
Abstract
Metastatic colorectal cancer (CRC) is a significant cause of morbidity and mortality around the world. Novel cytotoxic and biologic therapies have been developed; however, their optimal use in terms of patient selection, drug combinations, and regimen sequences must be better defined. The FDA-approved drugs include fluoropyrimidines (5-fluorouracil (5-FU) with or without leucovorin (LV), capecitabine), irinotecan, oxaliplatin, the vascular endothelial growth factor (VEGF) antibody bevacizumab, the epidermal growth factor receptor (EGFR) antibodies cetuximab and panitumumab for RAS wild-type patients, the VEGF receptors 1 and 2 fusion protein aflibercept and the multitarget tyrosine kinase inhibitor regorafenib. As a result, metastatic colorectal cancer median overall survival can now be as long as 33 months and up to 70 % of patients will receive at least two lines of treatment. Recent scientific and technologic advances in the field of metastatic colorectal cancer promise to elucidate the biological underpinnings of this disease and its therapies for the goal of improving personalized treatments for patients with metastatic colorectal cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Around one quarter of patients with CRC presents with metastatic disease at the time of diagnosis, and up to 40 % of patients will develop metastases during the course of their disease, resulting in a relatively high overall mortality rate associated with CRC [1]. Median overall survival (OS) as long as 33 months have been reported and up to 70 % of patients will receive at least two lines of treatment [2–7]. Several drugs as single agent or in various combinations are available for mCRC, including fluoropyrimidines (5-FU, 5-FU/LV, capecitabine), irinotecan, oxaliplatin, the vascular endothelial growth factor (VEGF) antibody bevacizumab, the epidermal growth factor receptor (EGFR) antibodies cetuximab and panitumumab for RAS wild-type patients, the VEGF receptors 1 and 2 fusion protein aflibercept and the multitarget tyrosine kinase inhibitor regorafenib. Moreover, secondary resection and/or ablation, e.g., by surgery or radiofrequency may contribute to long-term survival and even cure, or at least allow a relevant chemotherapy free interval [8, 9].
The prognosis for patients with metastatic disease without specific therapy is very poor, with a median survival of 5 to 6 months. For decades, standard first-line therapy consisted of 5-FU/LV, with response rates of approximately 20 % and a median survival of approximately 9 months. In the late 1990s and early 2000s, the addition of oxaliplatin and irinotecan to the backbone of 5-FU/LV resulted in an improvement in median survival to nearly 24 months when patients received active first-line and second-line therapy. The sequence of active regimens, FOLFIRI or FOLFOX, appears not to influence on efficacy results and therapy was tailored based on toxicity. Most recently, biologic agents, such as bevacizumab, cetuximab, and panitumumab, have further enhanced the efficacy of systemic medical therapy [10].
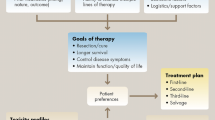
The availability of various active agents for the treatment of metastatic colorectal cancer has resulted in an abundance of therapeutic options that now demands a goal-oriented, strategic approach to maximize patient benefit. When treating a patient with metastatic colon cancer, the first determination is whether a patient with stage IV disease is potentially curable by a surgical resection of metastases either at the time of diagnosis or after downsizing initially unresectable metastases by neoadjuvant chemotherapy [10]. This will guide the choice and timing of chemotherapy because, in this scenario, the most appropriate treatment is conceivably the one that generates the highest response rates and carries the greatest potential to downsize metastases. If the patient does not appear curable, the main goals of systemic chemotherapy are to extend the duration of a patient’s life and to maintain quality of life as long as possible. In this scenario, treatment regimens that offer the longest progression-free and overall survivals, as well as a favorable toxicity profile, are preferred.
5-FU
Until 2000, standard first-line therapy for metastatic colon cancer was the fluoropyrimidine analog 5-FU and leucovorin (LV) was added as biomodulator and activator. LV forms a complex with 5-FU that permits prolonged inhibition of the enzyme thymidylate synthase, a key factor in the DNA synthesis. Response rates of 5-FU/LV are in the range of 15 to 25 %. More recently, the incorporation of irinotecan and oxaliplatin into 5-FU-based regimens has resulted in substantially better efficacy. This has shifted the paradigm for front-line treatment from 5-FU/LV alone to combination regimens incorporating these newer cytotoxic compounds.
Capecitabine
Capecitabine is an oral fluoropyrimidine, a prodrug of 5-FU, which is metabolized to its active form in three enzymatic steps. Its efficacy is similar to bolus 5-FU/LV, with higher response rates. Common side effects of capecitabine include diarrhea and hand-foot syndrome [11, 12]. Capecitabine has been used as backbone of combination regimens with both oxaliplatin and irinotecan, but overlapping toxicities specially diarrhea make a combination with irinotecan more difficult to tolerate than oxaliplatin. Although capecitabine has never been directly compared with infusional 5-FU/LV, oxaliplatin-based combination regimens with either capecitabine (CapeOX or XELOX) or infusional 5-FU/LV (FOLFOX) have shown to be of similar efficacy in the treatment of advanced colorectal cancer [13–15]. Dose-reducing capecitabine by approximately 20 % in combination regimens with oxaliplatin, however, does not appear to decrease the treatment efficacy but improves the side-effect profile [16].
Irinotecan
The first chemotherapy agent other than 5-FU that improved survival for metastatic colon cancer was irinotecan, which was used initially as a second-line treatment and then as a first line, in combination with 5-FU/LV [17–20]. Irinotecan is a camptothecin analog and acts on topoisomerase to prevent re-ligation of breaks in DNA. As a single-agent irinotecan yields approximately a 15 % response rate for patients with metastatic colon cancer refractory to 5-FU. Patients with 5-FU–refractory metastatic colon cancer were randomly selected to receive either best supportive care or single-agent irinotecan. The results of the trial demonstrated that irinotecan offers an approximate 3-month survival advantage as well as an improvement in quality of life [18]. In another second-line trial, irinotecan was also superior to infusional 5-FU [19]. Following this, three other trials were conducted to test the role of irinotecan in the front-line scenario. The trial comparing weekly bolus 5-FU/LV (Roswell Park regimen) to weekly bolus 5-FU/LV plus irinotecan (IFL), revealed a greater than 2-month survival advantage (14.8 vs. 12.6 months; p = 0.04) and an almost doubling of response rate (39 vs. 21 %; p < 0.001) for patients receiving the three-drug regimen compared with those receiving the regimen of bolus 5-FU/LV. This study established the three-drug regimen as the then-standard of care in the USA [20]. In Europe, two phase III trials were conducted in which 5-FU was given as an infusion in combination with irinotecan (FOLFIRI regimen). The results demonstrated a similar significant increase in response rate and time to disease progression for the three-drug regimen [21, 22]. However, only the trial reported by Douillard et al. demonstrated significant prolongation of overall survival (17.4 vs. 14.1 months; p = 0.031), likely because of the limited availability of active second- and third-line treatment options compared with the other European trial, which was conducted later [22].
The replacement of infusional 5FU by capecitabine in combination with irinotecan seems to cause more toxicity. BICC-C trial, CapeIRI showed worse median PFS than FOLFIRI (5.8 vs. 7.6 months; P = 0.015), and was considerably more toxic with higher rates of severe vomiting, diarrhea, and dehydration [40]. In this trial, the CapeIRI arm was discontinued. The EORTC study 40015 also compared FOLFIRI with CapeIRI and was discontinued after enrollment of only 85 patients because 7 deaths were determined to be treatment-related (5 in the CapeIRI arm) [78]. On the other hand, a randomized phase III HeCOG trial compared CapeIRI/Bev and FOLFIRI/Bev in the first-line metastatic setting and found no significant differences in efficacy between the regimens [79]. Despite the differing toxicity profiles reported, the toxicities seemed to be similar in both arms. However, because of the concerns about the toxicity of the CapeIRI combination, which may differ between American and European patients CapeIRI should not be recommended until new data can demonstrate safety and efficacy of this regimen.
The main toxicities of irinotecan are diarrhea, myelosuppression, and alopecia. Irinotecan is converted by an enzyme into its active metabolite SN-38, which is in turn inactivated by the enzyme UGT1A1 by glucuronidation. People with variants of the UGT1A1 called TA7, also express fewer UGT1A1 enzymes in their liver. During chemotherapy, they effectively receive a larger than expected dose because their bodies are not able to clear irinotecan, which can cause a higher incidence of severe neutropenia and diarrhea [23].
Oxaliplatin
Oxaliplatin is a platinum derivative which has activity in colorectal cancer, somewhat surprisingly given that cisplatin is inactive. The toxicities produced by oxaliplatin are also different from those seen with cisplatin. Nausea, vomiting, and renal impairment, which are dose limiting with cisplatin, are not such major problems with oxaliplatin. The principal and dose-limiting toxicity is a predominantly sensory peripheral neuropathy. Initially, these symptoms are transient and associated with cold, but after 4–5 months of treatment, the symptoms become constant. Slow improvement occurs after cessation of treatment. In addition to the peripheral neuropathy, a cold-related laryngopharyngeal dysaesthesia may occur which can be alarming to patients. Apart from the neuropathy, the main other toxicity associated with oxaliplatin is mild myelosuppression [24].
