
Abstract
Purpose of Review
Cardiovascular diseases are the leading cause of death worldwide, largely due to the limited regenerative capacity of the adult human heart. In contrast, teleost zebrafish hearts possess natural regeneration capacity by proliferation of pre-existing cardiomyocytes after injury. Hearts of mice can regenerate if injured in a few days after birth, which coincides with the transient capacity for cardiomyocyte proliferation. This review tends to elaborate the roles and mechanisms of Wnt/β-catenin signaling in heart development and regeneration in mammals and non-mammalian vertebrates.
Recent Findings
Studies in zebrafish, mice, and human embryonic stem cells demonstrate the binary effect for Wnt/β-catenin signaling during heart development. Both Wnts and Wnt antagonists are induced in multiple cell types during cardiac development and injury repair. In this review, we summarize composites of the Wnt signaling pathway and their different action routes, followed by the discussion of their involvements in cardiac specification, proliferation, and patterning. We provide overviews about canonical and non-canonical Wnt activity during heart homeostasis, remodeling, and regeneration.
Summary
Wnt/β-catenin signaling exhibits biphasic and antagonistic effects on cardiac specification and differentiation depending on the stage of embryogenesis. Inhibition of Wnt signaling is beneficial for cardiac wound healing and functional recovery after injury. Understanding of the roles and mechanisms of Wnt signaling pathway in injured animal hearts will contribute to the development of potential therapeutics for human diseased hearts.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The human heart is composed of four morphologically and functionally distinct chambers. Cardiomyocytes are the most prevalent cells in the heart with higher percentages in ventricles than atria [1]. Large-scale single-cell transcriptomes characterize the cellular heterogeneity of cardiomyocytes, endocardial cells, fibroblasts, and immune cells. These findings have identified distinct subsets of atrial and ventricular cells with diverse developmental origins and distinct lineage priming gene signatures [2, 3]. The ventricular region including apex and interventricular septum in adult human heart contains about 49.2% cardiomyocytes, 21.2% vascular smooth muscle cells, 15.5% fibroblasts, 7.8% endocardial cells, and 5.3% immune cells [1, 4, 5]. Unlike many other tissues, the adult cardiac muscle lacks stem cells and fails to meaningfully regenerate after massive cardiomyocyte loss [6,7,8,9]. Myocardial infarction (MI) is still a prominent cause of cardiovascular death with rising prevalence worldwide. Cardiomyocyte renewal in the injured heart has gained more attentions over the last decades. The field has reached a consensus on proliferation of pre-existing cardiomyocytes instead of stem cell as sources for new cardiomyocytes [10,11,12,13,14]. Neonatal mammals can fully regenerate heart following the left ventricular apex resection within a critical window period after birth [15,16,17]. Importantly, some lower vertebrates, such as newts and zebrafish, have a broad ability to achieve cardiac regeneration after adult amputation [18,19,20,21]. However, there is only 5% of cycling cardiomyocytes per year in adult mouse hearts, and most of them localize in the subendocardial muscle [7, 22,23,24]. In human heart, cardiomyocytes turn over at estimate of ~ 1% per year at age of 20, declining to ~ 0.4% per year at age of 75 [25,26,27]. The gradual loss of cardiomyocyte proliferative capacity in mammalian heart correlates with metabolic pattern switch from the glycolytic pathway at fetal stages to fatty acid β-oxidation at adulthoods [28, 29]. However, the limited capacity for cardiomyocyte regeneration is insufficient to repair injured mammalian hearts when a massive loss of cardiomyocytes occurs after MI or other injuries [9, 19, 22, 30].
Wnt/β-catenin signaling is a highly conserved pathway that plays crucial roles in various biological processes in mammals and non-mammalian vertebrates, such as embryonic development, organogenesis, neurological development, inflammation, and regeneration. The Wnt-secreted glycoproteins act as ligands and regulate a downstream effector β-catenin [31]. In mammals, the pathway is quiescent in many organs during adulthoods, which is reactivated during tissue injury and cardiac fibrosis [32]. Recent findings in mice and zebrafish, as well as human embryonic stem cells (hESCs), emphasize the critical importance of the Wnt/β-catenin pathway in the regulation of cardiac development, remodeling, injury repair, and regeneration, for which we summarize and provide overviews in this review.
The Wnt/β-Catenin Signaling Pathway
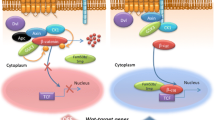
Nusse and Varmus identified the first member of the Wnt family as Wnt1(Int-1) 40 years ago [33, 34]. Since then, numerous studies have demonstrated that Wnt proteins are secreted signaling molecules present in all metazoans and are involved in diverse biological processes, including cell proliferation, differentiation, and tissue injury [35,36,37]. Currently, 19 different Wnt proteins, 10 frizzled receptors (Fzds), 5 secreted frizzled-related proteins (sFrps), and 4 Dickkopf (Dkk) have been identified, which constitute the highly complex signal transduction pathway [38,39,40]. Canonical Wnt signaling is mediated through intracellular β-catenin, whereas non-canonical Wnt signals function through the planar cell polarity (PCP) pathway and Ca2+ cascades [41, 42]. Wnt proteins secrete from cells and act as paracrine or autocrine manners, influencing target cell behavior by binding to Fzds, a family of G protein-coupled receptors (GPCRs) in the plasma membrane [43, 44] (Fig. 1). Wnts also bind the lipoprotein receptor-related5 (Lrp5) and Lrp6, stabilizing the Wnt/Fzd complex at the cell surface [45]. Wnt-Fzd-Lrp5/6 complex regulates β-catenin protein levels through a dedicated cytoplasmic destruction complex. The core of destruction complex contains a central scaffold protein AXIN that interacts with adenomatous polyposis coli (APC), glycogen synthase kinase-3β (GSK-3β), and casein kinase 1(CK1) [46]. In the absence of Wnt ligands, CK1α and GSK3β sequentially phosphorylate β-catenin at the N-terminus. The resulting phosphorylated β-catenin is subjected to ubiquitination by Skp1-Cul1-F-box (SCF) E3 ligase complex including β-Transducin repeat containing protein (β-TrCP1) and β-TrCP2, leading to subsequent degradation [47, 48].

Schematic representation of Wnt/β-catenin signaling pathway. In Wnt-producing cells, synthesized WNT protein is palmitoylated by the acyltransferase enzyme porcupine in the endoplasmic reticulum. Transport and secretion of the WNT protein is controlled by the multipass transmembrane protein WLS in secretory vesicles. After Wnt secretion, DKKs competitively bind to LRP5/6 receptors to antagonize the interaction of WNTs with LRP5/6. sFRPs and WIF1 can bind and scavenge extracellular WNT proteins, thereby preventing their interaction with the receptor complex. In the absence of Wnt ligands (Wnt-off), β-catenin is located in the adherent junction and the cytoplasm, where it is phosphorylated and targeted for proteasomal degradation by a destruction complex, comprising APC, AXIN, CK1α, β-TRCP, and GSK3β. When Wnt signaling is activated (Wnt on), Wnt ligands bind to the FZD and LRP5/LRP6 co-receptor complex. Subsequently, phosphorylated LRP6 recruits AXIN and DVL, which blocks phosphorylation and degradation of β-catenin by AXIN-mediated destruction complex. Dephosphorylated β-catenin translocates into the nucleus, where it binds to TCF/LEF transcriptional factors and controls the expression of target genes (CyclinD1, Axin2, c-Myc, etc.). Abbreviations: WLS, Wntless; DKK, Dickkopf; LRP5/6, low-density lipoprotein receptor-related protein 5/6; sFRPs, secreted frizzled-related proteins; WIF1, Wnt inhibitory factor1; APC, adenomatous polyposis coli; DVL, dishevelled; CK1α, casein kinase 1; GSK3β, glycogen synthase kinase-3β; FZD, frizzled; TCF, T cell factor; LEF, lymphoid enhancer-binding factor
Following Wnt ligand binding to Fzd and Lrp5/6 co-receptors, dishevelled (Dvl) is recruited and activated to inhibit the destruction complex and dephosphorylate β-catenin. Stabilized and dephosphorylated β-catenin translocates into the nucleus and binds to the T-cell factor/lymphoid enhancer factor (Tcf/Lef) transcriptional complex and coactivators containing p300, Cbp, Bcl9, and Pygo, promoting expressions of target genes Axin2, c-Myc, Ccdn1 (Cyclin-D1), Cd44, Mmp2/9, Vegf and others (Fig. 1) [49,50,51]. Axin2 serves as a negative regulator of the pathway as part of a protein destruction complex, which limits the duration and intensity of Wnt signaling [52,53,54]. CRISPR/Cas9 knockout screens have been used to identify Wnt signaling regulators, including ubiquitin-specific-processing protease 7 (Usp7) as a potent negative regulator and histone-lysine N-methyltransferase Setdb1 as a potent negative regulator. Previously unknown factors affecting Wnt signaling, such as DExH-box helicase 29 (Dhx29), have been identified to repress Wnt signaling [55].
