
Abstract
Microsatellite instability (MSI) refers to the biochemical detection of frameshifted microsatellite sequences from genomic DNA. Genesis of MSI is due to defective DNA mismatch repair (MMR) that fails to correct post-DNA replicative slippage mistakes at microsatellites. Most of the estimated 100,000 genomic microsatellites are non-coding; however, ∼150–300 microsatellites are coding such that, when frameshifted during the pathogenesis of an MSI tumor, they can generate immunogenic neopeptide antigens that limit the growth of tumor and prolong patient survival. In addition to the immune reaction and longer survival, patients with MSI colorectal cancers tend to have poorly differentiated tumors with mucinous features that are located in the right colon. Patients with MSI tumors are more resistant to 5-fluorouracil-based adjuvant chemotherapy but may be responsive to PD-1 immune checkpoint blockade. Specific defects of MMR function not only drive MSI but also elevate microsatellite alterations at selected tetranucleotide repeats that may further modify patient outcome.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
One of the most significant discoveries over the past two-and-a-half decades for the understanding of colorectal cancer (CRC) pathogenesis has been the identification of microsatellite instability (MSI), a biomarker from human tumor tissue [1, 2, 3•]. It is now recognized that MSI is likely one of the three pathways (the other two are chromosomal instability and the CpG island methylator phenotype, itself overlapping with MSI pathogenesis) that generates genomic driver mutations for the formation of CRC and is observed in ∼15% of all CRCs [1,2,3,4,5]. This discovery in human cancer was made possible by investigations originally made in bacterial systems (yielding the 2015 Nobel Prize for Chemistry) that showed that DNA microsatellite sequence fidelity after DNA replication is maintained by a specific system termed DNA mismatch repair (MMR) [6•]. Once it was recognized in humans, there was rapid association with the inherited cancer syndrome we now call Lynch syndrome, which is defined by a monoallelic germline mutation in a MMR gene [1, 7•, 8]. Furthermore, defective MMR became recognized as the driver pathway in a subset of sporadic CRCs, and additional studies demonstrated different biological behavior for MSI cancers that now informs daily medical practice [1, 2, 3•, 9].
Microsatellite DNA, defined as repeated sequences of 1–6 nucleotides in a repeated contiguous fashion of typically 6 to perhaps more than 40 times (e.g., mononucleotide A n , dinucleotide [CA] n , trinucleotide [CAG] n , tetranucleotide [AAAG] n , where n represents the number of repeats of the sequence), is present ∼100,000 times in the human genome [1, 2]. They are thought to be sites for homologous recombination. The vast majority of microsatellites are in non-coding DNA, with only ∼150–300 within coding sequences. Measurement for frameshift of any of these microsatellite sequences might be thought of collectively as microsatellite instability, but MSI is historically defined by frameshifts at mononucleotide and dinucleotide repeats [1]. When one examines a panel of at least 5 mono- and dinucleotide microsatellite markers, the presence of 20% or greater with frameshifts (termed MSI-high) fairly accurately corresponds with a defect in MMR function, particularly from the MMR proteins MSH2 or MLH1. Inactivation of MSH2 and MLH1, the most common proteins affected in Lynch syndrome, causes complete loss of MMR function; MLH1 is also the principal protein affected in sporadic MSI colorectal cancers [1, 2, 3•]. When between 0 and 20% of markers show frameshifts, this is termed MSI-low, and the frameshift is almost exclusively in the dinucleotide markers and not mononucleotide markers [1, 10•, 11]. Although MSH6 dysfunction can generate mono- and dinucleotide frameshifts, the absence of any mononucleotide frameshifts characteristic of MSI-low is most likely associated with isolated MSH3 dysfunction [10•, 11]. Lack of any mono- or dinucleotide markers with frameshifts defines microsatellite-stable (MSS) CRCs, equating to no observable defect in MMR function [1, 10•, 12]. Measurement of frameshifts from tri- or tetranucleotide microsatellites is exclusively a result of MSH3 dysfunction and at present is termed elevated microsatellite alterations at selected tetranucleotide repeats, or EMAST [3,13,14,•, 10•, 12–15].