Although oxaliplatin has very limited activity in colorectal cancer as a single agent, it shows enhanced clinical efficacy in combination with fluoropyrimidines, in particular with infusional 5-FU/LV. In three European phase III trials, combination protocols of infusional 5-FU/LV plus oxaliplatin (biweekly FOLFOX or weekly FUFOX) were compared with 5-FU/LV as first-line therapy for patients with advanced colorectal cancer [24–26]. In all three studies, a better antitumor activity was noted for the combination regimens, with response rates of approximately 50 % and progression-free survival in the range of 8 to 9 months. However, this higher efficacy did not translate into a significantly improved overall survival, most likely because of the availability of active salvage therapies for both treatment arms, which obscured the effects of the first-line chemotherapy on the overall survivals in the trials as the median overall survival achieved with FOLFOX was in the range of 17.5 to 20 months, the longest overall survival reported in phase III trials for advanced CRC at that time. Because no overall survival benefit was achieved in these first-line trials, FDA only approved FOLFOX in 2002, based on the results of a second-line study that showed prolonged progression-free survival and increased response rates compared with infusional 5-FU/LV for patients who experienced disease progression while receiving IFL as first-line therapy [27]. In this trial, the arm with oxaliplatin as a single agent, without 5-FU, did not show any relevant tumor activity, which means that oxaliplatin must be combined with another agent, preferably a fluoropyrimidine.
The combination of capecitabine and oxaliplatin, known as CapeOx or XELOX, has been studied as an active first-line therapy for patients with metastatic colorectal cancer. In a randomized phase III trial comparing CapeOx and FOLFOX in 2,034 patients, the regimens showed similar median PFS intervals of 8.0 and 8.5 months, respectively, and CapeOx was determined to be noninferior to FOLFOX as first-line treatment of metastatic disease [28]. A recent meta-analysis of 3,603 patients from seven randomized controlled trials also showed that CapeOx and FOLFOX had similar benefits for patients with metastatic colorectal cancer [29].
Irinotecan Versus Oxaliplatin-Based Regimens
North Central Cancer Treatment Group/Intergroup trial N9741 [19] was the pivotal and practice-changing trial that compared FOLFOX and the nonfluorouracil-containing combination of irinotecan and oxaliplatin (IROX), as well as with standard combination IFL at that time [27]. The results of N9741 clearly demonstrated the superiority of FOLFOX compared with IFL as first-line therapy for colorectal cancer regarding response rate (45 vs. 31 %; p = 0.002), progression-free survival (8.7 vs. 6.9 months; p = 0.0014), and overall survival (19.5 vs. 15.0 months; p = 0.0001). The toxicity profile also favored FOLFOX compared with IFL, with only neurotoxicity being more prevalent for patients receiving the oxaliplatin-based combination. Results for IROX were in between the two other arms (response rate, 35 %; progression-free survival, 6.5 months; overall survival, 17.4 months), and, as such, FOLFOX emerged as new standard first-line therapy with rapid and widespread adaptation in the USA. As a matter of fact, N9741 did not directly compare oxaliplatin and irinotecan; rather, it compared two different combination regimens with different 5-FU/LV backbones. The higher efficacy and better tolerability observed with infusional 5-FU/LV may have contributed to the differences in efficacy between IFL and FOLFOX. IROX seems to be an alternative for patients with contraindications to receive fluoropirimidines.
Two small trials compared directly FOLFOX versus FOLFIRI with the same 5-FU/LV backbone and both failed to show significant differences in activity of the two regimens [30, 31]. Despite the fact that FOLFOX showed significantly higher efficacy compared with irinotecan and bolus 5-FU (IFL), when administered in combination with infusional 5-FU and leucovorin, irinotecan (FOLFIRI), and oxaliplatin (FOLFOX) appear to be equally efficacious in patients with previously untreated metastatic colorectal cancer. The choice for first-line therapy is then often guided by the regimens’ adverse effects and institutional practice. A phase III trial by Tournigand and associates [32, 33] compared FOLFOX with FOLFIRI with crossover at progression. Response rates (56 versus 54 %), median progression-free survival (8.5 versus 8.0 months), and median overall survival (21.5 versus 20.6 months) were almost identical. Because the benefit of second-line therapy in colorectal cancer has been well established, patients should receive all active cytotoxic drugs in the course of their therapy to optimize outcome.
Combination of 5-FU/LV with Irinotecan and Oxaliplatin: FOLFOXIRI
The triple therapy showed a high activity but also increased toxicity in a phase III trial. A total of 244 patients were randomly assigned. An increase of grade 2 to 3 peripheral neurotoxicity (0 vs. 19 %; P < 0.001), and grade 3 to 4 neutropenia (28 vs. 50 %; P < 0.001) were observed in the FOLFOXIRI arm. The incidence of febrile neutropenia (3 vs. 5 %) and grade 3 to 4 diarrhea (12 vs. 20 %) were not significantly different. Responses, as assessed by investigators, were, for FOLFIRI and FOLFOXIRI, respectively, complete, 6 and 8 %; and partial, 35 and 58 %, (RR, 41 vs. 66 %; P = 0.0002). RR confirmed by an external panel was 34 versus 60 % (P < 0.0001). The R0 secondary resection rate of metastases was greater in the FOLFOXIRI arm (6 vs. 15 %; P = 0.033, among all 244 patients; and 12 vs. 36 %; P = 0.017 among patients with liver metastases only). Progression-free survival (PFS) and overall survival (OS) were both significantly improved in the FOLFOXIRI arm (median PFS, 6.9 vs. 9.8 months; hazard ratio [HR], 0.63; P = 0.0006; median OS, 16.7 vs. 22.6 months; HR, 0.70; P = 0.032) [34]. The use of these combinations should be reserved for specific situations, for instance, when dramatic tumor shrinkage as a prerequisite for a surgical approach toward borderline resectable is required.
Biologics in Colon Cancer
Antiangiogenic Agents
Blood vessel structure differs between tumor tissue and normal tissue. Normal blood vessels are well organized, have well-aligned endothelial cells, and do not require the presence of survival factor for their integrity. However, tumor blood vessels are contorted, have misaligned endothelial layers, and have holes in their walls that create a higher interstitial pressure, which inhibits the delivery of chemotherapy into the tumor. In addition, immature tumor blood vessels need the constant presence of vascular endothelial growth factor (VEGF). VEGF is key in the angiogenic process, driving endothelial cell migration, proliferation, apoptosis, and vascular permeability. The VEGF family is composed of a number of different ligands that bind to receptors on the surface of endothelial cells. Of these ligands, VEGF-A plays the largest role in tumor angiogenesis by binding to VEGF receptor 2. Preclinical studies have shown that VEGF overexpression increases the number of blood vessels. However, microvessel density was reduced in the presence of a VEGF inhibitor, allowing increased penetration of chemotherapeutic agents [35].
Bevacizumab
Bevacizumab is a humanized monoclonal antibody that targets VEGF-A, which is the key ligand to activate VEGF receptor 2 (KDR). It has a half-life of approximately 3 weeks, which has implications for surgery because angiogenesis is necessary for wound repair [36].
A phase III trial in previously untreated patients with metastatic colorectal cancer showed that patients receiving IFL and bevacizumab had increased median progression-free survival time by 4.5 months compared to patients receiving IFL and placebo (P < 0.0001) [37]. The overall RR was increased by 10 % in the group receiving bevacizumab compared to the placebo group (P < 0.005). Median PFS (10.6 vs. 6.2 months; HR 0.54; p < 0.00001) and median OS (20.3 vs. 15.6 months; HR 0.66; p = 0.00004) were also superior. Analysis of a third arm of this phase III study, which contained bolus 5-FU/LV in combination with bevacizumab, suggested that bevacizumab added more benefit to 5-FU and LV than irinotecan. This trial was the first phase III validation of an antiangiogenic agent as an effective treatment option in a human malignancy. In the recently reported phase III AVEX trial the addition of bevacizumab to capecitabine prolonged PFS from 5.1 to 9.1 mo (HR = 0.53; 95 % CI, 0.41–0.69; P < 0.0001) and showed a strong trend in OS with an acceptable tolerability profile in patients with at least 70 years of age [80]. Table 1 shows the pivotal trials of bevacizumab with combination chemotherapy in the treatment of first-line metastatic CRC.
Several meta-analyses have shown a benefit for the use of bevacizumab in first-line therapy for metastatic colorectal cancer [81, 82•, 83–85]. A recent meta-analysis of six randomized clinical trials (3,060 patients) that assessed the efficacy of bevacizumab in first-line treatment of metastatic colorectal cancer found that bevacizumab gave a PFS (HR, 0.72; 95 % CI, 0.66–0.78; P < 0.00001) and OS (HR, 0.84; 95 % CI, 0.77–0.91; P < 0.00001) advantage [86]. However, subgroup analyses showed that the advantage was limited to irinotecan-based regimens. In addition, a recent analysis of the SEER-Medicare database found that bevacizumab added a modest improvement to OS of patients with stage IV colorectal cancer diagnosed between 2002 and 2007 (HR, 0.85; 95 % CI, 0.78–0.93) [87]. The survival advantage was not evident when bevacizumab was combined with oxaliplatin-based chemotherapy, but was evident in irinotecan-based regimens. Overall, the addition of bevacizumab to first-line chemotherapy appears to offer a modest clinical benefit.
Subsequently, bevacizumab also has been shown to enhance the efficacy of oxaliplatin-based regimens in first- and second-line settings, as well as in combination with 5-FU/LV alone or with irinotecan [38–42]. It is important to note that bevacizumab does not appear to have significant single-agent activity in metastatic colorectal cancer [39]. Data of a second-line phase III trial in bevacizumab-naive patients who had progressed on irinotecan-based first-line therapy showed that the addition of bevacizumab to FOLFOX therapy increased overall survival, increased progression-free survival, and improved the overall response rate by approximately 10 %. Bevacizumab monotherapy was shown to be inferior to FOLFOX monotherapy in this study [42].