Studies have shown that the regulatory mechanisms regarding Wnt expression, modification, and secretion (Fig. 1). Lipidation of Wnts by the acyltransferase porcupine (Porcn) is essential for cellular secretion and biological activation. In the endoplasmic reticulum, Wntless (Wls) assists the transport and secretion of acylated Wnt proteins. Several synthetic Porcn inhibitors of this pathway have been developed to reduce Wnt secretion [56, 57]. After Wnt secretion, Dkks are effective antagonists of Wnt/β-catenin signaling, in which Dkks competitively bind to Lrp5/6 receptors to antagonize the interaction of Wnts with Lrp5/6 [58, 59]. The activity of Wnt signaling can also be modulated by Wnt inhibitory factor (Wif) and sFrps. These proteins can directly bind Wnts and prevent their interactions with the Fzd/Lrp receptor complex [60, 61].
Wnt Signaling in Heart Development
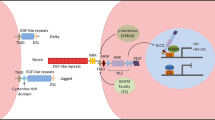
Wnt/β-catenin signaling regulates cardiac development during embryogenesis, including cardiac mesoderm specification, myocardial differentiation, and proliferation. The activation of Wnt signaling expands mesodermal cardiac progenitor cells (CPCs) and maintains them in an undifferentiated state [62,63,64]. Inhibition of Wnt signaling induces cardiac specification in mesoderm and promotes cardiomyocyte differentiation [65,66,67]. Wnt/β-catenin restriction by non-canonical Wnt11 is necessary for CPCs to differentiate into cardiomyocytes through caspase-mediated degradation [68]. sFrp1, sFrp2, and sFrp5 are closely related and play similar roles through Wnt signaling inhibition for the differentiation of CPCs into cardiomyocytes [61, 69]. Like sFrps, the secreted Wnt inhibitor Dkk1 enhances cardiac specification, whereas the addition of Dkk1 before CPC specification prevents cardiomyocyte differentiation [61, 70, 71]. Dkk1/Dkk2 double knockout mice display a variety of cardiac developmental defects including smaller hearts, suggesting a requirement for Dkks during cardiogenesis [72]. In murine embryonic stem cell (mESC) differentiation, activation of Wnt/β-catenin signaling after embryoid body formation inhibits myocardial differentiation while promoting the differentiation of endothelial and hematopoietic lineages [65]. During vertebrate embryo development, Wnt activation induces the specification of ESCs into the anterior and posterior lateral plate mesoderm (LPM) (Fig. 2). In the anterior LPM, Wnt inhibitor Dkk, secreting from the endoderm, prevents Wnts from binding to their receptors, leading to the induction of the cardiogenic mesoderm and the formation of CPCs. The inhibition of Wnt signaling cooperates with BMPs and Fgf8 to activate the expression of Mesp1 and Nkx2.5, which further induces the differentiation of CPCs into cardiomyocytes. In the LPM, Wnt signals that emanate from the neural tube instruct the posterior mesoderm to become hemangiogenic mesoderm. Both BMPs and Wnt signals operate together to promote the differentiation of hemangiogenic mesoderm into the blood and blood vessels [73,74,75,76]. Studies in zebrafish identify a novel small molecule Wnt inhibitor (named as Cardionogen) that enlarges the size of embryonic heart by inducting cardiomyocyte formation. Administration of Cardionogen during and after gastrulation promotes cardiomyogenesis, whereas its treatment before gastrulation inhibits heart formation [77]. These findings illustrate that Wnt/β-catenin signaling exhibits biphasic and antagonistic effects on cardiac differentiaiton, depending on the stage of development.

Wnt/β-catenin signaling exhibits biphasic effects on cardiogenesis. During early embryogenesis, Wnt/β-catenin signaling activation induces the mesoderm specification from embryonic stem (ES) cells. In the anterior mesoderm, Wnt inhibitor Dkk, secreting from the endoderm, prevents Wnt signaling from functioning and converts the anterior LPM into cardiogenic mesoderm. In collaboration with BMP and FGF8 signals, Wnt inhibition further induces the differentiation CPCs into cardiomyocytes. In the posterior mesoderm, Wnt signals, emanating from the neural tube, instruct the posterior LPM to become hemangiogenic mesoderm. The cooperation of BMPs with Wnt activation ultimately induces the differentiation of hemangiogenic mesoderm into the blood and blood vessels. Abbreviations: ES cell, embryonic stem cell; LPM, lateral plate mesoderm; BMP, bone morphogenetic protein; FGF8, fibroblast growth factor 8
Both in vivo and in vitro studies demonstrate that the effects of Wnt signaling oscillate between promoting and restricting cardiomyocytes formation during myocardial differentiation [62, 65, 66, 78, 79]. hESCs studies show that WNT3 and WNT8A via FZD7 regulate Brachyury expression and cardiac mesoderm induction. Subsequently, non-canonical WNT5A/5B via tyrosine-protein kinase transmembrane receptor (ROR2) controls MESP1 expression and CPC specification. During late hESC development, WNT2, WNT5A/5B, and WNT11, via FZD4 and FZD6, regulate cardiomyocyte differentiation through non-canonical Wnt signaling [80]. In human-induced pluripotent stem cell (hiPSC), shRNA knockdown of β-catenin during the initial differentiation stage fully blocks cardiac mesoderm specification, whereas GSK-3β inhibition enhances CPC formation [81]. Furthermore, GSK3β inhibition combined with the removal of cell–cell contacts enables the expansion of hiPSC-derived cardiomyocytes. Mechanistically, persistent cardiomyocyte proliferation requires both serine/threonine protein kinase Akt phosphorylation and Lef/Tcf activity that are independent of Yes-associated protein (YAP) signaling. At embryonic stages, Wnt signals can activate Akt, which in turn negatively regulates Gsk3β, thus promoting the expression of downstream target Cyclin D1. Gsk3β-mediated phosphorylation of cyclin D1 appears to be a central regulator of cell proliferation and differentiation in the developing heart [82].
During second heart field (SHF) development, Wnt/β-catenin signaling contributes to SHF expansion and right ventricle growth [83, 84]. Cells from SHF give rise to the right ventricle (RV), the outflow tract (OFT), and parts of the inflow tract (IFT). Wnt signaling is required for Islet1(Isl1)-expressing cardiac progenitor proliferation for SHF development. Conditional deletion of β-catenin using Isl1-Cre mice reduces the number of SHF progenitors, resulting in right ventricle formation defects. Inversely, constitutive expression of β-catenin in non-canonical Wnt5a and Wnt11 double-mutant mice leads to SHF expansion and OFT morphogenic defects [85,86,87,88]. Wnt signaling is negatively regulated by Notch1-mediated β-catenin phosphorylation within CPCs. The expression of cardiac transcription factors Isl1, Myocd, or Smyd1 is positively regulated by Notch1 but controlled negatively by β-catenin. This restriction by Notch1 is required for CPC transition from the expansive stage into the differentiated state [78]. Isl1 represses the CPC expansion but also induces the differentiation of CPCs, SHF cells, or the anterior foregut endodermal cells that are in close contact to the heart anlage. Forced activation of β-catenin in Isl1+ cells of mouse embryos decreases Isl1 expression in CPCs, enabling CPC expansion [78, 87, 89]. These evidence illustrate that Wnt/β-catenin signaling contributes to Isl1+ SHF progenitor expansion that is fine-tuned by proper differentiation.