Generation of Microsatellite Instability: Defective DNA Mismatch Repair
Human MMR is an evolutionarily conserved DNA repair system that is maximally operative after the replication of DNA in cells and functions to accurately and faithfully repair single nucleotide base mispairs and slippage mistakes at microsatellite sequences [1, 2, 3•, 6•]. Thus, when MMR is not functional, single base mutations and MSI result [1, 2]. MMR proteins are encoded for by MMR genes, and the proteins form heterodimers to properly function. MSH2 is a key protein that partners with MSH6 as well as MSH3, and MLH1 pairs with PMS2. The fidelity for repair of DNA lies with the two MSH2 complexes: MSH2-MSH6 recognizes single base mispairs as well as slippages at mono- and dinucleotide sequences, and MSH2-MSH3 recognizes slippages at dinucleotide or longer repeats. The MLH1-PMS2 complex binds to the MSH2-MSH6 and MSH2-MSH3 complexes and signals the cell to target repair (triggering use of an exonuclease that removes the affected bases from the newly synthesized DNA strand, followed by re-synthesis via DNA polymerase) or demise (if the DNA damage is overwhelming, thus maintaining the fidelity of DNA by preventing an inaccurate and changed genome for daughter cells) [1, 2, 3•]. Loss of MMR function through mutation or inactivation of any of the MMR genes or proteins can be detected by MSI assays and depends on which MMR gene/protein is affected. Figure 1 demonstrates the type of MSI that results with each dysfunctional MMR complex. For instance, MSH2, MLH1, and PMS2 defects show total absence of MMR at any of the microsatellite sequences, whereas MSH6 or MSH3 defects are more specific, with MSH6 dysfunction at mono- and dinucleotide sequences (as well as at single base pairs) and MSH3 defective at dinucleotide or longer repeats [1, 2, 3•, 10•, 13].

Spectrum of MMR gene and protein defects and category of frameshift microsatellite mutation to define MSI-H, MSI-L, and EMAST
With the understanding of the fidelity of the two MSH2 recognition complexes and the function of MMR, one can infer which MMR protein complex is dysfunctional based on the type of MSI detected biochemically. MSI-low, detected by generally 1 dinucleotide frameshift out of five mono- and dinucleotide markers, is consistent with MSH3 dysfunction in the absence of any mononucleotide marker frameshifts; this hypothesis has been experimentally confirmed [3•, 10•, 11, 13, 14]. EMAST detection using tetranucleotide markers is consistent with isolated MSH3 dysfunction [3,17,•, 6•, 11, 13, 14, 16–18]. The presence of both mono- and dinucleotide frameshifts can be attributed to MSH2, MLH1, PMS2, or MSH6 dysfunction [10•]. Dysfunction of MSH2, MLH1, and PMS2 not only causes tri- and tetranucleotide frameshifts, like those seen with MSH3 dysfunction, but also shows mononucleotide frameshifts (Fig. 1). The best way to detect which MMR protein complex is not functioning is to perform MSI analysis using mono-, di-, and tetranucleotide markers; however, most commercial entities only assay mononucleotide or mono- and dinucleotide markers.
The MMR complexes are heterodimers of two MMR proteins that keep each individual protein stable from decay [1, 2, 3•, 19]. With MSH2 mutated, for instance, its partners MSH6 and MSH3 become unstable since its binding partner is not present. However, if MSH6 alone is mutated, for instance, MSH2 can still partner with MSH3 and thus MSH2 remains stable. If MLH1 is not transcribed as a full protein, PMS2 becomes unstable. MLH1 can also bind to one or more other MMR proteins that are not thought to function in pre-mitotic MMR; thus, PMS2 mutation may not cause loss of MLH1. These MMR protein patterns can be seen by immunohistochemistry of CRCs to help determine which MMR protein is not expressed.
MSI Presence in Human Colorectal Cancer
Microsatellite instability (MSI) is generated when MMR function is compromised. There are five human conditions described to date in which MMR is dysfunctional: two germline causes and three somatic causes (Table 1) [6•, 7•, 8, 20•]. Among germline causes, autosomal dominant transmission of a monoallelic germline mutation in a MMR gene causes Lynch syndrome, the most common inherited form of CRC, and found in 3% of all CRC patients based on population studies [6•, 7•, 20•]. Lynch families may demonstrate CRC and other gastrointestinal tract cancers such gastric tumors, biliary tract cancers, cancers of the female reproductive tract (particularly endometrial and ovarian cancers), cancers of the urinary tract, specific skin tumors, and glioblastomas. These patients often present 10–30 years younger than sporadic CRC patients. Lynch families with MSH6 or PMS2 mutations tend to present at later ages (50s to 60s) as compared to families with MSH2 or MLH1 mutations that are more common (40s) [7•, 20•]. Germline mutation of MSH3 as a cause of Lynch syndrome has yet to be described; such patients likely have a presentation phenotype indistinguishable from sporadic CRC patients [10•]. Another germline cause for MSI is the autosomal recessive inheritance of the constitutional mismatch repair deficiency (CMMRD) syndrome (Table 1). These very rare patients inherit biallelic germline MMR gene mutations, one from each parent, to cause a virulent and early presentation of CRC associated with café au lait spots, typically <10 years of age [6•, 21, 22]. Most commonly, patients carry biallelic PMS2 or MSH6 mutations, as their Lynch syndrome parents may have childbeared prior to knowledge of carrying a mutation. Biallelic MSH3 mutations have been described; consistent with the mild (or null) presentation of patients with sporadic monoallelic MSH3 mutation, patients with biallelic MSH3 germline mutation present after age 30 years—much later in age than CMMRD caused by PMS2 or MSH6 biallelic mutations—and demonstrate colonic adenomatous polyposis [23•]. Lynch CRCs and some adenomas demonstrate MSI, whereas in CMMRD both tumors and normal tissue demonstrate MSI [6•, 21, 22]. Those CMMRD patients with biallelic MSH3 germline mutations show EMAST and lack mononucleotide frameshifts [23•].