The adverse events associated with bevacizumab therapy in phase III study included gastrointestinal perforation (1.5 to 2 % of the patients), arterial as arterial thrombotic events (4 to 5 % of the patients) and grade 3 hypertension. Hypertension appears to be a class effect of VEGF inhibitors that is manageable with antihypertensive therapy. Rates of grade 3 or 4 bleeding, venous thromboembolism, and grade 3 proteinuria were not significantly different between treatment groups [43]. In addition to arterial thrombotic events, a meta-analysis identified a 33 % higher incidence of venous thrombotic events in patients receiving bevacizumab compared with the those not receiving it in randomized trials, [41] although a more recent analysis refuted this claim [44]. Based on its well-documented efficacy and relative moderate toxicity, bevacizumab has emerged as a standard component of first-line chemotherapy for advanced colorectal cancer.
Bevacizumab Plus Folfoxiri
Results of the GONO group’s phase III TRIBE trial found that FOLFOXIRI/bevacizumab significantly increased PFS (12.2 vs. 9.7 months; P = 0.0012). A total of 508 patients were randomly assigned, with similar baseline characteristics among the treatment groups. After a median follow-up of 32.3 months, 439 patients (86 %) had progressed and 286 (56 %) had died. A significantly higher incidence of grade 3–4 neurotoxicity, diarrhea, stomatitis, and neutropenia was observed (p < 0.05) in patients receiving FOLFOXIRI/bevacizumab; the incidence of febrile neutropenia, serious adverse events, and treatment-related deaths were similar among the two groups. In the primary analysis, FOLFOXIRI/bevacizumab had significantly greater PFS (median 12.1 months) compared with FOLFIRI/bevacizumab (9.7 months, stratified hazard ratio [HR] 0.75, 95 % CI [0.62, 0.9]; p = 0.003). In subgroup analyses of PFS, no significant interactions between treatment groups and stratification factors of performance status, site of primary tumor, liver-only disease, type of metastases, resection of primary, and Kohne score were detected. However, a significant difference in PFS favoring FOLFOXIRI/bevacizumab was observed in patients who did not have prior adjuvant chemotherapy (HR 0.70; p = 0.006) compared with those who had (HR 1.3; p = 0.039) and response rate (65 vs. 53 %; P = 0.006) compared to FOLFIRI/ bevacizumab in patients with unresectable metastatic colorectal cancer [45, 46]. Subgroup analyses indicated that no benefit to the addition of oxaliplatin was seen in patients who received prior adjuvant therapy. Diarrhea, stomatitis, neurotoxicity, and neutropenia were significantly more prevalent in the FOLFOXIRI arm.
Results from the randomized phase II OLIVIA trial, which compared mFOLFOX6/bevacizumab to FOLFOXIRI/bevacizumab in patients with unresectable colorectal liver metastases, were recently reported [47]. Improvement in R0 resection rate was seen in the FOLFOXIRI/bevacizumab arm (49 vs. 23 %; P = 0.017). Because of toxicity this aggressive combination only should be used in very select patients who could potentially be converted to a resectable state.
Bevacizumab Plus Anti-EGFR
Initial reports suggested an over-additive activity when bevacizumab was combined with cetuximab in a salvage-therapy setting [48] but subsequent larger, randomized first-line trials, however, suggested an antagonistic effect of the combination of EGFR antibodies with bevacizumab in the context of concurrent chemotherapy [49, 50]. Thus, combinations of bevacizumab and EGFR antibodies should not be used in clinical practice unless other new data suggest the opposite.
Ziv-Aflibercept
Ziv-aflibercept is a recombinant protein that has part of the human VEGF receptors 1 and 2 fused to the Fc portion of human IgG1. It is designed to function as a VEGF trap to prevent activation of VEGF receptors and thus inhibit angiogenesis [96]. The VELOUR trial tested second-line ziv-aflibercept in patients with metastatic colorectal cancer that progressed after one regimen containing oxaliplatin. The trial met its primary endpoint with a small improvement in OS (13.5 months for FOLFIRI/ziv-aflibercept vs. 12.1 months for FOLFIRI/placebo; HR, 0.82; 95 % CI, 0.71–0.94; P = 0.003). A prespecified subgroup analysis from the VELOUR trial found that median OS in the ziv-aflibercept arm versus the placebo arm was 12.5 months (95 % CI, 10.8–15.5) versus 11.7 months (95 % CI, 9.8–13.8) in patients with prior bevacizumab treatment and 13.9 months (95 % CI, 12.7–15.6) versus 12.4 (95 % CI, 11.2–13.5) in patients with no prior bevacizumab treatment [97]. Ziv-aflibercept has only shown activity when given in conjunction with FOLFIRI in FOLFIRI-naïve patients. No data suggest activity of FOLFIRI plus ziv-aflibercept in patients who progressed on FOLFIRI plus bevacizumab or vice-versa, and no data suggest activity of single- agent ziv-aflibercept. The drug now has been tested as a maintenance therapy and in combination with FOLFOX as a first-line therapy of advanced disease.
Regorafenib
Regorafenib is a small molecule inhibitor of multiple kinases (including VEGF receptors, fibroblast growth factor [FGF] receptors, platelet- derived growth factor [PDGF] receptors, BRAF, KIT, and RET) that are involved with various processes including tumor growth and angiogenesis [98]. The phase III CORRECT trial randomized 760 patients who progressed on standard therapy to best supportive care with placebo or regorafenib [99]. The trial met its primary endpoint of OS (6.4 months for regorafenib vs. 5.0 months for placebo; HR, 0.77; 95 % CI, 0.64–0.94; P = 0.005). PFS was also significantly but modestly improved (1.9 vs. 1.7 months; HR, 0.49; 95 % CI, 0.42–0.58; P < 0.000001). The most common grade 3 or higher adverse events in the regorafenib arm of the CORRECT trial were hand-foot skin reaction (17 %), fatigue (10 %), hypertension (7 %), diarrhea (7 %), and rash/desquamation (6 %). Severe and fatal liver toxicity occurred in 0.3 % of 1,100 patients treated with regorafenib across all trials [98].
Currently, regorafenib is being investigated in combination with other agents for the treatment of mCRC. One open-label, international phase II trial evaluated the efficacy and safety of regorafenib when given with modified FOLFOX (mFOLFOX6) as first-line treatment in patients with metastatic CRC [100]. The study showed that regorafenib, when combined with mFOLFOX6, has an acceptable safety profile. While this study reported some efficacy for regorafenib and FOLFOX, definitive conclusions cannot be made from a single arm study. Another ongoing Phase II study is the randomized, multicenter, placebo- controlled US trial of regorafenib in combination with FOLFIRI as second-line treatment in patients with mCRC [101]. This trial is designed to compare PFS between regorafenib plus FOLFIRI versus placebo plus FOLFIRI in patients with mCRC who were previosly treated with a FOLFOX regimen.
Biological Therapy
Anti-EGFR Antibodies
Cetuximab and Panitumumab
Cetuximab and panitumumab are monoclonal antibodies directed against EGFR that inhibit its downstream signaling pathways. Panitumumab is a fully human monoclonal antibody, whereas cetuximab is a chimeric monoclonal antibody. Both monoclonal antibodies against the EGFR, cetuximab and panitumumab have single-agent efficacy in advanced colorectal cancer. Two US phase II trials confirmed the activity of cetuximab for the treatment of patients who had experienced disease progression on prior irinotecan-based therapy [51]. The single-agent response rate of approximately 10 % noted with cetuximab alone was in the same range as previously noted with FOLFOX in the same setting. A large international randomized phase III trial comparing cetuximab with cetuximab plus irinotecan confirmed the findings with almost identical results [52]. For patients who experienced progressive disease while receiving irinotecan-based therapy (with approximately two thirds of patients also refractory to oxaliplatin), cetuximab monotherapy induced responses for approximately 11 % of patients. When irinotecan was added, response rate and time to progression were significantly increased (HR 0.54, 95 % CI [0.42, 0.71]; p < 0.001). These data served as the basis for the initial approval of cetuximab as a treatment option for patients with metastatic colorectal cancer who were pretreated with irinotecan-based regimens.
Single-agent panitumumab was tested against best supportive care in a large international phase III trial in an extensively pretreated population; crossover was optional upon progression [53]. Panitumumab demonstrated similar single-agent activity to cetuximab, with an approximate 10 % response rate when used as salvage therapy after failure of standard chemotherapy. In comparison with best supportive care, it significantly prolonged progression-free survival (HR 0.54, 95 % CI [0.44, 0.66]; p < 0.0001). Overall survival was not increased, presumably because 75 % of patients crossed over from best supportive care to the panitumumab arm. Based on these data, panitumumab was approved as a single-agent salvage therapy option in the USA in 2006. A similar last-line trial comparing cetuximab with best supportive care (without crossover) showed almost identical results in terms of response rate and progression-free survival, but also with survival benefit for the cetuximab arm [54].
The main toxicities of anti-EGFR antibodies are skin rash, hypomagnesemia, diarrhea, and hypersensitivity reactions, which is particularly relevant for the chimeric antibody cetuximab. The incidence and severity of skin reactions with cetuximab and panitumumab seems to be very similar. Furthermore, the presence and severity of skin rash in patients receiving either of these drugs have been shown to predict increased response and survival [55]. Anaphylactic reactions to cetuximab have been correlated to the presence of preexisting serum IgE antibodies to an oligosaccharide, galactose-alpha-1,3-galactose, which is present on the Fab portion of the cetuximab heavy chain. Data from various clinical trials and translational studies now have opened the door toward individualized treatment approaches in colorectal cancer by identifying patients who are most likely to benefit from antibodies against the EGFR, cetuximab, and panitumumab.