Wnt signaling also plays a pivotal role during endocardial cushion formation and atrioventricular valve development. At E12.5, Wnt2 is expressed in the cushion mesenchyme, whereas Wnt4 and Wnt9b are predominantly present in overlying endothelial cells. At E17.5, both Wnt3a and Wnt7b are expressed in the remodeling atrioventricular (AV) and semilunar valves [90]. The cushion myocardium exhibits high Wnt activities during endocardial cushion growth [91, 92]. The deletion of β-catenin in myocardium results in hypoplastic endocardial cushions with a reduction of mesenchymal cell proliferation. The loss of β-catenin reduces Bmp2 expression in myocardium and Smad signaling in cushion mesenchyme [93, 94]. Studies in zebrafish indicate that an application of Wnt8 after gastrulation fails to form the endocardial cushion [66]. Overexpression of Apc or Dkk1 also blocks endocardial cushion formation. In Apc-truncated-hearts, proliferation and epithelial–mesenchymal transition (EMT) that is normally restricted to endocardial cushion occurs throughout the endocardium [95]. Furthermore, the disruption of Tbx20 results in aberrant Wnt/β-catenin signaling in the endocardial cushion, causing a severe valve elongation defect and an impaired cardiac function [96]. Overall, these findings demonstrate the crucial regulation of endocardial cushion formation by Wnt/β-catenin signaling during heart valve development.
Wnt Signaling in Heart Homeostasis and Wound Healing
Recent studies report that Wnt/β-catenin signaling governs metabolic regulatory programs and sustains metabolic plasticity during adult heart homeostasis. In the heart of human over 45 years old, the expression of β-catenin is stronger than younger individuals, implying that Wnt signaling involves in the regulation of adult heart homeostasis in the process of age-related cardiac function [97]. Myocardial-specific deletion of exon 3 of the Ctnnb1 gene (β-catenin) from P14 to 4 weeks in murine heart by removing GSK3β phosphorylation sites stabilizes β-catenin that activates the Wnt/β-catenin pathway. Although the post-mitotic cardiomyocytes overexpressing stabilized β-catenin re-enter the cell cycle and express cytokinesis genes, these cardiomyocytes fail to increase in cardiomyocytes numbers. In contrast, the deletion of exons 8–13 of Ctnnb1 that expresses the inactive truncated β-catenin causes the increased expression of genes involved in oxidative phosphorylation. The short-term β-catenin expression from P14 to 4 weeks in these adult cardiomyocytes emphasizes the potential role for this pathway in mitochondrial oxidative phosphorylation during cardiac homeostasis [98]. Consistently, activation of Wnt signaling in C2C12 myoblasts is reported to stimulate mitochondrial proliferation and mitochondrial oxidative phosphorylation [99]. Furthermore, active β-catenin restores mitochondrial function in the brain of parkinsonian rats, but hepatocyte-specific expression of constitutively active β-catenin leads to early lethality due to mitochondrial dysfunction. Recent studies report that cardiac-specific β-catenin ablation in 1-day-old mice disrupts the energy substrate shift that is essential for postnatal heart maturation, leading to perinatal lethality of mice [100]. Transcriptomic analyses show that β-catenin deficiency at postnatal mice leads to mitochondria dysfunction via the downregulation of Sirt1/Ppargc-1α pathway, a master transcriptional activator complex for controlling the expression of metabolism regulators and mitochondrial biogenesis genes. Further chromatin immunoprecipitation sequencing (Chip-seq) analyses in Gsk3 inhibitor CHIR99021 (CHIR)-treated adult hearts indicate that β-catenin fails to re-engage neonatal proliferative gene network despite partial transcriptional re-activation of a neonatal glycolytic gene program [101•]. Notably, β-catenin haplo-insufficient mice subjected to endurance training disturbs the activity of mitochondrial oxidative phosphorylation complexes and enhances Ampk, PI3K–Akt, and Mapk/Erk1/2 signaling pathways, leading to attenuation of cardiomyocytes hypertrophic growth [102].
There is increasing evidence that both Wnts and Wnt antagonists are induced in multiple cell types during cardiac injury repair and wound healing process. In mouse MI model with left anterior descending artery (LAD) ligation, the increased expression of canonical Wnt1, Wnt10b, and non-canonical Wnt2, Wnt4, and Wnt11 was observed in the epicardium and fibroblasts near-injured areas. Moreover, the increased expression of Dkk1 and Dkk2, as well as Fzd1, Fzd2, Fzd5, and Fzd10, is detectable in injured hearts [103,104,105]. In human dilated cardiomyopathy or coronary heart disease (CHD), the mRNA levels of sFRP3 and sFRP4 but not of sFRP1 and sFRP2 are elevated [106]. In the same context, sFRP1 levels are shown to be downregulated in patients suffering from heart failure but this effect is reversed following left ventricular assist device (LVAD) support [107]. Nuclear levels of β-catenin, Lef1, and Tcf1/3/4 are shown to be induced in the epicardium at the end-stage pediatric allografts [108]. Studies using Axin2 reporter mice and TOPGAL reporter mice reveal the expression of Wnt pathway components in endothelial cells, fibroblasts, leukocytes, and Sca+ progenitor cells in the border zone of the infarct heart. During human right ventricular remodeling and failure, the fetal Wnt gene program, especially non-canonical pathway, is robustly reactivated that upregulates ROR2/Ca2+ responsive protease calpain-μ and increases cleavage of calpain-target cytoskeletal proteins [83]. The accumulation of β-catenin in endothelial cells of newly formed blood vessels is also observed in the neovascularization of the infarct area [109].
Most evidence support that inhibition of Wnt signaling is beneficial for injury repair and recovery of cardiac function. Conditional depletion of β-catenin in myocardium using the Myh6-Cre driver line improves left ventricular function and survival rate after ligation of LAD in mice. β-catenin-depleted infarct hearts show increased numbers of cTnT+ cardiomyocytes in subepicardial and subendocardial positions [110]. Further transcriptomic analysis of β-catenin-overexpressing hearts shows a strong recapitulation of cardiac developmental program and cytoskeletal remodeling, in which transcription factor-7 like 2 (Tcf7L2) co-occupies genomic regions with Nkx2.5 and Gata4. Conversely, preventing β-catenin activation in post-pressure-overload TAC model results in a downregulation of Tcf7L2/Nkx2.5/Gata4 developmental reprogramming, preventing heart failure [111]. Consistently, mice with cardiac-specific Dkk3 deficiency increases infarct size and exacerbates left ventricular dysfunction after MI. Inversely, Dkk3 overexpression in infarct hearts leads to the opposite phenotype with improved cardiac function recovery [112]. Similarly, the injection of Dkk2 enhances the neovascularization of the infarct area with concomitant improved cardiac function [113]. On the other hand, myocardium-specific deletion of Gsk3β that elevates Wnt/β-catenin activity results in an increase in numbers of BrdU+/TnI+ cells and a reduction in infarct sizes after LAD ligation, suggesting beneficial for cardiac wound healing [114]. These findings contrast to the observation that inhibition of Wnt/β-catenin signaling is ameliorative for cardiac injury repair and function recovery. It has been noted that GSK3β does not exclusively function in Wnt/β-catenin signaling but is also involved in other signaling pathways, such as AMPK-TSC2-mTOR pathway that plays an important role in cellular energy homeostasis. Furthermore, GSK3β modulates intracellular signaling molecules downstream from Wnts that are independent of β-catenin [115]. Therefore, it is likely that the beneficial effect for cardiac wound healing by Gsk3β inhibition might be caused by its Wnt-independent activities.
Wnt/β-Catenin Pathway in Cardiac Fibrosis
Cardiac fibroblasts (CF) comprise approximately two-thirds of cells in the healthy adult heart but only a minor fraction of the heart volume [116]. CFs are interstitial mesenchymal like cells with intermediate filaments and highly heterogeneous, embedding in extracellular matrix of connective tissue [117]. During development, most CFs are of epicardial origin and some of them are endocardial [118, 119]. CFs transform into cardiac myofibroblasts and secrete excessive extracellular matrix (ECM) in response to cardiac stress such as MI and inflammation. ECM and growth factors (FGF23, FGF21, PDGFα, and TNFα) secreted from CFs are essential for cardiac remodeling and fibrotic scar formation [120,121,122,123,124]. Wnt-dependent interactions between CFs and cardiomyocytes play crucial roles during cardiac injury and regeneration. Epicardial cells that express Wnt1 undergo EMT to adopt fibroblast fates in a β-catenin-dependent manner following injury [104, 125]. Wnt trafficking gene Wntless (Wls) regulates noncanonical Wnt signaling between CFs and cardiomyocytes. Wls deletion decreases the secretion of noncanonical Wnt5a and Wnt9a from cardiomyocytes to CFs, leading to CF activation and the impairment of neonatal heart regeneration [126••]. On the other hand, noncanonical Wnt5a can stimulate fibroblasts to secrete pro-inflammatory cytokine interleukin-6 (IL-6) and tissue inhibitor of metalloproteinase-1 (TIMP-1), promoting myocardial inflammation and fibrosis [127]. In cultured endothelial cells, treatment with GSK3β inhibitor BIO that activates β-catenin transcription is sufficient to induce EndMT and CF formation [119]. In human hearts, the loss of cardiac endogenous Klotho, an antiaging substance with pleiotropic actions including regulation of mineral metabolism, facilitates TGF-β1 signaling and enables vigorous cardiac fibrosis through upregulating Wnt signaling [128].