Somatic MMR dysfunction is seen in (a) sporadic MSI CRCs; (b) double somatic mutation of MMR genes, sometime referred to Lynch-like syndrome; and (c) EMAST CRCs. Sporadic MSI CRCs, about 12–15% of all CRCs, demonstrate biallelic hypermethylation of the MLH1 gene promoter, preventing the transcription of MLH1 [1,25,26,27,, 2, 3•, 24–28]. This completely inactivates MMR function to manifest MSI. Sporadic MSI CRCs may show mutations in the oncogene BRAF in ∼25% of cases, distinguishing themselves from Lynch CRCs which lack BRAF mutations (Table 1) [3•, 7•, 8, 9]. Sporadic MSI CRC patients more often, but not exclusively, present past 70 years of age and trend towards female gender [2, 3•, 29]. Lynch-like patients present on average in their 50s, younger than sporadic MSI CRC patients but older than Lynch syndrome patients. These patients lack a germline MMR mutation, but the CRCs show two somatic hits to any of the MMR genes, typically by mutation in one allele and loss of heterozygosity of the non-mutated MMR gene allele, and may be seen in ∼1% of all CRCs [3•, 8, 30•, 31]. EMAST CRCs are driven by any pathogenic pathway (MSI, chromosomal instability, or CpG island methylator phenotype) [3•, 6•, 9, 10•, 12, 17, 18]. Best studied in MSS CRC patients (eliminating the effects of total complete loss of MMR function) [6•, 10•, 16, 32•, 33•], EMAST CRCs demonstrate isolated loss of MSH3 function, not by mutation or epimutation, but by change in MSH3 cellular location shifting from the nucleus (where MMR acts to survey and repair DNA) to the cytosol [13, 34•]. Loss of MSH3 from the nucleus allows genomic frameshifts at tetranucleotide sequences to be detected [13, 34•]. EMAST CRCs are the most common demonstration of loss of MMR among all CRCs, occurring in ∼50% of all tumors (Table 1). The trigger for the nuclear-to-cytosol shift appears to be inflammation and oxidative stress and, in particular, interleukin-6 signaling [3•, 13, 34•].
MSI the Biomarker
MSI-H CRCs are a result of loss of function of MMR (particularly MSH2, MLH1, MSH6, and PMS2 proteins). The presence of MSI-H in a CRC is associated with certain histological and patient characteristics. MSI-H CRCs tend to be poorly differentiated and demonstrate mucin production and subepithelial lymphoid aggregates (sometimes referred to as a “Crohns-like reaction” as it simulates the non-caseating granuloma formation in this disease) [1, 2, 3•, 35]. Approximately 70% of all MSI-H CRCs are located in the right colon (transverse, ascending, and cecum portions) [1, 2, 3•, 6•, 7•, 35]. Overall survival is higher in patients with MSI-H CRCs as compared to patients with MSS CRCs [1, 2, 3•, 36, 37, 38•].