The RAS/RAF/MAPK pathway is downstream of EGFR; mutations in components of this pathway are being studied in search of predictive markers for efficacy of these therapies. It increasingly appears that patients with advanced colorectal cancer must have tumor with wild-type KRAS for EGFR antibodies to be effective [56–61, 62•, 63, 64]. KRAS is a phosphorylated signal transducer that self-inactivates via intrinsic guanosine triphosphatase (GTPase) activity. The frequency of KRAS mutations in colorectal cancer is about 40 % [66]. A recent retrospective study from De Roock et al. [71] raised the possibility that codon 13 mutations (G13D) may not be absolutely predictive of non-response. However, a recent retrospective analysis of three randomized controlled phase III trials concluded that patients with KRAS G13D mutations were unlikely to respond to panitumumab [72].
More recently, mutated BRAF, encoding a protein downstream of KRAS, has also been correlated to lack of efficacy of EGFR antibodies in colorectal cancer, as well as with a poor prognosis, regardless the treatment. [61]. The frequency of BRAF mutations in colorectal cancer is approximately 5 to 9 %. Since KRAS and BRAF mutations are mutually exclusive [65], testing for BRAF and KRAS mutations can exclude about 50 % of patients with colorectal cancer from an ineffective but potentially harmful (and expensive) therapy with cetuximab and panitumumab. The validity of BRAF mutations as a negative predictive marker for the activity of EGFR antibodies has recently been challenged when the addition of cetuximab to FOLFIRI in a first-line setting demonstrated a numeric, but not statistically significant, improvement in response rate and time-related endpoints in BRAF-mutated colorectal cancers [57, 58]. All results obtained on BRAF demonstrate, however, that patients with BRAF mutations have a very poor prognosis, with overall survival in the range of 12 to 14 months in spite of modern systemic therapy [57, 58].
The clinical importance of KRAS mutations as predictors for efficacy of EGFR-targeted agents in colorectal cancer was highlighted in an analysis of the two phase III trials conducted with panitumumab and cetuximab as single-agent salvage therapy in colorectal cancer [60, 63]. Patients with KRAS-mutated cancers did not derive any benefit from EGFR antibody monotherapy. In contrast, cetuximab almost doubled overall survival compared with best supportive care for patients with wild-type KRAS colorectal cancer (9.5 vs. 4.8 months; HR for death 0.55, 95 % CI [0.41, 0.74]; p < 0.001)0.247 In randomized trials testing the addition of cetuximab or panitumumab to standard first-line therapy (FOLFIRI or FOLFOX), again, only patients with KRAS wild-type tumors benefited from EGFR antibodies [56, 59, 62•].
Extended RAS Analysis: NRAS and Other KRAS Mutations
Beyond mutations in exon 2, additional recurrent mutations lead to constitutive KRAS activation, including mutations in exon 3 (codon 61) and exon 4 (codons 117 and 146). It was recently reported that 17 % of 641 patients from the PRIME trial without KRAS exon 2 mutations were found to have mutations in exons 3 and 4 of KRAS or mutations in exons 2, 3, and 4 of NRAS. A predefined retrospective subset analysis revealed that PFS (HR, 1.31; 95 % CI, 1.07–1.60; P = 0.008) and OS (HR, 1.21; 95 % CI, 1.01-1.45; P = 0.04) were decreased in patients with any KRAS or NRAS mutation who received panitumumab plus FOLFOX compared to those who received FOLFOX alone [67]. These results show that panitumumab does not benefit patients with KRAS or NRAS mutations and may even have a detrimental effect in these patients. Updated analysis of the FIRE-3 trial (see below further discussion) which compared FOLFIRI plus either cetuximab or bevacizumab as first-line treatment in patients with metastatic CRC was recently presented and updated. When all RAS (KRAS/NRAS) mutations were considered, PFS was significantly worse in RAS-mutant patients receiving FOLFIRI plus cetuximab than RAS-mutant patients receiving FOLFIRI plus bevacizumab (6.1 vs. 12.2 months; P = 0.004). On the other hand, KRAS/NRAS wild-type patients showed no difference in PFS between the regimens (10.4 vs. 10.2 months; P = 0.54) but superior median OS (33.1 vs. 25.6 months) and ORR (55.5 vs. 72.5; P = 0.0063) [68, 69]. This result indicates that cetuximab likely has a detrimental effect in patients with KRAS or NRAS mutations. The FDA indication for panitumumab was recently updated to state that panitumumab is not indicated for the treatment of patients with KRAS or NRAS mutation-positive disease in combination with oxaliplatin-based chemotherapy [70].
Cetuximab with FOLFIRI
Use of cetuximab as initial therapy for metastatic disease was investigated in the CRYSTAL trial, in which patients were randomly assigned to receive FOLFIRI with or without cetuximab [57, 58]. Retrospective analyses of the subset of patients with known KRAS exon 2 tumor status showed a statistically significant improvement in median PFS with the addition of cetuximab in the group with disease characterized by KRAS wild-type exon 2 (9.9 vs. 8.7 months; HR, 0.68; 95 % CI, 0.50–0.94; P = 0.02) and the statistically significant benefit in PFS for patients with KRAS exon 2 wild-type tumors receiving cetuximab was confirmed in a recent publication of an updated analysis of the CRYSTAL data [59]. This recent study included a retrospective analysis of OS in the KRAS exon 2 wild-type population and found an improvement with the addition of cetuximab (23.5 vs. 20.0 months, P = 0.009). Importantly, the addition of cetuximab did not affect the quality of life of participants in the CRYSTAL trial [72].
Panitumumab with FOLFOX
Panitumumab in combination with FOLFOX has been studied in the first-line treatment of patients with metastatic colorectal cancer. Results from the large, open-label, randomized PRIME trial comparing panitumumab plus FOLFOX versus FOLFOX alone in patients with KRAS/NRAS wild-type advanced CRC [66]. In this prospective–retrospective analysis, efficacy and safety of panitumumab plus FOLFOX4 was compared with FOLFOX4 alone, according to RAS (KRAS or NRAS) or BRAF mutation status. A total of 639 patients who had metastatic colorectal cancer without KRAS mutations in exon 2 had results for at least one of the following: KRAS exon 3 or 4; NRAS exon 2, 3, or 4; or BRAF exon 15. The overall rate of ascertainment of RAS status was 90 %. Among 512 patients without RAS mutations, median PFS was 10.1 months with panitumumab–FOLFOX4 versus 7.9 months with FOLFOX4 alone (hazard ratio for progression or death with combination therapy, 0.72; 95 % confidence interval [CI], 0.58 to 0.90; P = 0.004). Median OS was 26.0 months in the panitumumab– FOLFOX4 group versus 20.2 months in the FOLFOX4-alone group (hazard ratio for death, 0.78; 95 % CI, 0.62 to 0.99; P = 0.04). A total of 108 patients (17 %) with non- mutated KRAS exon 2 had other RAS mutations. These mutations were associated with inferior progression-free survival and overall survival with panitumumab–FOLFOX4 treatment, which was consistent with the findings in patients with KRAS mutations in exon 2. BRAF mutations were a negative prognostic factor. No new safety signals were identified..
Cetuximab with FOLFOX or CapeOX
In a recent randomized phase III Medical Research Council (MRC) COIN trial, no benefit in OS (17.9 vs. 17.0 months; P = 0.067) or PFS (8.6 months in both groups; P = 0.60) was seen with the addition of cetuximab to FOLFOX or CapeOX as first-line treatment of patients with locally advanced or metastatic colorectal cancer and wild-type KRAS exon 2 [73]. Exploratory analyses of the COIN trial, however, suggest that there may be a benefit to the addition of cetuximab in patients who received FOLFOX instead of CapeOX [77]. Similarly, a recent pooled analysis of the COIN and OPUS studies found that a benefit was suggested in response rate and PFS with the addition of cetuximab to FOLFOX in KRAS exon 2 wild-type patients, although there was no OS benefit [74]. Notably, more recent trials examining the efficacity of the addition of cetuximab to oxaliplatin-containing regimens in the first-line treatment of patients with advanced or metastatic colorectal cancer and wild-type KRAS exon 2 have not shown any benefit. The addition of cetuximab to the Nordic FLOX regimen showed no benefit in OS or PFS in this population of patients in the randomized phase III NORDIC VII study of the Nordic Colorectal Cancer Biomodulation Group [75].
However, in a recent trial from the Cancer and Leukemia Group B/South West Oncology Group (CALGB/SWOG- Study 80405) suggested that chemotherapy with either FOLFOX or FOLFIRI was acceptable in combination with bevacizumab or cetuximab [76]. Overall survival of 29 or more months in both arms (see below further discussion).
Cetuximab or Bevacizumab Plus Chemotherapy in First-Line Treatment?