sFrps have emerged as key regulators by antagonizing Wnt signaling during cardiac fibrosis [60]. CFs lacking sFrp-1 increase α-smooth muscle actin and collagen deposition [129]. Injection of sFrp1, sFrp2, and sFrp4 into the rat MI models inhibits injury-induced collagen and left ventricular fibrosis. Bmp1 is a key enzyme involved in regulating collagen biosynthesis and maturation, which can be inhibited by high concentrations of sFrp2 [126••]. Although the anti-fibrotic effect of sFrp2 on cardiac remodeling has been reported, many studies report a pro-fibrotic role for sFrp2. Kobayashi et al. found that sFrp2 is primarily expressed on CFs in the region of injury, which enhances pro-collagen C-proteinase activity and promotes the conversion of collagen from pro-collagen. Mice deficient in sFrp2 exhibit a reduction in fibrosis after MI, leading to marked preservation in post-injury cardiac function [130]. Similarly, a sFrp2-neutralizing antibody by the intraperitoneal delivery in cardiomyopathy hamsters is beneficial to reducing fibrosis and improving cardiac function [131]. The exact reason for the differential effect of sFrp2 on myocardial infarct is unclear. It could be related to the concentration of sFrp2 in the infarcted heart and the degree of sFrp2-mediated Wnt antagonism [126••].
Inhibition of Wnt/β-catenin signaling is capable of reducing cardiac fibrosis. Cardiac pressure overload resulting from TAC in mice leads to increased Wnt/β-catenin signaling in CFs and cardiac fibrosis. β-catenin is specifically required in resident CFs for ECM gene expression after cardiac injury. CFs lacking β-catenin after TAC significantly reduces interstitial fibrosis and preserves cardiac function but does not alter the number of activated CFs [132]. Interruption of Wnt signaling in mice lacking Dvl-1 attenuates the onset of pressure overload-induced cardiac hypertrophy and interstitial fibrosis [133]. Together, these findings illustrate that Wnt/β-catenin signaling promotes cardiac fibrosis by transition to the mesenchymal state from epicardial and endothelial cell fates in response to injury.
Intervention of Wnt Signaling in Cardiac Repair and Regeneration
Intervention in Wnt signaling has been employed as potential therapeutic strategies following cardiac injury at different levels or steps in Wnt signaling pathway, including the position at Wnt secretion, ligand, receptor, destruction complex, or the nucleus (Table 1). A large number of studies focus on the roles of sFrp family proteins in cardiac injury treatment. Genetic overexpression of sFrp1 is shown to reduce Mmp2/Mmp9 activity, collagen deposition, and infarct size, improving cardiac function [105]. Injection of sFrp2 protein into the infarct area of rat ventricle inhibits MI-induced cardiac fibrosis, prevents anterior wall thinning, and improves cardiac function recovery [134]. Intramuscular injection of recombinant sFrp4 reduces fibrosis scar size and ameliorates cardiac function after ischemic/reperfusion (I/R) injury [135]. In the same direction, the deletion of sFrp5 results in a significant increase in infarct size. sFrp5 limits the magnitude of the inflammatory response, thereby reducing infarct size following I/R injury [136, 137]. Wif-1, a secreted antagonist of Wnt signaling, has been shown to significantly attenuate the monocyte response and improve cardiac function through AAV9-mediated overexpression. In contrast, Wif1 knockout mice develops severe and unwanted cardiac remodeling 4 weeks after MI, manifesting the increased scar size and decreased ejection fraction [138]. Non-canonical Wnt11 administration via rAAV9 confers cardioprotective effects in Coxsackievirus B3 (CVB3)-induced myocarditis model by reducing cardiomyocyte necrosis, infiltration of inflammatory cells, and inflammatory cytokine expression [139]. In contrast, cardiac injection of recombinant canonical Wnt3a (rWnt3a) leads to a substantial enhancement in infarct size and cardiac remodeling after the induction of MI [140]. On the other hand, canonical Wnt10b gain-of-function improves cardiac repair by arteriole formation and attenuation of fibrosis through stimulating NF-kB signaling in endothelial cells and vascular smooth muscle cells (VSMCs) [141]. Notably, insulin-like growth factor binding protein 4 (Igfbp4) prevents MI-induced cardiomyocyte death and improves the recovery of heart function by inhibiting β-catenin stability, providing a molecular link between IGF signaling and Wnt signaling in heart repair [142]. Similarly, when infarct mice expose to Gata4, Mef2c, Tbx5(GMT) and TGF-β inhibitor, together with Tankyrase inhibitor XAV939 that stimulates β-catenin degradation by stabilizing Axin, manifest significantly improved cardiac reprogramming of fibroblasts and cardiac function compared to those exposed to only GMT [143, 144]. In cultured adult mouse CFs, XAV939 administration abrogates CF activation and ECM production in response to angiotensin II [132].
Porcupine, an acyltransferase capable of secreting Wnt ligands, has been shown to be a highly druggable target for inhibiting Wnt signaling pathway (Table 1). Wnt974, a chemical Porcn inhibitor, improves the recovery of heart function by reducing collagen production and fibrotic scarring [145]. The treatment of infarct hearts with CGX1321, another Porcn inhibitor, also causes suppression of fibrotic depositions [146]. Notably, the administration of CGX1321 enhances cardiomyocyte generation in the border zone of infarct heart [146, 147]. Myocardial injection of pyrvinium, a CK1α agonist that is known to block Wnt signaling, can also increase cell proliferation in the injured myocardium and mitigates adverse cardiac remodeling [148]. Cardiomogen (CDMG), a novel Wnt inhibitor, was identified to promote cardiomyocyte formation during zebrafish embryogenesis. Administration of CDMG in mice following LAD ligation reduces fibrotic scar tissue and enhances cardiomyocyte generation in the infarct border zone [149]. These studies suggest that the development of Wnt inhibitors may ultimately aid in the design of therapeutic approaches to promote cardiac repair and regeneration in response to injury.
Zebrafish heart possesses innate repair and regeneration capacity after cardiac injury. Recent studies report that cardiac apex resection induces the expression and secretion of Wnt inhibitors Dkk3/sFrp1 from the epicardium and fibroblasts, Notumb1/Wif1 from the endocardium, and Dkk1/sFrp2 in the myocardium [150, 151••]. Inversely, expressions of Wnt ligands, including Wnt4a, Wnt6b, and Wnt8a, are reduced in injured zebrafish hearts. However, the expression of non-canonical Wnt2bb is increased after cardiac injury [150, 152], suggesting that canonical Wnt signaling needs to be restrained to enable innate heart regeneration. Importantly, blocking Wnt signaling via heat-shock induced Dkk1 or non-canonical Wnt2bb overexpression enhances cardiomyocyte generation and reduces fibrotic scarring following cardiac resection [150, 152]. On the contrary, ectopic activation of canonical Wnt8 blunts injury-induced cardiomyocyte dedifferentiation and proliferation and increases fibrotic scarring [151••]. Consistently, dampen myocardial Wnt signaling by the endocardial Notch signaling promotes cardiomyocyte formation and zebrafish heart regeneration [150]. On the other hand, it has been shown that myocardial overexpression of Axin1 that negatively regulates Wnt signaling perturbs cardiomyocyte proliferation and heart regeneration [153]. This impairment of cardiac regeneration by Axin might be due to its involvement in non-canonical Wnt signaling or Wnt-independent molecular events [154]. For example, Axin1 scaffolding protein bridges Daxx to activate P53-dependent cell death [155]. Axin family members also cooperate with other signaling proteins including JNK, TGF-β, and AMPK to regulate diverse cellular processes [156]. Alternatively, current studies suggest that cardiac regeneration undergoes dedifferentiation of existing-cardiomyocytes, following by proliferation and re-differentiation of these cells [19]. It is likely that Wnt activation and inhibition are needed respectively for heart regeneration at different stages, analogous to its biphasic signaling during heart development.