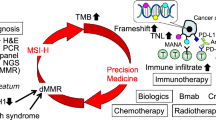
MSI-H CRCs are hypermutated, meaning that the cancer has accumulated hundreds to a thousand somatic mutations in its genome, as compared to MSS CRCs, which accumulate only tens of mutations [3•, 4]. As expected from a defect in MMR, there are single base mutations as well as frameshift mutations in the cancer genome [3•, 4]. Whether a patient inherits a germline mutation in a MMR gene, acquires methylation of the MLH1 promoter, or acquires two somatic MMR gene hits in the CRC, a colonic crypt stem cell must be affected to propagate the defect, coupled with disruption of Wnt signaling to generate an adenoma (Fig. 2) [1, 2, 3•]. With defective MMR, the ability to mutate is accelerated greatly, and it is believed that this rapid mutation ability shortens the typical time frame for adenoma-to-carcinoma formation from 1–2 decades to 1–2 years [1, 2, 3•, 5, 8, 39]. Sporadic MSI-H CRCs, but not Lynch CRCs, may further acquire activating mutations in the oncogene BRAF [3•, 7•, 8, 9]. Following inactivation of MMR genes, mutations continually accumulate in the cancer genome, as there is no ability to repair post-DNA replicative base substitutions or slippage mistakes at microsatellite sequences (Fig. 2) [1, 2, 3•, 6•]. Although most frameshift mutations that accumulate are in non-coding regions, as many as 300 microsatellites are in coding regions [1, 2, 3•]. This fact is believed to be the driver of improved outcome for patients with MSI-H CRCs. The frameshifted genes are transcribed and translated as shorter novel peptides due to the frameshift and new stop codon (Fig. 2) [1,41,42,43,44,, 2, 3•, 40–45]. These neopeptides are immunogenic and are recognized by the immune system, which causes the development of subepithelial lymphoid aggregates. Lymphocytes that respond to the neopeptides then contain and/or limit the spread of the tumor [1, 3•, 46]. Essentially, MSI-H CRC patients immunize themselves with the neopeptides, which is likely the principal mechanism that extends patient survival [3,47,•, 46–48]. It is probable that some of the frameshifted genes are driver genes for the pathogenesis of MSI-H CRCs (e.g., ACVR2, TGFBR2) [1, 2, 3•, 40, 41], while others are passenger genes, with both driver and passenger frameshifted genes contributing to the immune induction [46,47,48].

Genetic path of colonic neoplasia with loss of MMR function
EMAST CRCs with isolated MSH3 protein dysfunction show a markedly different histological and patient characteristic pattern than MSI-H CRCs. EMAST CRCs demonstrate a high prevalence of intraepithelial lymphocytes, showing an intimate association with the epithelial cancer cells, which supports the notion of localized interleukin-6 ligand generation and interaction [3,17,•, 6•, 10•, 11, 16–18]. EMAST CRCs also show a decreased heterogeneous expression of MSH3 from within the nuclei of CRC cells [10•, 13, 15, 17, 18]. Patients with EMAST CRCs are associated with a higher prevalence of metastases and decreased patient survival [3•, 6•, 12, 16, 33•, 39]. The prevalence of EMAST is higher among African American CRC patients than in Caucasian patients [3•, 6•, 10•, 16, 29, 39, 49]. Available evidence suggests that once a CRC is formed, inflammation may occur to generate interleukin-6, triggering nuclear-to-cytosol shift of MSH3 within the cancer cell (accounting for the observed decreased nuclear expression), causing a loss-of-function of MSH3 (generating EMAST and DNA double strand breaks), which ultimately causes more aggressive cell behavior and generation of metastasis and contributes to poor patient survival [10,13,14,15,•, 12–16, 18, 33•, 34•, 50, 51].
It should be noted that the biochemical detection of tetranucleotide frameshifts (EMAST) can be observed with complete loss of MMR (i.e., MLH1, MSH2, PMS2 inactivation) in addition to the isolated loss-of-function of MSH3 (Fig. 1). Patient outcome is determined by which of these scenarios is present within the tumor. As depicted in Fig. 3, an MSI-H CRC generates neopeptides that immunize the patient against the tumor, and even if inflammation occurs to shift MSH3 protein out of the nucleus, the MSI-H phenotype is more important for patient outcome, with improved survival over patients with MSS CRCs and no apparent effect from any MSH3 nuclear shift. However, a MSS CRC does not generate immunogenic neopeptides like the MSI-H CRC, and if inflammation occurs that can cause MSH3 to shift out of the nucleus, the CRC acquires the EMAST phenotype that increases metastatic behavior and worsens patient outcome (Fig. 3) [6•, 10•].

Schema showing the effect of inflammation on MMR-defective and MMR-intact cancers
Clinically, immunohistochemistry is commonly used in most pathology labs to determine the presence of the MMR proteins MSH2, MLH1, MSH6, and PMS2 [1, 2, 3•, 7•, 9, 20•]. The absence of one or more of these proteins may be caused by either a sporadic or germline mutation. MSI can be detected in certain specialty laboratories commercially, and typically tests for mono- and dinucleotide repeat instability only [1, 2, 3•, 7•, 9, 20•]. MSH3 protein is not routinely assayed for in pathology labs, and tetranucleotide microsatellites are not typically tested for in MSI assays. Thus, EMAST is not assayed for outside of the research setting at present.