Cetuximab plus chemotherapy and bevacizumab plus chemotherapy were compared in two trials that have the results recently presented. The randomized, open-label, multicenter FIRE-3 trial from the German AIO group compared the efficacy of FOLFIRI plus cetuximab to FOLFIRI plus bevacizumab in first-line, KRAS exon 2 wild-type, metastatic disease [67]. The results presented were from a preplanned analysis that evaluated the effect of KRAS mutations in exon 3 (codon 59/61), exon 4 (codon 117/146), NRAS exon 2 (cordons 12/13), exon 3 (cordons 59/61), exon 4 (codons 117/146), and BRAF (V600E) on the overall response rate (ORR), PFS and OS on treatment arms A and B of the FIRE-3 trial. Mutational analyses were done by pyrosequencing in 592 patients who were wild type for KRAS exon 2. A total of 444 (75 %) patients had available tumour tissue; of these, sequencing of all RAS mutations was possible in 396 patients. Greater benefit was demonstrated with FOLFIRI plus cetuximab in the overall intent to treat population of 592 patients with KRAS wild type disease; ORR was 62.0 and 58.0 % in arm A and B, respectively (p = 0.183 [Fisher’s one-sided test]). Median OS was 28.7 months with FOLFIRI/cetuximab versus 25.0 months with FOLFIRI/bevacizumab (logrank p = 0.017). Median PFS was similar in both study arms; 10.0 months in arm A versus 10.3 months in arm B (p = 0.547). However, more recent analysis at the 16th World Congress on Gastrointestinal Cancer (WCGC) [68] showed that 301 patients within the wild type RAS subgroup (wild-type KRAS exon 2/3/4 and NRAS exon 2/3/4) has shown significantly higher overall RR (72.5 vs. 55.5 %; p = 0.0063) and rate of early tumour shrinkage (ETS) (69.2 vs. 47.4 %) (p = 0.0006) in the subset of patients within FIRE-3 with RAS wild-type metastatic colorectal cancer (mCRC) with cetuximab plus chemotherapy (n = 120) compared to bevacizumab plus chemotherapy (n = 137) (p = 0.0006); this represents a 45.9 % increase in ETS in the cetuximab plus FOLFIRI arm compared to the bevacizumab plus FOLFIRI. ETS is defined as the reduction in tumour diameter by at least 20 % at first assessment after baseline (week 6), whereas DpR is defined as the maximum tumour percent shrinkage observed in a patient greater ORR of 76.0 % with FOLFIRI/cetuximab over patients who received FOLFIRI/bevacizumab (ORR = 65.2 %; Fischer’s one-sided p = 0.026, Fischer’s two-sided p = 0.044). KRAS/NRAS wild-type patients showed no difference in PFS between the regimens (10.4 vs. 10.2 months; P = 0.54) but superior median OS which was even higher (33.1 vs. 25.6 months) [69]. This result indicates that cetuximab likely has a detrimental effect in patients with KRAS or NRAS mutations. Although several criticisms have raised about this trial, including regarding the lack of third-party review and low rates of second-line therapy, these data represent the first phase III findings demonstrating the influence of KRAS exon 3,4 and NRAS exon 2,3,4 mutations on the efficacy of first-line treatment with FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab in patients with mCRC and wild type KRAS (exon 2).
The second trial was the CALGB/SWOG 80405, which was designed in 2004, involving 14 amendments and 11 interim analyses as the field of colorectal cancer treatment evolved over 10 years. At the time it was designed, bevacizumab and cetuximab had been only recently approved [76]. The study involved 1,137 previously untreated KRAS wild-type (codons 12, 13) metastatic colorectal cancer patients. Patients were randomly assigned to chemotherapy plus either bevacizumab (5 mg/kg every 2 weeks) or cetuximab (400 mg/m2 initial dose, then 250 mg/m2 weekly). The selection of chemotherapy was based on physician preference: 73.4 % received FOLFOX and 26.6 % FOLFIRI at a median follow-up of 24 months, no significant differences were observed in either overall survival or progression-free survival between the treatment groups, which was 29 and 10.8 months, respectively, with bevacizumab/chemotherapy and 29.9 and 10.4 months, respectively, with cetuximab/chemotherapy. In the FOLFOX arm, median overall survival was 30.1 months with cetuximab and 26.9 months with bevacizumab (P = 0.09). Since three quarters of the physicians chose FOLFOX as the chemotherapy backbone, the small subset receiving FOLFIRI limits any comparison of the chemotherapy backbone. No new toxicities emerged in this study. With bevacizumab, common side effects were hypertension, headache, mucositis, nosebleed, diarrhea, rectal bleeding, loss of appetite, fatigue, and weakness, while cetuximab patients were more likely to have acneiform rash, pruritus, changes in fingernails and toenails, infections, fatigue, and low serum electrolyte levels. FOLFOX was associated with more neuropathy, whereas FOLFIRI caused more alopecia and diarrhea.
The overall quality of life for patients on either drug was similar, though, as expected, cetuximab-treated patients reported less “skin satisfaction” with that drug. The results suggested that either is a reasonable first-line option, according to the presenter. Expanded RAS was tested in all wild type RAS exon 2 using beaming technology including KRAS exon 3,4 and NRAS exon 2, 3 and 4 with a detection sensitivity of 0.01 %. The primary endpoint was OS. In expanded RAS wild type population, the median OS was pushed beyond 30 months. However, there was no significant difference between the cetuximab and bevacizumab in combination with chemotherapy (32 vs. 31.2 months). There was no difference in the progression-free survival. However, there was higher response achieved in the cetuximab arm in the expanded RAS population, 68.6 vs. 53.6 % (p < 0.01) [76].
Treatment Duration: Stop and Go or Maintenance Therapy
Optimal duration of chemotherapy is crucial because it has a direct influence on quality of life, toxicity and cost, as well as its potential relationship with survival. Use of oxaliplatin has been associated with an increased incidence of peripheral sensory neuropathy and this approach can reduce its severity and duration. To perform an induction chemotherapy (3–6 months of treatment) and then to stop until disease progression, or even to include chemotherapy holidays, is a very attractive and extensive practice among oncologists, although not very well validated. This approach could only be considered reasonable when PFS and OS are not compromised [88, 89]. In any case, it is necessary to keep in mind that the objective of the chemotherapy is palliative and the potential toxicity of the drugs in some cases, cumulative. Different strategies have been used to answer the question about optimal duration of chemotherapy in the palliative setting. The main goal in the majority of the studies is to use an induction chemotherapy for several months followed by free-intervals of chemotherapy (chemo- holidays) of all agents or only some of them. As a matter of fact, two approaches are possible: “Stop and go” strategy: stopping all chemotherapy agents after a pre-fixed number of cycles and restart on progression and maintenance chemotherapy stopping only some agents [88, 89].
Results of the OPTIMOX1 study showed that a “stop-and-go” approach using oxaliplatin-free intervals resulted in decreased neurotoxicity but did not affect OS in patients receiving FOLFOX as initial therapy for metastatic disease. Three hundred ninety-six other trials have also addressed the question of treatment breaks, with or without maintenance therapy, and found that toxicity can be minimized with minimal or no effect on survival [90]. Therefore the schedule/timing of the administration of this drug should be adjusted as a means of limiting this adverse effect. Discontinuation of oxaliplatin from FOLFOX or CapeOx should be strongly considered after 3 months of therapy, or sooner for unacceptable neurotoxicity, with other drugs in the regimen maintained for the entire 6 months or until time of tumor progression. Patients experiencing neurotoxicity on oxaliplatin should not receive subsequent oxaliplatin therapy until and unless they experience near-total resolution of that neurotoxicity.
In the phase II OPTIMOX2 trial, patients were randomized to receive either an OPTIMOX1 approach (discontinuation of oxaliplatin after 6 cycles of FOLFOX to prevent or reduce neurotoxicity with continuance of 5-FU/LV followed by reintroduction of oxaliplatin on disease progression) or an induction FOLFOX regimen (6 cycles) followed by discontinuation of all chemotherapy until tumor progression reached baseline, followed by reintroduction of FOLFOX.407 Results of the study showed no difference in OS for patients receiving the OPTIMOX1 approach compared with those undergoing an early, pre-planned, chemotherapy-free interval (median OS 23.8 vs. 19.5 months; P = 0.42). However, the median duration of disease control, which was the primary endpoint of the study, reached statistical significance at 13.1 months in patients undergoing maintenance therapy and 9.2 months in patients with a chemotherapy-free interval (P = 0.046) [91].
Use of Bevacizumab As a Maintenance Treatment
MACRO-TTD trial [92] is a Spanish multicenter, randomized, phase III study aimed to evaluate in 480 patients, the efficacy and tolerability of 6 cycles of bevacizumab + CapeOX followed by maintenance of CapeOX-bevacizumab or single agent bevacizumab. There were not statistically significant differences between the two arms in ORR (62 vs. 59 %), PFS (10.3 vs. 9.7 m) and OS (23.4 vs. 21.6 m). G3-G4 toxicities were inferior in the maintenance arm concerning hand-foot syndrome, asthenia and neuropathy. This study suggests that maintenance therapy with single agent bevacizumab is an appropriate option following the induction CapeOX-bevacizumab in patients with metastatic CRC.
More recently, final results from the phase III CAIRO3 trial also provide guidance to clinicians who are uncertain about just how big a treatment holiday to give to their patients with metastatic CRC following bevacizumab-containing induction therapy [93]. In a comparison of maintenance therapy with capecitabine and bevacizumab versus observation after six cycles of first-line CapeOX plus bevacizumab, the maintenance approach markedly improved PFS and yielded OS benefits in select patient subgroups. The 558 patients in this trial with stable disease or better after CapeOX-bevacizumab were randomly assigned to undergo observation or to receive 625 mg/m2 of oral capecitabine twice daily in combination with 7.5 mg/kg of intravenous bevacizumab every 3 weeks on a continuous basis. At the time of first disease progression (PFS1), patients in both arms were retreated with CapeOX-bevacizumab until second progression (PFS2) occurred. Over the median follow-up period of 48 months, CapeOX-bevacizumab was reintroduced in 60 % of patients in the observation arm as compared with 47 % in the maintenance arm. The findings revealed a significant improvement in median PFS2—the primary study endpoint—with maintenance treatment versus observation (11.7 vs. 8.5 months; stratified hazard ratio [HR] 0.67, 95 % CI [0.56–0.81]; p < 0.0001). Maintenance therapy also conferred a significant increase in median PFS1 as compared with observation (8.5 vs. 4.1 months; stratified HR 0.43, 95 % CI [0.36–0.52]; p < 0.0001) and significantly improved the median time to second progression (13.9 vs. 11.1 months; stratified HR 0.68, 95 % CI [0.57–0.82]; p < 0.0001). Importantly, the benefits observed with combination maintenance capecitabine and bevacizumab did not come at the expense of patient quality of life. Mean quality-of-life scores remained similar between the arms over the course of the study, without any clinically relevant differences. The improvements in disease progression timing did not translate into a statistically significant OS benefit in the total study population (median OS: 21.6 months with maintenance therapy vs. 18.1 months with observation; stratified HR 0.89, 95 % CI [0.73–1.07]; p = 0.22). However, this could have been affected by the greater proportion of patients with synchronous lesions in the maintenance arm versus the observation arm (79 vs. 68 %). Subsequent multivariate analysis evaluating the interactions between treatment and baseline covariates found that maintenance therapy independently conferred a marked OS improvement in two groups of patients: those with synchronous disease who underwent resection of the primary tumor (median OS, 25.0 months with maintenance therapy vs. 18.0 months with observation; log-rank p value < 0.0001), and those who attained a complete or partial response to induction therapy prior to randomization (median OS, 24.1 months with maintenance therapy vs. 18.8 months with observation; log-rank p value < 0.0001).