Conclusion
Wnt/β-catenin signaling plays highly conserved roles in various biological processes in mammals and non-mammalian vertebrates. Studies in mice, zebrafish, and human embryonic stem cells demonstrate an elegant and binary role for Wnt/β-catenin signaling during heart development. Recent studies illustrate the critical importance of canonical and non-canonical Wnt activities in the regulation of cardiac homeostasis, fibrosis, injury repair, and regeneration. A general picture has emerged that inhibition of Wnt signaling is advantageous for wound healing and cardiac repair. A couple of remarks need to be presented, since the effects of Wnt signaling on infarct healing are sometimes variable and inconsistent. Cardiac healing is a complex and dynamic process and many factors can affect the outcomes of repair, including diverse injury manipulations and different wound stages. Genetic intervention and deletion of a specific genes have been also used in many cardiac injury studies. This may lead to redundancy and adaptation of organisms to specific gene deletions, considering Wnt pathway has many different levels of homologues in signaling cascades. Many cross-regulatory mechanisms (e.g., Notch, ROS, NF-κB, TGFβ, YAP/TAZ, RAS, and VEGF signaling) can modulate Wnt signaling transduction activities that can further influence the outcomes. Moreover, many key components in the Wnt pathway have non-canonical Wnt activities or participate in Wnt-independent molecular pathways. For example, Gsk3β, β-catenin, Axin, and sFrp are involved in a plethora of Wnt-independent cellular processes. These non-canonical or Wnt-independent activities may add to the effects on cardiac repair and wound healing. It is intriguing to note, in the most recent publications, that cardiac regeneration has been implicated as the strength underlying the beneficial effects of the Wnt inhibition on cardiac wound healing. In the future, new experimental technology, such as spatial transcriptome, metabolomics, and combined multi-omics, will be required for a higher-level systematic analysis for complex cellular processes such as injury repair and heart regeneration. Highly selective Wnt antagonists without affecting other molecular pathways will be imperative to develop and optimize. Hopefully, the multi-cellular level experimental strategies in different animal models combined with higher-level systems analysis will transform the outcome of basic studies into clinical medicine, ultimately allowing the evaluation of Wnt-dependent therapeutic intervention in human cardiovascular diseases.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Litvinukova M, Talavera-Lopez C, Maatz H, Reichart D, Worth CL, Lindberg EL, et al. Cells of the adult human heart. Nature. 2020;588(7838):466–72.
Tucker NR, Chaffin M, Fleming SJ, Hall AW, Parsons VA, Bedi KC Jr, et al. Transcriptional and cellular diversity of the human heart. Circulation. 2020;142(5):466–82.
Cui Y, Zheng Y, Liu X, Yan L, Fan X, Yong J, et al. Single-cell transcriptome analysis maps the developmental track of the human heart. Cell Rep. 2019;26(7):1934–50 e5.
Chen L, Hua K, Zhang N, Wang J, Meng J, Hu Z, et al. Multifaceted spatial and functional zonation of cardiac cells in adult human heart. Circulation. 2022;145(4):315–8.
Gao L, Zhang H, Cui J, Pei L, Huang S, Mao Y, et al. Single-cell transcriptomics of cardiac progenitors reveals functional subpopulations and their cooperative crosstalk in cardiac repair. Protein Cell. 2021;12(2):152–7.
Maliken BD, Molkentin JD. Undeniable evidence that the adult mammalian heart lacks an endogenous regenerative stem cell. Circulation. 2018;138(8):806–8.
Li Y, He L, Huang X, Bhaloo SI, Zhao H, Zhang S, et al. Genetic lineage tracing of nonmyocyte population by dual recombinases. Circulation. 2018;138(8):793–805.
Elhelaly WM, Cardoso AC, Pereira AHM, Elnawasany A, Ebrahimi S, Nakada Y, et al. C-kit cells do not significantly contribute to cardiomyogenesis during neonatal heart regeneration. Circulation. 2019;139(4):559–61.
Wang YX, Blau HM. Reversing aging for heart repair. Science. 2021;373(6562):1439–40.
He L, Nguyen NB, Ardehali R, Zhou B. Heart regeneration by endogenous stem cells and cardiomyocyte proliferation: controversy, fallacy, and progress. Circulation. 2020;142(3):275–91.
Jopling C, Sleep E, Raya M, Marti M, Raya A, Izpisua Belmonte JC. Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature. 2010;464(7288):606–9.
Garbern JC, Lee RT. Heart regeneration: 20 years of progress and renewed optimism. Dev Cell. 2022;57(4):424–39.
Bongiovanni C, Sacchi F, Da Pra S, Pantano E, Miano C, Morelli MB, et al. Reawakening the intrinsic cardiac regenerative potential: molecular strategies to boost dedifferentiation and proliferation of endogenous cardiomyocytes. Front Cardiovasc Med. 2021;8: 750604.
Vagnozzi RJ, Molkentin JD, Houser SR. New myocyte formation in the adult heart: endogenous sources and therapeutic implications. Circ Res. 2018;123(2):159–76.
Cai W, Tan J, Yan J, Zhang L, Cai X, Wang H, et al. Limited regeneration potential with minimal epicardial progenitor conversions in the neonatal mouse heart after injury. Cell Rep. 2019;28(1):190-201.e3.
Sereti KI, Nguyen NB, Kamran P, Zhao P, Ranjbarvaziri S, Park S, et al. Analysis of cardiomyocyte clonal expansion during mouse heart development and injury. Nat Commun. 2018;9(1):754.
Porrello ER, Mahmoud AI, Simpson E, Johnson BA, Grinsfelder D, Canseco D, et al. Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proc Natl Acad Sci U S A. 2013;110(1):187–92.
Poss KD, Wilson LG, Keating MT. Heart regeneration in zebrafish. Science. 2002;298(5601):2188–90.
Tzahor E, Poss KD. Cardiac regeneration strategies: staying young at heart. Science. 2017;356(6342):1035–9.
Cano-Martinez A, Vargas-Gonzalez A, Guarner-Lans V, Prado-Zayago E, Leon-Oleda M, Nieto-Lima B. Functional and structural regeneration in the axolotl heart (Ambystoma mexicanum) after partial ventricular amputation. Arch Cardiol Mex. 2010;80(2):79–86.
Flink IL. Cell cycle reentry of ventricular and atrial cardiomyocytes and cells within the epicardium following amputation of the ventricular apex in the axolotl, Amblystoma mexicanum: confocal microscopic immunofluorescent image analysis of bromodeoxyuridine-labeled nuclei. Anat Embryol (Berl). 2002;205(3):235–44.
Senyo SE, Steinhauser ML, Pizzimenti CL, Yang VK, Cai L, Wang M, et al. Mammalian heart renewal by pre-existing cardiomyocytes. Nature. 2013;493(7432):433–6.
Liu X, Pu W, He L, Li Y, Zhao H, Li Y, et al. Cell proliferation fate mapping reveals regional cardiomyocyte cell-cycle activity in subendocardial muscle of left ventricle. Nat Commun. 2021;12(1):5784.
Pu W, Zhang M, Liu X, He L, Li J, Han X, et al. Genetic proliferation tracing reveals a rapid cell cycle withdrawal in preadolescent cardiomyocytes. Circulation. 2022;145(5):410–2.
Lazar E, Sadek HA, Bergmann O. Cardiomyocyte renewal in the human heart: insights from the fall-out. Eur Heart J. 2017;38(30):2333–42.
Bergmann O, Zdunek S, Felker A, Salehpour M, Alkass K, Bernard S, et al. Dynamics of cell generation and turnover in the human heart. Cell. 2015;161(7):1566–75.
Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabe-Heider F, Walsh S, et al. Evidence for cardiomyocyte renewal in humans. Science. 2009;324(5923):98–102.
Nakada Y, Canseco DC, Thet S, Abdisalaam S, Asaithamby A, Santos CX, et al. Hypoxia induces heart regeneration in adult mice. Nature. 2017;541(7636):222–7.
Cardoso AC, Lam NT, Savla JJ, Nakada Y, Pereira AHM, Elnwasany A, et al. Mitochondrial substrate utilization regulates cardiomyocyte cell cycle progression. Nat Metab. 2020;2(2):167–78.
Xin M, Olson EN, Bassel-Duby R. Mending broken hearts: cardiac development as a basis for adult heart regeneration and repair. Nat Rev Mol Cell Biol. 2013;14(8):529–41.
Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149(6):1192–205.
Bastakoty D, Saraswati S, Joshi P, Atkinson J, Feoktistov I, Liu J, et al. Temporary, systemic inhibition of the WNT/beta-catenin pathway promotes regenerative cardiac repair following myocardial infarct. Cell Stem Cells Regen Med. 2016;2(2). https://doi.org/10.16966/2472-6990.111.
Nusse R, Varmus HE. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell. 1982;31(1):99–109.