MSI and Modification of Treatment Approaches for Patients With CRC
In addition to MSI being a prognostic marker, MSI can be used to direct patient therapy. The MMR recognition complexes recognize and bind to ingested or parenteral 5-fluorouracil (5FU) and are then converted and incorporated into DNA. Both MSH2-MSH6 and MSH2-MSH3 complexes can bind 5FU within DNA and trigger cell death [52,53,54,55,56,57,58,59, 60•]. When the MMR recognition complexes are not present, cell death does not occur and cells may proceed with mitosis with 5FU-induced and other novel mutations contained within DNA. The ability of MMR to trigger cell death after 5FU recognition is 30-fold higher than when MMR is absent [52]. Retrospective and prospective studies on stage II and III MSI-H CRC patients as well as Lynch syndrome CRC patients confirm there is no increased survival time with adjuvant 5FU therapy, as compared to increased survival among MSS CRC patients [53, 54]. Current clinical practice refrains from using 5FU monotherapy for stage II MSI-H CRC patients [55, 56]. At present, stage III MSI-H CRC patients are offered adjuvant 5FU in combination with oxliplatin and/or irinotecan (both of which show activity against MSI-H CRC cells in culture), but generally not 5FU alone or solely with leucovorin. Interestingly, the nature of an MMR-deficient, MSI-H tumor may modify the resistance to 5FU in some cases. MSI-H CRC cells in which the DNA glycosylase MBD4 is frameshift mutated (which removes its glycosylase domain while keeping its DNA-binding domain and occurs in 20–35% of MSI-H CRCs) re-acquires some sensitivity to 5FU through a yet-to-be-determined mechanism [57, 58].
Patients with stage II/III EMAST CRCs (i.e., those with MSS tumors and isolated MSH3 dysfunction) respond to adjuvant 5FU therapy to the same degree as non-EMAST MSS CRC patients. This is likely because MSH2-MSH6 is fully present and functional to bind 5FU within DNA to trigger cell death despite the MSH2-MSH3 dysfunction [32•].
The immunogenic nature of MSI-H CRCs goes beyond the generation of neopeptides from frameshifted coding microsatellite sequences as a consequence of defective MMR and activation of T lymphocytes. The majority of MSI-H CRCs acquire expression of the immune checkpoint molecule PD-1, which prevents immune destruction of the cancer cell by cytotoxic lymphocytes when engaged with PD-L1 [47•]. This makes MSI-H CRCs more susceptible to immune checkpoint blockade with PD-1 antibodies [48, 59, 60•]. Indeed, patients with deficient MMR tumors showed 78% progression-free survival as compared to 11% for patients with proficient MMR tumors and anti-PD-1 more than doubled the average survival of MSI-H patients [59, 60•]. Thus, immune checkpoint blockade could further improve the overall good survival of MSI-H CRC patients, in contrast to the limited efficacy of adjuvant 5FU.
Conclusions
Since its discovery as a part of human disease, MSI is proving to be a valuable biomarker for the diagnosis, prognosis, and determination of treatment approaches in CRC patients. Defective MMR generates MSI and hypermutated cancers, and detection via immunohistochemistry and mono-, di-, tri-, and tetranucleotide microsatellite frameshifts helps determine which germline or somatic MMR-defective condition is present (although only mono- and dinucleotide microsatellites are assayed currently commercially). Immunorecognition of neopeptides improves survival of MSI-H CRC patients; in contrast, isolated MSH3 dysfunction that causes EMAST worsens outcome for MSS patients. This outcome difference depends on if the MMR defect drives an immune response, or if acquired inflammation causes disruption of MMR via MSH3 cellular mislocation. Loss of MMR function not only prevents repair at single nucleotides and microsatellite sequences but also prevents recognition of altered nucleotides such as 5FU that gets incorporated into DNA. MSI-H CRCs often acquire immune checkpoint molecule expression which may be exploited therapeutically to prolong patient survival.
References
Papers of particular interest, published recently, have been highlighted as: • of importance
Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. 2010;138:2073–87.
Grady WM, Carethers JM. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology. 2008;135:1079–99.
• Carethers JM, Jung BH. Genetics and genetic biomarkers in sporadic colorectal cancer. Gastroenterology. 2015;149:1177–90. Recent review on pathways for sporadic colorectal cancer. Includes how biomarkers are formed during the pathways and how they might be used clinically for prognostic, diagnostic, and therapeutic approaches.
Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7.
Fearon ER, Carethers JM. Molecular subtyping of colorectal cancer—time to explore both intertumoral and intratumoral heterogeneity to evaluate patient outcome. Gastroenterology. 2015;148:10–3.
• Carethers JM. Hereditary, sporadic and metastatic colorectal cancers are commonly driven by specific spectrums of defective DNA mismatch repair components. Trans Am Clin Climatol Assoc. 2016;127:81–97. Review on five human conditions with defective DNA mismatch repair.
• Carethers JM, Stoffel EM. Lynch syndrome and Lynch syndrome mimics: the growing complex landscape of hereditary colon cancer. World J Gastroenterol. 2015;21:9253–61. Review of several mimics of Lynch syndrome and how to differentiate them based on genetic biomarkers.