Selecting the Optimal Treatment for First-Line Metastatic CCR Based on Clinical Stratification. Summary of the Recommendations
Decision of treatment intensity for first-line treatment should be based on clinical presentation at diagnosis, considering factors like patients’ characteristics independent from the malignant disease, tumour-related symptoms, patients’ preferences, localizations of metastases, and the general treatment aim. Current ESMO guidelines stratify patients according to these factors in clinical groups with different treatment intensities (Table 2) [94••]. Four groups are defined: ESMO group 0 comprising patients with clearly resectable liver metastases, group 1 with potentially resectable disease after achieving tumour response, group 2 symptomatic patients or high tumour load with risk of rapid deterioration and finally group 3 with asymptomatic, low tumour burden and severe co-morbidity.
For ESMO group 0 patients with clearly R0 resectable colorectal liver metastases surgery is the treatment of choice due to the proven chance of cure, whereas the sequence type of regimen (FOLFOX or others) and intensity of perioperative chemotherapy is controversial. Patients with unresectable disease (ESMO groups 1, 2, or 3) should receive upfront systemic chemotherapy, apart from the small group of asymptomatic patients with low tumour burden eligible for and complying with a watch and wait approach. Whereas groups 1 and 2 patients urge for intensive upfront chemotherapy to either ensure secondary resectability or allow for rapid symptom control, group 3 could be treated with a sequential treatment approach, starting with a low toxic single agent or two-drug combination regimen. Patients with asymptomatic, but surely unresectable disease due to location or overall extent and without relevant co-morbidity may not be ideally stratified in ESMO group 3, but rather treated with upfront intensive chemotherapy. Moreover, current available phase III trials included patients irrespective of ESMO grouping, thus limiting the potential prognostic or predictive value of upfront patient stratification. Although grouping patients might be helpful for guidance of treatment strategy beyond induction treatment, e.g., secondary resection, main systemic treatment options are either intensive three- to four-drug regimens or “sequential” one- to two-drugs regimens (see the main protocol regimens in Table 3).
Conclusions
Treatment of metastatic CRC is complex and highly individualized taking into account disease and patient characteristics, molecular, and biochemical markers and thus enabling a personalized management in terms of selecting the most appropriate measures and sequences of systemic treatment. As far as the current data unresectable patients with RAS wild-type should receive an EGFR antibody based chemotherapy, whereas patients with RAS mutation should receive two- or three-drug chemotherapy in combination with bevacizumab, if an intensive treatment approach is chosen. For patients with a non-intense or sequential approach fluoropyrimidine and bevacizumab seems to be an efficacious and low toxic treatment option.
Future research will help to further tailor anti EGFR treatment, excluding patients deriving no benefit from EGFR inhibition and other molecular-based approaches. Moreover, close timely information such as acquired by liquid biopsies about the current molecular tumor situation and potentially developing resistance might be helpful to orient treatment during the course of disease.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Malvezzi M, Bertuccio P, Levi F, La Vecchia C, Negri E. European cancer mortality predictions for the year 2012. Ann Oncol. 2012;23:1044–52. doi:10.1093/annonc/mds024.
Stintzing S, Fischer von Weikersthal L, Decker T, Vehling-Kaiser U, Jäger E, Heintges T, et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer-subgroup analysis of patients with KRAS: mutated tumours in the randomised German AIO study KRK-0306. Ann Oncol. 2012;23:1693–9.
Van Cutsem E, Köhne CH, Láng I, Folprecht G, Nowacki MP, Cascinu S, et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol. 2011;29:2011–9. doi:10.1200/JCO.2010.33.5091.
Douillard JY, Siena S, Cassidy J, Tabernero J, Burkes R, Barugel M, et al. Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: the PRIME study. J Clin Oncol. 2010;28:4697–705. doi:10.1200/JCO.2009.27.4860.
Kopetz S, Chang GJ, Overman MJ, Eng C, Sargent DJ, Larson DW, et al. Improved survival in metastatic colorectal cancer is associated with adoption of hepatic resection and improved chemotherapy. J Clin Oncol. 2009;27:3677–83. doi:10.1200/JCO.2008.20.5278.
Falcone A, Cremolini C, Masi G. FOLFOXIRI/bevacizumab (bev) versus FOLFIRI/bev as first-line treatment in unresectable metastatic colorectal cancer (mCRC) patients (pts): results of the phase III TRIBE trial by GONO group. J Clin Oncol. 2013;31suppl:3505 [abstract].
Heinemann V, von Weikersthal LF, Decker T, Kiani A, Vehling-Kaiser U, Al-Batran SE, et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): a randomised, open-label, phase 3 trial. Lancet Oncol. 2014. doi:10.1016/S1470-2045(14)70330-4.
Folprecht G, Gruenberger T, Bechstein WO, Raab HR, Lordick F, Hartmann JT, et al. Tumour response and secondary resectability of colorectal liver metastases following neoadjuvant chemotherapy with cetuximab: the CELIM randomised phase 2 trial. Lancet Oncol. 2010;11:38–47. doi:10.1016/S1470-2045(09)70330-4.
Morris EJ, Forman D, Thomas JD, Quirke P, Taylor EF, Fairley L, et al. Surgical management and outcomes of colorectal cancer liver metastases. Br J Surg. 2010;97:1110–8. doi:10.1002/bjs.7032.
Grothey A, Marshall JL. Optimizing palliative treatment of metastatic colorectal cancer in the era of biologic therapy. Oncology (Williston Park). 2007;21:553–64.
Hoff PM, Ansari R, Batist G, et al. Comparison of oral capecitabine versus intravenous fluorouracil plus leucovorin as first-line treatment in 605 patients with metastatic colorectal cancer: results of a randomized phase III study. J Clin Oncol. 2001;19:2282–92.
Arkenau HT, Arnold D, Cassidy J, et al. Efficacy of oxaliplatin plus capecitabine or infusional fluorouracil/leucovorin in patients with metastatic colorectal cancer: a pooled analysis of randomized trials. J Clin Oncol. 2008;26:5910–7.
Cassidy J, Clarke S, Diaz-Rubio E, et al. Randomized phase III study of capecitabine plus oxaliplatin compared with fluorouracil/folinic acid plus oxaliplatin as first-line therapy for metastatic colorectal cancer. J Clin Oncol. 2008;26:2006–12.
Porschen R, Arkenau HT, Kubicka S, et al. Phase III study of capecitabine plus oxaliplatin compared with fluorouracil and leucovorin plus oxaliplatin in metastatic colorectal cancer: a final report of the AIO Colorectal Study Group. J Clin Oncol. 2007;25:4217–23.
Haller DG, Cassidy J, Clarke SJ, et al. Potential regional differences for the tolerability profiles of fluoropyrimidines. J Clin Oncol. 2008;26:2118–23.
Van Cutsem E, Findlay M, Osterwalder B, et al. Capecitabine, an oral fluoropyrimidine carbamate with substantial activity in advanced colorectal cancer: results of a randomized phase II study. J Clin Oncol. 2000;18:1337–45.
Hochster HS, Hart LL, Ramanathan RK, et al. Safety and efficacy of oxaliplatin and fluoropyrimidine regimens with or without bevacizumab as first-line treatment of metastatic colorectal cancer: results of the TREE Study. J Clin Oncol. 2008;26:3523–39.
Cunningham D, Pyrhonen S, James RD, et al. Randomised trial of irinotecan plus supportive care versus supportive care alone after fluorouracil failure for patients with metastatic colorectal cancer. Lancet. 1998;352:1413–8.
Rougier P, Van Cutsem E, Bajetta E, et al. Randomised trial of irinotecan versus fluorouracil by continuous infusion after fluorouracil failure in patients with metastatic colorectal cancer. Lancet. 1998;352:1407–12.
Saltz LB, Cox JV, Blanke C, et al. Irinotecan plus fluorouracil and leucovorin for metastatic colorectal cancer. Irinotecan Study Group. N Engl J Med. 2000;343:905–14.
Kohne CH, van Cutsem E, Wils J, et al. Phase III study of weekly high-dose infusional fluorouracil plus folinic acid with or without irinotecan in patients with metastatic colorectal cancer: European Organisation for Research and Treatment of Cancer Gastrointestinal Group Study 40986. J Clin Oncol. 2005;23:4856–65.
Douillard JY, Cunningham D, Roth AD, et al. Irinotecan combined with fluorouracil compared with fluorouracil alone as first-line treatment for metastatic colorectal cancer: a multicentre randomised trial. Lancet. 2000;355:1041–7.
Innocenti F, Undevia SD, Iyer L, et al. Genetic variants in the UDP-glucuronosyltransferase 1A1 gene predict the risk of severe neutropenia of irinotecan. J Clin Oncol. 2004;22:1382–8.
de Gramont A, Figer A, Seymour M, et al. Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment in advanced colorectal cancer. J Clin Oncol. 2000;18:2938–47.