Nusse R, Varmus H. Three decades of Wnts: a personal perspective on how a scientific field developed. EMBO J. 2012;31(12):2670–84.
MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17(1):9–26.
Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127(3):469–80.
Nusse R, Clevers H. Wnt/beta-catenin signaling, disease, and emerging therapeutic modalities. Cell. 2017;169(6):985–99.
Gao C, Chen YG. Dishevelled: The hub of Wnt signaling. Cell Signal. 2010;22(5):717–27.
Giles RH, van Es JH, Clevers H. Caught up in a Wnt storm: Wnt signaling in cancer. Biochim Biophys Acta. 2003;1653(1):1–24.
Kikuchi A, Yamamoto H, Sato A, Matsumoto S. New insights into the mechanism of Wnt signaling pathway activation. Int Rev Cell Mol Biol. 2011;291:21–71.
Veeman MT, Axelrod JD, Moon RT. A second canon. Functions and mechanisms of beta-catenin-independent Wnt signaling. Dev Cell. 2003;5(3):367–77.
Mattes B, Dang Y, Greicius G, Kaufmann LT, Prunsche B, Rosenbauer J, et al. Wnt/PCP controls spreading of Wnt/beta-catenin signals by cytonemes in vertebrates. Elife. 2018;7: e36953.
Zhang X, Dong S, Xu F. Structural and druggability landscape of frizzled G protein-coupled receptors. Trends Biochem Sci. 2018;43(12):1033–46.
Foulquier S, Daskalopoulos EP, Lluri G, Hermans KCM, Deb A, Blankesteijn WM. WNT signaling in cardiac and vascular disease. Pharmacol Rev. 2018;70(1):68–141.
Niehrs C, Shen J. Regulation of Lrp6 phosphorylation. Cell Mol Life Sci. 2010;67(15):2551–62.
Song X, Wang S, Li L. New insights into the regulation of Axin function in canonical Wnt signaling pathway. Protein Cell. 2014;5(3):186–93.
de Man SM, Zwanenburg G, van der Wal T, Hink MA, van Amerongen R. Quantitative live-cell imaging and computational modeling shed new light on endogenous WNT/CTNNB1 signaling dynamics. Elife. 2021;10: e66440.
Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997;16(13):3797–804.
Herbst A, Jurinovic V, Krebs S, Thieme SE, Blum H, Goke B, et al. Comprehensive analysis of beta-catenin target genes in colorectal carcinoma cell lines with deregulated Wnt/beta-catenin signaling. BMC Genomics. 2014;15:74.
Valenta T, Hausmann G, Basler K. The many faces and functions of beta-catenin. EMBO J. 2012;31(12):2714–36.
Cadigan KM, Waterman ML. TCF/LEFs and Wnt signaling in the nucleus. Cold Spring Harb Perspect Biol. 2012;4(11): a007906.
Moshkovsky AR, Kirschner MW. The nonredundant nature of the Axin2 regulatory network in the canonical Wnt signaling pathway. Proc Natl Acad Sci U S A. 2022;119(9): e2108408119.
Hulin A, Moore V, James JM, Yutzey KE. Loss of Axin2 results in impaired heart valve maturation and subsequent myxomatous valve disease. Cardiovasc Res. 2017;113(1):40–51.
Lustig B, Jerchow B, Sachs M, Weiler S, Pietsch T, Karsten U, et al. Negative feedback loop of Wnt signaling through upregulation of conductin/axin2 in colorectal and liver tumors. Mol Cell Biol. 2002;22(4):1184–93.
Evron T, Caspi M, Kazelnik M, Shor-Nareznoy Y, Armoza-Eilat S, Kariv R, et al. A CRISPR knockout screen reveals new regulators of canonical Wnt signaling. Oncogenesis. 2021;10(9):63.
Tuladhar R, Yarravarapu N, Ma Y, Zhang C, Herbert J, Kim J, et al. Stereoselective fatty acylation is essential for the release of lipidated WNT proteins from the acyltransferase Porcupine (PORCN). J Biol Chem. 2019;294(16):6273–82.
Langton PF, Kakugawa S, Vincent JP. Making, exporting, and modulating Wnts. Trends Cell Biol. 2016;26(10):756–65.
Bao J, Zheng JJ, Wu D. The structural basis of DKK-mediated inhibition of Wnt/LRP signaling. Sci Signal. 2012;5(224):pe22.
Joiner DM, Ke J, Zhong Z, Xu HE, Williams BO. LRP5 and LRP6 in development and disease. Trends Endocrinol Metab. 2013;24(1):31–9.
Cruciat CM, Niehrs C. Secreted and transmembrane wnt inhibitors and activators. Cold Spring Harb Perspect Biol. 2013;5(3): a015081.
Hsueh YC, Hodgkinson CP, Gomez JA. The role of Sfrp and DKK proteins in cardiomyocyte development. Physiol Rep. 2021;9(3): e14678.
Qyang Y, Martin-Puig S, Chiravuri M, Chen S, Xu H, Bu L, et al. The renewal and differentiation of Isl1+ cardiovascular progenitors are controlled by a Wnt/beta-catenin pathway. Cell Stem Cell. 2007;1(2):165–79.
Noseda M, Peterkin T, Simoes FC, Patient R, Schneider MD. Cardiopoietic factors: extracellular signals for cardiac lineage commitment. Circ Res. 2011;108(1):129–52.
Gessert S, Kuhl M. The multiple phases and faces of wnt signaling during cardiac differentiation and development. Circ Res. 2010;107(2):186–99.
Naito AT, Shiojima I, Akazawa H, Hidaka K, Morisaki T, Kikuchi A, et al. Developmental stage-specific biphasic roles of Wnt/beta-catenin signaling in cardiomyogenesis and hematopoiesis. Proc Natl Acad Sci U S A. 2006;103(52):19812–7.
Ueno S, Weidinger G, Osugi T, Kohn AD, Golob JL, Pabon L, et al. Biphasic role for Wnt/beta-catenin signaling in cardiac specification in zebrafish and embryonic stem cells. Proc Natl Acad Sci U S A. 2007;104(23):9685–90.
Schneider VA, Mercola M. Wnt antagonism initiates cardiogenesis in Xenopus laevis. Genes Dev. 2001;15(3):304–15.
Touma M, Kang X, Gao F, Zhao Y, Cass AA, Biniwale R, et al. Wnt11 regulates cardiac chamber development and disease during perinatal maturation. JCI Insight. 2017;2(17): e94904.
Gomez JA, Payne A, Pratt RE, Hodgkinson CP, Dzau VJ. A role for Sfrp2 in cardiomyogenesis in vivo. Proc Natl Acad Sci U S A. 2021;118(33): e2103676118.
Yamashita JK, Takano M, Hiraoka-Kanie M, Shimazu C, Peishi Y, Yanagi K, et al. Prospective identification of cardiac progenitors by a novel single cell-based cardiomyocyte induction. FASEB J. 2005;19(11):1534–6.
Yang Y, Mlodzik M. Wnt-Frizzled/planar cell polarity signaling: cellular orientation by facing the wind (Wnt). Annu Rev Cell Dev Biol. 2015;31:623–46.
Phillips MD, Mukhopadhyay M, Poscablo C, Westphal H. Dkk1 and Dkk2 regulate epicardial specification during mouse heart development. Int J Cardiol. 2011;150(2):186–92.
David R, Brenner C, Stieber J, Schwarz F, Brunner S, Vollmer M, et al. MesP1 drives vertebrate cardiovascular differentiation through Dkk-1-mediated blockade of Wnt-signalling. Nat Cell Biol. 2008;10(3):338–45.
Tzahor E, Lassar AB. Wnt signals from the neural tube block ectopic cardiogenesis. Genes Dev. 2001;15(3):255–60.
Marvin MJ, Di Rocco G, Gardiner A, Bush SM, Lassar AB. Inhibition of Wnt activity induces heart formation from posterior mesoderm. Genes Dev. 2001;15(3):316–27.
Olson EN. Development. The path to the heart and the road not taken. Science. 2001;291(5512):2327–8.
Ni TT, Rellinger EJ, Mukherjee A, Xie S, Stephens L, Thorne CA, et al. Discovering small molecules that promote cardiomyocyte generation by modulating Wnt signaling. Chem Biol. 2011;18(12):1658–68.
Kwon C, Qian L, Cheng P, Nigam V, Arnold J, Srivastava D. A regulatory pathway involving Notch1/beta-catenin/Isl1 determines cardiac progenitor cell fate. Nat Cell Biol. 2009;11(8):951–7.