Carethers JM. Differentiating Lynch-like from Lynch syndrome. Gastroenterology. 2014;146:602–4.
Carethers JM. DNA testing and molecular screening for colon cancer. Clin Gastroenterol Hepatol. 2014;12:377–81.
• Carethers JM, Koi M, Tseng-Rogenski S. EMAST is a form of microsatellite instability that is initiated by inflammation and modulates colorectal cancer progression. Genes. 2015;6:185–205. Only published review of EMAST and how it is generated and its implications as a biomarker.
Huang SC, Lee JK, Smith EJ, Doctolero R, Tajima A, Beck SE, et al. Evidence for an hMSH3 defect in familial hamartomatous polyps. Cancer. 2011;117:492–500.
Garcia M, Choi C, Kim HR, Daoud Y, Toiyama Y, Takahashi M, et al. Association between recurrent metastasis from stage II and III primary colorectal tumors and moderate microsatellite instability. Gastroenterology. 2012;143:48–50.
Tseng-Rogenski S, Chung H, Wilk MB, Zhang S, Iwaizumi M, Carethers JM. Oxidative stress induces nuclear-to-cytosol shift of hMSH3, a potential mechanism for EMAST in colorectal cancer cells. PLoS One. 2012;7(11):e50616.
Campregher C, Schmid G, Ferk F, Knasmüller S, Khare V, Kortüm B, et al. MSH3-deficiency initiates EMAST without oncogenic transformation of human colon epithelial cells. PLoS One. 2012;7:e50541.
Haugen AC, Goel A, Yamada K, Marra G, Nguyen TP, Nagasaka T, et al. Genetic instability caused by loss of MutS homologue 3 in human colorectal cancer. Cancer Res. 2008;68:8465–72.
Devaraj B, Lee A, Cabrera BL, et al. Relationship of EMAST and microsatellite instability among patients with rectal cancer. J Gastrointest Surg. 2010;14:1521–8.
Lee S-Y, Chung H, Devaraj B, Iwaizumi M, Han HS, Hwang D-Y, et al. Elevated microsatellite alterations at selected tetranucleotide repeats are associated with morphologies of colorectal neoplasia. Gastroenterology. 2010;139:1519–25.
Lee S-Y, Miyai K, Han HS, Hwang D-Y, Seong MK, Chung H, et al. Microsatellite instability, EMAST, and morphology associations with T cell infiltration in colorectal neoplasia. Dig Dis Sci. 2012;57:72–8.
Chang DK, Ricciardiello L, Goel A, Chang CL, Boland CR. Steady-state regulation of the human DNA mismatch repair system. J Biol Chem. 2000;275:29178.
• Stoffel EM, Boland CR. Genetics and genetic testing in hereditary colorectal cancer. Gastroenterology. 2015;149:1191–203. Updated review on common inherited syndromes that lead to colorectal cancer.
Kratz CP, Holter S, Etzler J, Lauten M, Pollett A, Niemeyer CM, et al. Rhabdomyosarcoma in patients with constitutional mismatch-repair-deficiency syndrome. J Med Genet. 2009;46:418–20.
Jasperson KW, Samowitz WS, Burt RW. Constitutional mismatch repair-deficiency syndrome presenting as colonic adenomatous polyposis: clues from the skin. Clin Genet. 2011;80:394–7.
• Adam R, Spier I, Zhao B, Kloth M, Marquez J, Hinrichsen I, et al. Exome sequencing identifies biallelic MSH3 germline mutations as a recessive subtype of colorectal adenomatous polyposis. Am J Hum Genet. 2016;99:337–51. Very first description of biallelic MSH3 mutations, with contributor parents having monoallelic MSH3 mutations, but no phenotype.
Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, et al. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997;57:808–11.
Herman JG, Umar A, Polyak K, Graff JR, Ahuja N, Issa JP, et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci U S A. 1998;95:6870–5.
Veigl ML, Kasturi L, Olechnowicz J, Ma AH, Lutterbaugh JD, Periyasamy S, et al. Biallelic inactivation of hMLH1 by epigenetic gene silencing, a novel mechanism causing human MSI cancers. Proc Natl Acad Sci U S A. 1998;95:8698–702.
Miyakura Y, Sugano K, Konishi F, Ichikawa A, Maekawa M, Shitoh K, et al. Extensive methylation of hMLH1 promoter region predominates in proximal colon cancer with microsatellite instability. Gastroenterology. 2001;121:1300–9.