Giacchetti S, Perpoint B, Zidani R, et al. Phase III multicenter randomized trial of oxaliplatin added to chronomodulated fluorouracil-leucovorin as first-line treatment of metastatic colorectal cancer. J Clin Oncol. 2000;18:136–47.
Grothey A, Deschler B, Kroening H, et al. Phase III study of bolus 5-fluorouracil (5-FU)/ folinic acid (FA) (Mayo) vs. weekly high-dose 24h 5-FU infusion/ FA + oxaliplatin (OXA) in advanced colorectal cancer (ACRC). Proc Am Soc Clin Oncol. 2001;21:(abstr 512).
Rothenberg ML, Oza AM, Bigelow RH, et al. Superiority of oxaliplatin and fluorouracil-leucovorin compared with either therapy alone in patients with progressive colorectal cancer after irinotecan and fluorouracil-leucovorin: interim results of a phase III trial. J Clin Oncol. 2003;21:2059–69.
Ducreux M, Bennouna J, Hebbar M, et al. Capecitabine plus oxaliplatin (XELOX) versus 5-fluorouracil/leucovorin plus oxaliplatin (FOLFOX-6) as first-line treatment for metastatic colorectal cancer. Int J Cancer. 2011;128:682–90.
Zhang C, Wang J, Gu H, et al. Capecitabine plus oxaliplatin compared with 5-fluorouracil plus oxaliplatin in metastatic colorectal cancer: meta-analysis of randomized controlled trials. Oncol Lett. 2012;3:831–8.
Goldberg RM, Sargent DJ, Morton RF, et al. A randomized controlled trial of fluorouracil plus leucovorin, irinotecan, and oxaliplatin combinations in patients with previously untreated metastatic colorectal cancer. J Clin Oncol. 2004;22:23–30.
Colucci G, Gebbia V, Paoletti G, et al. Phase III randomized trial of FOLFIRI versus FOLFOX4 in the treatment of advanced colorectal cancer: a multicenter study of the Gruppo Oncologico Dell’Italia Meridionale. J Clin Oncol. 2005;23:4866–75.
Tournigand C, Andre T, Achille E, et al. FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: a randomized GERCOR study. J Clin Oncol. 2004;22:229–37.
Grothey A, Sargent D, Goldberg RM, et al. Survival of patients with advanced colorectal cancer improves with the availability of fluorouracil-leucovorin, irinotecan, and oxaliplatin in the course of treatment. J Clin Oncol. 2004;22:1209–14.
Falcone A, Ricci S, Brunetti I, et al. Phase III trial of infusional fluorouracil, leucovorin, oxaliplatin, and irinotecan (FOLFOXIRI) compared with infusional fluorouracil, leucovorin, and irinotecan (FOLFIRI) as first-line treatment for metastatic colorectal cancer: the Gruppo Oncologico Nord Ovest. J Clin Oncol. 2007;25:1670–6.
Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol. 2005;23:1011–27.
Presta LG, Chen H, O’Connor SJ, et al. Humanization of an anti-vascular endothelial growth factor monoclonal anti- body for the therapy of solid tumors and other disorders. Cancer Res. 1997;57:4593–9.
Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–42.
Kabbinavar FF, Hambleton J, Mass RD, et al. Combined analysis of efficacy: the addition of bevacizumab to fluorouracil/leucovorin improves survival for patients with metastatic colorectal cancer. J Clin Oncol. 2005;23:3706–12.
Saltz LB, Clarke S, Diaz-Rubio E, et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol. 2008;26:2013–9.
Fuchs CS, Marshall J, Mitchell E, et al. Randomized, controlled trial of irinotecan plus infusional, bolus, or oral fluoropyrimidines in first-line treatment of metastatic colorectal cancer: results from the BICC-C Study. J Clin Oncol. 2007;25:4779–86.
Grothey A. Recognizing and managing toxicities of molecular targeted therapies for colorectal cancer. Oncology (Williston Park). 2006;20:21–8.
Giantonio BJ, Catalano PJ, Meropol NJ, et al. Bevacizumab in combination with oxaliplatin, fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer: results from the Eastern Cooperative Oncology Group Study E3200. J Clin Oncol. 2007;25:1539–44.
Nalluri SR, Chu D, Keresztes R, et al. Risk of venous thromboembolism with the angiogenesis inhibitor bevacizumab in cancer patients: a meta-analysis. JAMA. 2008;300:2277–85.
Hurwitz HI, Saltz LB, Van Cutsem E, et al. Venous thromboembolic events with chemotherapy plus bevacizumab: a pooled analysis of patients in randomized phase II and III studies. J Clin Oncol. 2011;29:1757–64.
Falcone A, Cremolini C, Masi G, et al. FOLFOXIRI/bevacizumab (bev) versus FOLFIRI/bev as first-line treatment in unresectable metastatic colorectal cancer (mCRC) patients (pts): results of the phase III TRIBE trial by GONO group [abstract]. ASCO Meet Abstr. 2013;31:3505.
Loupakis F, Cremolini C, Masi G, et al. FOLFOXIRI plus bevacizumab (bev) versus FOLFIRI plus bev as first-line treatment of metastatic colorectal cancer (MCRC): results of the phase III randomized TRIBE trial [abstract]. ASCO Meet Abstr. 2013;31:336.
Gruenberger T, Bridgewater JA, Chau I, et al. Randomized, phase II study of bevacizumab with mFOLFOX6 or FOLFOXIRI in patients with initially unresectable liver metastases from colorectal cancer: resectability and safety in OLIVIA [abstract]. ASCO Meet Abstr. 2013;31:3619.
Saltz LB, Lenz HJ, Kindler HL, et al. Randomized phase II trial of cetuximab, bevacizumab, and irinotecan compared with cetuximab and bevacizumab alone in irinotecan-refractory colorectal cancer: the BOND-2 study. J Clin Oncol. 2007;25:4557–61.
Hecht JR, Mitchell E, Chidiac T, et al. A randomized phase IIIB trial of chemotherapy, bevacizumab, and panitumumab compared with chemotherapy and bevacizumab alone for metastatic colorectal cancer. J Clin Oncol. 2009;27:672–80.
Tol J, Koopman M, Cats A, et al. Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer. N Engl J Med. 2009;360:563–72.
Saltz LB, Meropol NJ, Loehrer Sr PJ, et al. Phase II trial of cetuximab in patients with refractory colorectal cancer that expresses the epidermal growth factor receptor. J Clin Oncol. 2004;22:1201–8.
Cunningham D, Humblet Y, Siena S, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351:337–45.
Van Cutsem E, Peeters M, Siena S, et al. Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol. 2007;25:1658–64.
Jonker DJ, O’Callaghan CJ, Karapetis CS, et al. Cetuximab for the treatment of colorectal cancer. N Engl J Med. 2007;357:2040–8.
Chung CH, Mirakhur B, Chan E, et al. Cetuximab-induced anaphylaxis and IgE specific for galactose-alpha-1,3-galactose. N Engl J Med. 2008;358:1109–17.
Douillard JY, Siena S, Cassidy J, et al. Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: the PRIME study. J Clin Oncol. 2010;28:4697–705.
Van Cutsem E, Kohne CH, Hitre E, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360:1408–17.
Van Cutsem E, Kohne CH, Lang I, et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol. 2011;29:2011–9.
Amado RG, Wolf M, Peeters M, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626–34.
Artale S, Sartore-Bianchi A, Veronese SM, et al. Mutations of KRAS and BRAF in primary and matched metastatic sites of colorectal cancer. J Clin Oncol. 2008;26:4217–9.
Bokemeyer C, Bondarenko I, Hartmann JT, et al. KRAS status and efficacy of first-line treatment of patients with metastatic colorectal cancer (mCRC) with FOLFOX with or without cetuximab: the OPUS experience. J Clin Oncol. 2008; 26(May 20 suppl; abstr 4000).
Karapetis CS, Khambata-Ford S, Jonker DJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359:1757–65. The authors analyzed tumor samples, obtained from 394 patients with colorectal cancer who were randomly assigned to receive cetuximab plus best supportive care or best supportive care alone, to look for activating mutations in exon 2 of the K-ras gene and showed that mutated K-ras did not benefit from cetuximab, whereas patients with a tumor bearing wild-type K-ras did benefit from cetuximab.
Lievre A, Bachet JB, Le Corre D, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66:3992–5.
Rajagopalan H, Bardelli A, Lengauer C, et al. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature. 2002;418:934.
Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. 2003;3:459–65.
Douillard JY, Oliner KS, Siena S, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369:1023–34.
Stintzing S, Jung A, Rossius L, et al. Analysis of KRAS/NRAS and BRAF mutations in FIRE-3: a randomized phase III study of FOLFIRI plus cetuximab or bevacizumab as first-line treatment for wild-type KRAS (exon 2) metastatic colorectal cancer patients [abstract]. ESMO Eur Cancer Congr 2013;E17-7073.
Heinemann et al. Independent radiological evaluation of objective response, early tumor shrinkage, and depth of response in FIRE-3. Oral abstract present at WCGC 2014.
Package Insert. Vectibix® (Panitumumab). Thousand Oaks, CA: Amgen Inc.; 2013. Available at: http://dailymed.nlm.nih.gov/dailymed/lookup.cfm?setid=e0fa4bca-f245-4d92-ae29-b0c630a315c2. Accessed 26 Feb 2013.
De Roock W, Jonker DJ, Di Nicolantonio F, et al. Association of KRAS p.G13D mutation with outcome in patients with chemotherapy- refractory metastatic colorectal cancer treated with cetuximab. JAMA. 2010;304:1812–20.
Peeters M, Douillard JY, Van Cutsem E, et al. Mutant KRAS codon 12 and 13 alleles in patients with metastatic colorectal cancer: assessment as prognostic and predictive biomarkers of response to panitumumab. J Clin Oncol. 2013;31:759–65.