Tzahor E. Wnt/beta-catenin signaling and cardiogenesis: timing does matter. Dev Cell. 2007;13(1):10–3.
Mazzotta S, Neves C, Bonner RJ, Bernardo AS, Docherty K, Hoppler S. Distinctive roles of canonical and noncanonical Wnt signaling in human embryonic cardiomyocyte development. Stem Cell Reports. 2016;7(4):764–76.
Lian X, Hsiao C, Wilson G, Zhu K, Hazeltine LB, Azarin SM, et al. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc Natl Acad Sci U S A. 2012;109(27):E1848–57.
Buikema JW, Lee S, Goodyer WR, Maas RG, Chirikian O, Li G, et al. Wnt activation and reduced cell-cell contact synergistically induce massive expansion of functional human iPSC-derived cardiomyocytes. Cell Stem Cell. 2020;27(1):50–63 e5.
Edwards JJ, Brandimarto J, Hu DQ, Jeong S, Yucel N, Li L, et al. Noncanonical WNT activation in human right ventricular heart failure. Front Cardiovasc Med. 2020;7: 582407.
Ai D, Fu X, Wang J, Lu MF, Chen L, Baldini A, et al. Canonical Wnt signaling functions in second heart field to promote right ventricular growth. Proc Natl Acad Sci U S A. 2007;104(22):9319–24.
Li D, Sinha T, Ajima R, Seo HS, Yamaguchi TP, Wang J. Spatial regulation of cell cohesion by Wnt5a during second heart field progenitor deployment. Dev Biol. 2016;412(1):18–31.
Sinha T, Li D, Theveniau-Ruissy M, Hutson MR, Kelly RG, Wang J. Loss of Wnt5a disrupts second heart field cell deployment and may contribute to OFT malformations in DiGeorge syndrome. Hum Mol Genet. 2015;24(6):1704–16.
Cohen ED, Miller MF, Wang Z, Moon RT, Morrisey EE. Wnt5a and Wnt11 are essential for second heart field progenitor development. Development. 2012;139(11):1931–40.
Li D, Angermeier A, Wang J. Planar cell polarity signaling regulates polarized second heart field morphogenesis to promote both arterial and venous pole septation. Development. 2019;146(20): dev181719.
Klaus A, Saga Y, Taketo MM, Tzahor E, Birchmeier W. Distinct roles of Wnt/beta-catenin and Bmp signaling during early cardiogenesis. Proc Natl Acad Sci U S A. 2007;104(47):18531–6.
Alfieri CM, Cheek J, Chakraborty S, Yutzey KE. Wnt signaling in heart valve development and osteogenic gene induction. Dev Biol. 2010;338(2):127–35.
Bosada FM, Devasthali V, Jones KA, Stankunas K. Wnt/beta-catenin signaling enables developmental transitions during valvulogenesis. Development. 2016;143(6):1041–54.
Combs MD, Yutzey KE. Heart valve development: regulatory networks in development and disease. Circ Res. 2009;105(5):408–21.
Wang Y, Lu P, Wu B, Riascos-Bernal DF, Sibinga NES, Valenta T, et al. Myocardial beta-Catenin-BMP2 signaling promotes mesenchymal cell proliferation during endocardial cushion formation. J Mol Cell Cardiol. 2018;123:150–8.
Saxon JG, Baer DR, Barton JA, Hawkins T, Wu B, Trusk TC, et al. BMP2 expression in the endocardial lineage is required for AV endocardial cushion maturation and remodeling. Dev Biol. 2017;430(1):113–28.
Hurlstone AF, Haramis AP, Wienholds E, Begthel H, Korving J, Van Eeden F, et al. The Wnt/beta-catenin pathway regulates cardiac valve formation. Nature. 2003;425(6958):633–7.
Cai X, Zhang W, Hu J, Zhang L, Sultana N, Wu B, et al. Tbx20 acts upstream of Wnt signaling to regulate endocardial cushion formation and valve remodeling during mouse cardiogenesis. Development. 2013;140(15):3176–87.
Kasacka I, Piotrowska Z, Niezgoda M, Lewandowska A, Lebkowski W. Ageing-related changes in the levels of beta-catenin, CacyBP/SIP, galectin-3 and immunoproteasome subunit LMP7 in the heart of men. PLoS ONE. 2020;15(3): e0229462.
Olcum M, Cheedipudi SM, Rouhi L, Fan S, Jeong HH, Zhao Z, et al. The WNT/beta-catenin pathway regulates expression of the genes involved in cell cycle progression and mitochondrial oxidative phosphorylation in the postmitotic cardiac myocytes. J Cardiovasc Aging. 2022;2(2):15.
Yoon JC, Ng A, Kim BH, Bianco A, Xavier RJ, Elledge SJ. Wnt signaling regulates mitochondrial physiology and insulin sensitivity. Genes Dev. 2010;24(14):1507–18.
Balatskyi VV, Vaskivskyi VO, Myronova A, Avramets D, Nahia KA, Macewicz LL, et al. Cardiac-specific beta-catenin deletion dysregulates energetic metabolism and mitochondrial function in perinatal cardiomyocytes. Mitochondrion. 2021;60:59–69.
• Quaife-Ryan GA, Mills RJ, Lavers G, Voges HK, Vivien CJ, Elliott DA, et al. beta-Catenin drives distinct transcriptional networks in proliferative and nonproliferative cardiomyocytes. Development. 2020;147(22): dev193417. Findings from this study suggest that Wnt/β-catenin signalling in adult mice is cardioprotective but fails to induce cardiomyocyte proliferation, depending on the metabolic status of cardiomyocytes.
Balatskyi VV, Palchevska OL, Bortnichuk L, Gan AM, Myronova A, Macewicz LL, et al. Beta-catenin regulates cardiac energy metabolism in sedentary and trained mice. Life (Basel). 2020;10(12):357.
Aisagbonhi O, Rai M, Ryzhov S, Atria N, Feoktistov I, Hatzopoulos AK. Experimental myocardial infarction triggers canonical Wnt signaling and endothelial-to-mesenchymal transition. Dis Model Mech. 2011;4(4):469–83.
Duan J, Gherghe C, Liu D, Hamlett E, Srikantha L, Rodgers L, et al. Wnt1/betacatenin injury response activates the epicardium and cardiac fibroblasts to promote cardiac repair. EMBO J. 2012;31(2):429–42.
Barandon L, Couffinhal T, Ezan J, Dufourcq P, Costet P, Alzieu P, et al. Reduction of infarct size and prevention of cardiac rupture in transgenic mice overexpressing FrzA. Circulation. 2003;108(18):2282–9.
Schumann H, Holtz J, Zerkowski HR, Hatzfeld M. Expression of secreted frizzled related proteins 3 and 4 in human ventricular myocardium correlates with apoptosis related gene expression. Cardiovasc Res. 2000;45(3):720–8.
Felkin LE, Lara-Pezzi EA, Hall JL, Birks EJ, Barton PJ. Reverse remodelling and recovery from heart failure are associated with complex patterns of gene expression. J Cardiovasc Transl Res. 2011;4(3):321–31.
Ye B, Ge Y, Perens G, Hong L, Xu H, Fishbein MC, et al. Canonical Wnt/beta-catenin signaling in epicardial fibrosis of failed pediatric heart allografts with diastolic dysfunction. Cardiovasc Pathol. 2013;22(1):54–7.
Blankesteijn WM, van Gijn ME, Essers-Janssen YP, Daemen MJ, Smits JF. Beta-catenin, an inducer of uncontrolled cell proliferation and migration in malignancies, is localized in the cytoplasm of vascular endothelium during neovascularization after myocardial infarction. Am J Pathol. 2000;157(3):877–83.
Zelarayan LC, Noack C, Sekkali B, Kmecova J, Gehrke C, Renger A, et al. Beta-Catenin downregulation attenuates ischemic cardiac remodeling through enhanced resident precursor cell differentiation. Proc Natl Acad Sci U S A. 2008;105(50):19762–7.
Iyer LM, Nagarajan S, Woelfer M, Schoger E, Khadjeh S, Zafiriou MP, et al. A context-specific cardiac beta-catenin and GATA4 interaction influences TCF7L2 occupancy and remodels chromatin driving disease progression in the adult heart. Nucleic Acids Res. 2018;46(6):2850–67.
Bao MW, Cai Z, Zhang XJ, Li L, Liu X, Wan N, et al. Dickkopf-3 protects against cardiac dysfunction and ventricular remodelling following myocardial infarction. Basic Res Cardiol. 2015;110(3):25.