Nakagawa H, Nuovo GJ, Zervos EE, Martin Jr EW, Salovaara R, Aaltonen LA, et al. Age-related hypermethylation of the 5′ region of MLH1 in normal colonic mucosa is associated with microsatellite-unstable colorectal cancer development. Cancer Res. 2001;61:6991–5.
Carethers JM, Murali B, Yang B, Doctolero RT, Tajima A, Basa R, et al. Influence of race on microsatellite instability and CD8+ T cell infiltration in colon cancer. PLoS One. 2014;9:e100461.
• Mensenkamp AR, Vogelaar IP, van Zelst-Stams WA, Goossens M, Ouchene H, Hendriks-Cornelissen SJ, et al. Somatic mutations in MLH1 and MSH2 are a frequent cause of mismatch-repair deficiency in Lynch syndrome-like tumors. Gastroenterology. 2014;146:643–646.e8. One of the first descriptions of double somatic hits to cause the genotype of microsatellite instability, but in the absence of Lynch syndrome and in absence of MLH1 hypermethylation.
Haraldsdottir S, Hampel H, Tomsic J, Frankel WL, Pearlman R, de la Chapelle A, et al. Colon and endometrial cancers with mismatch repair deficiency can arise from somatic, rather than germline, mutations. Gastroenterology. 2014;147:1308–16.
• Hamaya Y, Guarinos C, Tseng-Rogenski SS, Iwaizumi M, Das R, Jover R, et al. Efficacy of 5-fluorouracil adjuvant therapy for patients with EMAST-positive stage II/III colorectal cancers. PLoS One. 2015;10:e0127591. Retrospective analysis of more than 200 patients characterized by EMAST status, showing 5-fluorouracil is still effective in EMAST-positive patients.
• Koi M, Garcia M, Choi C, Kim H-R, Koike J, Hemmi H, et al. Microsatellite alterations with allelic loss on 9p24.2 signify less aggressive colorectal cancer metastasis. Gastroenterology. 2016;150:944–55. First description of potential modulators of EMAST, identified from metachronous liver metastasis of EMAST-positive stage II/III colorectal cancer patients.
• Tseng-Rogenski S, Hamaya Y, Choi D, Carethers JM. Interleukin 6 alters localization of hMSH3, leading to DNA mismatch repair defects in colorectal cancer cells. Gastroenterology. 2015;148:579–89. Elegant finding of MSH3 mislocalization in the presence of IL-6 induced inflammation.
Carethers JM. One colon lumen but two organs. Gastroenterology. 2011;141:411–2.
Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol. 2005;23:609–18.
Sinicrope FA, Shi Q, Smyrk TC, Thibodeau SN, Dienstmann R, Guinney J, et al. Molecular markers identify subtypes of stage III colon cancer associated with patient outcomes. Gastroenterology. 2015;148:88–99.
• Phipps AI, Limburg PJ, Baron JA, Burnett-Hartman AN, Weisenberger DJ, Laird PW, et al. Association between molecular subtypes of colorectal cancer and patient survival. Gastroenterology. 2015;148:77–87. Description of outcome of over 2000 patients based on genotyping colorectal cancer patients into 5 genetic categories.
Carethers JM. Screening for colorectal cancer in African Americans: determinants and rationale for an earlier age to commence screening. Dig Dis Sci. 2015;60:711–21.
Jung B, Doctolero RT, Tajima A, Nguyen AK, Keku T, Sandler RS, et al. Loss of activin receptor type 2 protein expression in microsatellite unstable colon cancers. Gastroenterology. 2004;126:654–9.
Jung BH, Beck SE, Cabral J, Chau E, Cabrera BL, Fiorino A, et al. Activin type 2 receptor restoration in MSI-H colon cancer suppresses growth and enhances migration with activin. Gastroenterology. 2007;132:633–44.
Chung H, Young DJ, Lopez C, Le T-AT, Lee JK, Ream-Robinson D, et al. Mutation rates of TGFBR2 and ACVR2 coding microsatellites in human cells with defective DNA mismatch repair. PLoS One. 2008;3:e3463.
Chung H, Lopez CG, Young DJ, Lai JF, Holmstrom J, Ream-Robinson D, et al. Flanking sequence specificity determines coding microsatellite heteroduplex and mutation rates with defective DNA mismatch repair. Oncogene. 2010;29:2172–80.
Chung H, Lopez CG, Holmstrom J, Young DJ, Lai JF, Ream-Robinson D, et al. Both microsatellite length and sequence context determine frameshift mutation rates in defective DNA mismatch repair. Hum Mol Genet. 2010;19:2638–4.
Chung H, Chaudhry J, Lai JF, Young DJ, Carethers JM. Flanking nucleotide specificity for DNA mismatch repair-deficient frameshifts within activin receptor 2 (ACVR2). Mutat Res. 2012;729:73–80.