Lang I, Kohne CH, Folprecht G, et al. Quality of life analysis in patients with KRAS wild-type metastatic colorectal cancer treated first- line with cetuximab plus irinotecan, fluorouracil and leucovorin. Eur J Cancer. 2013;49:439–48.
Maughan TS, Adams RA, Smith CG, et al. Addition of cetuximab to oxaliplatin-based first-line combination chemotherapy for treatment of advanced colorectal cancer: results of the randomised phase 3 MRC COIN trial. Lancet. 2011;377:2103–14.
Taieb J, Maughan T, Bokemeyer C, et al. Cetuximab combined with infusional 5-fluorouracil/folinic acid (5-FU/FA) and oxaliplatin in metastatic colorectal cancer (mCRC): a pooled analysis of COIN and OPUS study data [abstract]. ASCO Meet Abstr. 2012;30:3574.
Tveit KM, Guren T, Glimelius B, et al. Phase III trial of cetuximab with continuous or intermittent fluorouracil, leucovorin, and oxaliplatin (Nordic FLOX) versus FLOX alone in first-line treatment of metastatic colorectal cancer: the NORDIC-VII study. J Clin Oncol. 2012;30:1755–62.
Venook AP, Niedzwiecki D, Lenz H-J, et al. Abstract 501O—CALGB/SWOG 80405: PHASE III trial of irinotecan/5-FU/leucovorin (FOLFIRI) or oxaliplatin/5-FU/leucovorin (mFOLFOX6) with bevacizumab (BV) or cetuximab (CET) for patients (pts) with expanded ras analyses untreated metastatic adenocarcinoma of the colon or rectum (MCRC). ESMO Annual Meeting. Presented September 29, 2014.
Adams RA, Meade AM, Seymour MT, Wilson RH, Madi A, Fisher D, et al. Intermittent versus continuous oxaliplatin and fluoropyrimidine combination chemotherapy for first-line treatment of advanced colorectal cancer: results of the randomised phase 3 MRC COIN trial. Lancet Oncol. 2011;12(7)642–53. doi:10.1016/S1470-2045(11)70102-4.
Kohne CH, De Greve J, Hartmann JT, et al. Irinotecan combined with infusional 5-fluorouracil/folinic acid or capecitabine plus celecoxib or placebo in the first-line treatment of patients with metastatic colorectal cancer. EORTC study 40015. Ann Oncol. 2008;19:920–6.
Pectasides D, Papaxoinis G, Kalogeras K, et al. XELIRI- bevacizumab versus FOLFIRI-bevacizumab as first-line treatment in patients with metastatic colorectal cancer: a Hellenic Cooperative Oncology Group phase III trial with collateral biomarker analysis. BMC Cancer. 2012;12:271.
Cunningham D, Lang I, Marcuello E, Lorusso V, Ocvirk J, Shin DB, et al. Bevacizumab plus capecitabine versus capecitabine alone in elderly patients with previously un-treated metastatic colorectal cancer (AVEX): an open-label, randomised phase 3 trial. Lancet Oncol. 2013;14:1077–85.
Cao Y, Tan A, Gao F, et al. A meta-analysis of randomized controlled trials comparing chemotherapy plus bevacizumab with chemotherapy alone in metastatic colorectal cancer. Int J Colorectal Dis. 2009;24:677–85.
Hurwitz HI, Tebbutt NC, Kabbinavar F, et al. Efficacy and safety of bevacizumab in metastatic colorectal cancer: pooled analysis from seven randomized controlled trials. Oncologist. 2013. This analysis pooled individual patient data from randomized controlled trials (RCTs) to more thoroughly examine clinical outcomes when adding bevacizumab to chemotherapy for patients with metastatic colorectal cancer.
Loupakis F, Bria E, Vaccaro V, et al. Magnitude of benefit of the addition of bevacizumab to first-line chemotherapy for metastatic colorectal cancer: meta-analysis of randomized clinical trials. J Exp Clin Cancer Res. 2010;29:58.
Lv C, Wu S, Zheng D, et al. The efficacy of additional bevacizumab to cytotoxic chemotherapy regimens for the treatment of colorectal cancer: an updated meta-analysis for randomized trials. Cancer Biother Radiopharm. 2013;28:501–9.
Welch S, Spithoff K, Rumble RB, Maroun J. Bevacizumab combined with chemotherapy for patients with advanced colorectal cancer: a systematic review. Ann Oncol. 2010;21:1152–62.
Macedo LT, da Costa Lima AB, Sasse AD. Addition of bevacizumab to first-line chemotherapy in advanced colorectal cancer: a systematic review and meta-analysis, with emphasis on chemotherapy subgroups. BMC Cancer. 2012;12:89.
Meyerhardt JA, Li L, Sanoff HK, et al. Effectiveness of bevacizumab with first-line combination chemotherapy for Medicare patients with stage IV colorectal cancer. J Clin Oncol. 2012;30:608–15.
Grothey A. Optimized treatment of metastatic colorectal cancer. Educational Book. ASCO. 2009;209–11.
Holash J, Davis S, Papadopoulos N, et al. VEGF-Trap: a VEGF blocker with potent antitumor effects. Proceedings of the National Academy of Sciences of the United States of America website. April 19, 2002. Accessed 20 Aug 2012.
Tournigand C, Cervantes A, Figer A, et al. OPTIMOX1: a randomized study of FOLFOX4 or FOLFOX7 with oxaliplatin in a stop-and-go fashion in advanced colorectal cancer–a GERCOR study. J Clin Oncol. 2006;24:394–400.
Chibaudel B, Maindrault-Goebel F, Lledo G, et al. Can chemotherapy be discontinued in unresectable metastatic colorectal cancer? The GERCOR OPTIMOX2 Study. J Clin Oncol. 2009;27:5727–33.
Díaz-Rubio E, Gómez-España A, Massutí B, Sastre J, Abad A, Valladares M, et al. First-line XELOX plus bevacizumab followed by XELOX plus bevacizumab or single-agent bevacizumab as maintenance therapy in patients with metastatic colorectal cancer: the phase III MACRO TTD study. Oncologist. 2012;17:15–25.
Koopman M, Simkens L, May AM, et al. Final results and subgroup analyses of the phase 3 CAIRO3 study: maintenance treatment with capecitabine + bevacizumab versus observation after induction treatment with chemotherapy + bevacizumab in metastatic colorectal cancer (mCRC). Presented at the 2014 American Society of Clinical Oncology (ASCO) Annual Meeting; Abstract 3504.
Schmoll HJ, Van Cutsem E, Stein A, Valentini V, Glimelius B, Haustermans K, et al. ESMO Consensus Guidelines for management of patients with colon and rectal cancer. a personalized approach to clinical decision mak- ing. Ann Oncol. 2012;23:2479–516. Treatment decisions must be based on the available evidence, which has been the basis for this consensus conference-based guideline delivering a clear proposal for diagnostic and treatment measures in each stage of rectal and colon cancer and the individual clinical situations. This ESMO guideline is recommended to be used as the basis for treatment and management decisions.
Rose LJ. Colon Cancer Treatment Protocols. Medscape. Updated May 29, 2014. Available at: http://emedicine.medscape.com/article/2005487-overview. Updates in treatment protocols for colon cancer are provided in this Medscape overview, including adjuvant and neoadjuvant therapy for resectable disease and chemotherapy for advanced or metastatic colon cancer.
Package Insert. ZALTRAP® (ziv-aflibercept). Bridgewater: Regeneron Pharmaceuticals, Inc. / sanofi-aventis U.S. LLC; 2013. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/125418s02 0lbl.pdf. Accessed 15 Aug 2014.
Tabernero J, Van Cutsem E, Lakomy R, et al. Aflibercept versus placebo in combination with fluorouracil, leucovorin and irinotecan in the treatment of previously treated metastatic colorectal cancer: prespecified subgroup analyses from the VELOUR trial. Eur J Cancer. 2014;50:320–31. Available at: http://www.ncbi.nlm.nih.gov/pubmed/24140268.
Package Insert. STIVARGA (regorafenib) tablets, oral. Wayne: Bayer HealthCare Pharmaceuticals; 2013. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/203085s001lbl.pdf. Accessed 15 Aug 2014.
Grothey A, Van Cutsem E, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo- controlled, phase 3 trial. Lancet. 2013;381:303–12. Available at: http://www.ncbi.nlm.nih.gov/pubmed/23177514.
Argiles G, Troiani T, Rivera F, et al. First-line treatment with regorafenib in combination with mFOLFOX6 for metastatic colorectal cancer: a single-arm, open-label phase II clinical trial; Presented at the European Society of Medical Oncology 2013 Congress; September 27–October 1, 2013; Amsterdam, the Netherlands. Abstract 2367.
UNC Lineberger Comprehensive Cancer Center. ClinicalTrialsgov [website on the Internet] Bethseda, MD: US National Library of Medicine; 2011. [Accessed November 1, 2011]. Regorafenib+FOLFIRI Versus Placebo+FOLFIRI as 2nd Line Tx in Metastatic Colorectal Cancer. [updated August 20, 2013]. Available from: http://clinicaltrial.gov/show/NCT01298570. NLM identifier: NCT01298570.
Compliance with Ethics Guidelines
Conflict of Interest
Andre M. Murad and Lucas S. Murad declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Personalized Medicine in Colorectal Cancer
Rights and permissions
About this article

Cite this article
Murad, A.M., Murad, L.S. First-Line Therapy in Metastatic Colorectal Cancer Patients Not Candidates for Curative Surgery. Curr Colorectal Cancer Rep 11, 54–69 (2015). https://doi.org/10.1007/s11888-015-0259-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11888-015-0259-4