diabetes Association Innovation Award 7-12-IN-11 (P.A.D.), and American Heart Association Scientist Development Grant 12SDG8960025 (J.A.-V).
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
Joseph F. Arboleda-Velasquez, Cammi Valdez, Christina Kaiser Marko, and Patricia A. D’Amore declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Microvascular Complications—Retinopathy
Rights and permissions
About this article

Cite this article
Arboleda-Velasquez, J.F., Valdez, C.N., Marko, C.K. et al. From Pathobiology to the Targeting of Pericytes for the Treatment of Diabetic Retinopathy. Curr Diab Rep 15, 5 (2015). https://doi.org/10.1007/s11892-014-0573-2
Published:
DOI: https://doi.org/10.1007/s11892-014-0573-2