Schwitalle Y, Kloor M, Eiermann S, Linnebacher M, Kienle P, Knaebel HP, et al. Immune response against frameshift-induced neopeptides in HNPCC patients and healthy HNPCC mutation carriers. Gastroenterology. 2008;134:988–97.
• Mlecnik B, Bindea G, Angell HK, Maby P, Angelova M, Tougeron D, et al. Integrative analyses of colorectal cancer show immunoscore is a stronger predictor of patient survival than microsatellite instability. Immunity. 2016;44:698–711. Comprehensive immunogenic analysis of colorectal cancers. Highlights acquirement of microsatellite unstable tumors for PD-1 expression.
Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015;5:43–51.
Ashktorab H, Ahuja S, Kannan L, Llor X, Ellis N, Xicola RM, et al. A meta-analysis of MSI frequency and race in colorectal cancer. Oncotarget. 2016;7:34546–57.
van Oers JM, Edwards Y, Chahwan R, Zhang W, Smith C, Pechuan X, et al. The MutSβ complex is a modulator of p53-driven tumorigenesis through its functions in both DNA double-strand break repair and mismatch repair. Oncogene. 2014;33:3939–46.
Dietlein F, Thelen L, Jokic M, Jachimowicz RD, Ivan L, Knittel G, et al. A functional cancer genomics screen identifies a druggable synthetic lethal interaction between MSH3 and PRKDC. Cancer Discov. 2014;4:592–605.
Carethers JM, Chauhan DP, Fink D, Nebel S, Bresalier RS, Howell SB, et al. Mismatch repair proficiency and in vitro response to 5-fluorouracil. Gastroenterology. 1999;117:123–31.
Ribic CM, Sargent DJ, Moore MJ, Thibodeau SN, French AJ, Goldberg RM, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med. 2003;349:247–57.
Carethers JM, Smith EJ, Behling CA, Nguyen L, Tajima A, Doctolero RT, et al. Use of 5-fluorouracil and survival in patients with microsatellite unstable colorectal cancer. Gastroenterology. 2004;126:394–401.
Srivastava G, Renfro LA, Behrens RJ, Lopatin M, Chao C, Soori GS, et al. Prospective multicenter study of the impact of oncotype DX colon cancer assay results on treatment recommendations in stage II colon cancer patients. Oncologist. 2014;19:492–7.
Tajima A, Hess MT, Cabrera BL, Kolodner RD, Carethers JM. The mismatch repair complex hMutS recognizes 5-fluorouracil-modified DNA: implications for chemosensitivity and resistance. Gastroenterology. 2004;127:1678–84.
Suzuki S, Iwaizumi M, Tseng-Rogenski S, Hamaya Y, Miyajima H, Kanaoka S, et al. Production of truncated MBD4 protein by frameshift mutation in DNA mismatch repair-deficient cells enhances 5-fluorouracil sensitivity that is independent of hMLH1 status. Cancer Biol Ther. 2016;17:760–80.
Iwaizumi M, Tseng-Rogenski S, Carethers JM. DNA mismatch repair proficiency executing 5-fluorouracil cytotoxicity in colorectal cancer cells. Cancer Biol Ther. 2011;12:756–64.
Tajima A, Iwaizumi M, Tseng-Rogenski S, Cabrera BL, Carethers JM. Both hMutS and hMutS complexes participate in 5-fluorouracil cytotoxicity. PLoS One. 2011;6:e28117.
• Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–20. First-ever description of PD-1 blockade showing increased survival in patients with microsatellite unstable tumors.
Acknowledgements
This work was supported by the US Public Health Service (R01 DK067287, U01 CA162147, and R01 CA206010) and the A. Alfred Taubman Medical Research Institute of the University of Michigan. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The author declares that he has no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by the author.
Additional information
This article is part of the Topical Collection on Basic Science Foundations in Colorectal Cancer
Rights and permissions
About this article

Cite this article
Carethers, J.M. Microsatellite Instability Pathway and EMAST in Colorectal Cancer. Curr Colorectal Cancer Rep 13, 73–80 (2017). https://doi.org/10.1007/s11888-017-0352-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11888-017-0352-y
Keywords
- Microsatellite instability
- EMAST (elevated microsatellite alterations at selected tetranucleotide repeats)
- DNA mismatch repair
- Lynch syndrome
- Constitutional mismatch repair deficiency syndrome
- Sporadic
- Familial
- Colorectal cancer
- Lynch-like syndrome
- DNA microsatellites
- DNA repair
- Patient outcome
- Patient survival
- MLH1 hypermethylation
- Mononucleotide repeats
- Dinucleotide repeats
- Trinucleotide repeats
- Tetranucleotide repeats
- MSH3