Min JK, Park H, Choi HJ, Kim Y, Pyun BJ, Agrawal V, et al. The WNT antagonist Dickkopf2 promotes angiogenesis in rodent and human endothelial cells. J Clin Invest. 2011;121(5):1882–93.
Woulfe KC, Gao E, Lal H, Harris D, Fan Q, Vagnozzi R, et al. Glycogen synthase kinase-3beta regulates post-myocardial infarction remodeling and stress-induced cardiomyocyte proliferation in vivo. Circ Res. 2010;106(10):1635–45.
Wu D, Pan W. GSK3: a multifaceted kinase in Wnt signaling. Trends Biochem Sci. 2010;35(3):161–8.
Deb A, Ubil E. Cardiac fibroblast in development and wound healing. J Mol Cell Cardiol. 2014;70:47–55.
Klesen A, Jakob D, Emig R, Kohl P, Ravens U, Peyronnet R. Cardiac fibroblasts : active players in (atrial) electrophysiology? Herzschrittmacherther Elektrophysiol. 2018;29(1):62–9.
Furtado MB, Nim HT, Boyd SE, Rosenthal NA. View from the heart: cardiac fibroblasts in development, scarring and regeneration. Development. 2016;143(3):387–97.
Moore-Morris T, Guimaraes-Camboa N, Banerjee I, Zambon AC, Kisseleva T, Velayoudon A, et al. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J Clin Invest. 2014;124(7):2921–34.
Lee TW, Chung CC, Lee TI, Lin YK, Kao YH, Chen YJ. Fibroblast growth factor 23 stimulates cardiac fibroblast activity through phospholipase C-mediated calcium signaling. Int J Mol Sci. 2021;23(1):166.
Voloshenyuk TG, Hart AD, Khoutorova E, Gardner JD. TNF-α increases cardiac fibroblast lysyl oxidase expression through TGF-β and PI3Kinase signaling pathways. Biochem Biophys Res Commun. 2011;413(2):370–5.
Fan D, Takawale A, Lee J, Kassiri Z. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis Tissue Repair. 2012;5(1):15.
Ivey MJ, Kuwabara JT, Riggsbee KL, Tallquist MD. Platelet-derived growth factor receptor-alpha is essential for cardiac fibroblast survival. Am J Physiol Heart Circ Physiol. 2019;317(2):H330–44.
Li S, Zhu Z, Xue M, Yi X, Liang J, Niu C, et al. Fibroblast growth factor 21 protects the heart from angiotensin II-induced cardiac hypertrophy and dysfunction via SIRT1. Biochim Biophys Acta Mol Basis Dis. 2019;1865(6):1241–52.
Deb A. Cell-cell interaction in the heart via Wnt/beta-catenin pathway after cardiac injury. Cardiovasc Res. 2014;102(2):214–23.
•• Liu S, Tang L, Zhao X, Nguyen B, Heallen TR, Li M, et al. Yap promotes noncanonical wnt signals from cardiomyocytes for heart regeneration. Circ Res. 2021;129(8):782–97. Findings from this study indicate that Wntless, a direct target of Yap in cardiomyocyte, is required for neonatal murine heart regeneration through regulating noncanonical Wnt signaling from cardiomyocytes to cardiac fibroblasts.
Abraityte A, Vinge LE, Askevold ET, Lekva T, Michelsen AE, Ranheim T, et al. Wnt5a is elevated in heart failure and affects cardiac fibroblast function. J Mol Med (Berl). 2017;95(7):767–77.
Liu Q, Zhu L-J, Waaga-Gasser AM, Ding Y, Cao M, Jadhav SJ, et al. The axis of local cardiac endogenous Klotho-TGF-β1-Wnt signaling mediates cardiac fibrosis in human. J Mol Cell Cardiol. 2019;136:113–24.
Sklepkiewicz P, Shiomi T, Kaur R, Sun J, Kwon S, Mercer B, et al. Loss of secreted frizzled-related protein-1 leads to deterioration of cardiac function in mice and plays a role in human cardiomyopathy. Circ Heart Fail. 2015;8(2):362–72.
Kobayashi K, Luo M, Zhang Y, Wilkes DC, Ge G, Grieskamp T, et al. Secreted Frizzled-related protein 2 is a procollagen C proteinase enhancer with a role in fibrosis associated with myocardial infarction. Nat Cell Biol. 2009;11(1):46–55.
Mastri M, Shah Z, Hsieh K, Wang X, Wooldridge B, Martin S, et al. Secreted Frizzled-related protein 2 as a target in antifibrotic therapeutic intervention. Am J Physiol Cell Physiol. 2014;306(6):C531–9.
Xiang FL, Fang M, Yutzey KE. Loss of beta-catenin in resident cardiac fibroblasts attenuates fibrosis induced by pressure overload in mice. Nat Commun. 2017;8(1):712.
van de Schans VA, van den Borne SW, Strzelecka AE, Janssen BJ, van der Velden JL, Langen RC, et al. Interruption of Wnt signaling attenuates the onset of pressure overload-induced cardiac hypertrophy. Hypertension. 2007;49(3):473–80.
He W, Zhang L, Ni A, Zhang Z, Mirotsou M, Mao L, et al. Exogenously administered secreted frizzled related protein 2 (Sfrp2) reduces fibrosis and improves cardiac function in a rat model of myocardial infarction. Proc Natl Acad Sci U S A. 2010;107(49):21110–5.
Matsushima K, Suyama T, Takenaka C, Nishishita N, Ikeda K, Ikada Y, et al. Secreted frizzled related protein 4 reduces fibrosis scar size and ameliorates cardiac function after ischemic injury. Tissue Eng Part A. 2010;16(11):3329–41.
Nakamura K, Sano S, Fuster JJ, Kikuchi R, Shimizu I, Ohshima K, et al. Secreted frizzled-related protein 5 diminishes cardiac inflammation and protects the heart from ischemia/reperfusion injury. J Biol Chem. 2016;291(6):2566–75.
Huang X, Yan Y, Zheng W, Ma Y, Wang X, Gong W, et al. Secreted frizzled-related protein 5 protects against cardiac rupture and improves cardiac function through inhibiting mitochondrial dysfunction. Front Cardiovasc Med. 2021;8: 682409.
Meyer IS, Jungmann A, Dieterich C, Zhang M, Lasitschka F, Werkmeister S, et al. The cardiac microenvironment uses non-canonical WNT signaling to activate monocytes after myocardial infarction. EMBO Mol Med. 2017;9(9):1279–93.
Aoyama Y, Kobayashi K, Morishita Y, Maeda K, Murohara T. Wnt11 gene therapy with adeno-associated virus 9 improves the survival of mice with myocarditis induced by coxsackievirus B3 through the suppression of the inflammatory reaction. J Mol Cell Cardiol. 2015;84:45–51.
Oikonomopoulos A, Sereti KI, Conyers F, Bauer M, Liao A, Guan J, et al. Wnt signaling exerts an antiproliferative effect on adult cardiac progenitor cells through IGFBP3. Circ Res. 2011;109(12):1363–74.
Paik DT, Rai M, Ryzhov S, Sanders LN, Aisagbonhi O, Funke MJ, et al. Wnt10b gain-of-function improves cardiac repair by arteriole formation and attenuation of fibrosis. Circ Res. 2015;117(9):804–16.
Wo D, Peng J, Ren DN, Qiu L, Chen J, Zhu Y, et al. Opposing roles of Wnt inhibitors IGFBP-4 and Dkk1 in cardiac ischemia by differential targeting of LRP5/6 and beta-catenin. Circulation. 2016;134(24):1991–2007.
Mohamed TM, Stone NR, Berry EC, Radzinsky E, Huang Y, Pratt K, et al. Chemical enhancement of in vitro and in vivo direct cardiac reprogramming. circulation. 2017;135(10):978–95.
Huang SM, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA, et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature. 2009;461(7264):614–20.
Moon J, Zhou H, Zhang LS, Tan W, Liu Y, Zhang S, et al. Blockade to pathological remodeling of infarcted heart tissue using a porcupine antagonist. Proc Natl Acad Sci U S A. 2017;114(7):1649–54.
Jiang J, Lan C, Li L, Yang D, Xia X, Liao Q, et al. A novel porcupine inhibitor blocks WNT pathways and attenuates cardiac hypertrophy. Biochim Biophys Acta Mol Basis Dis. 2018;1864(10):3459–67.
Yang D, Fu W, Li L, Xia X, Liao Q, Yue R, et al. Therapeutic effect of a novel Wnt pathway inhibitor on cardiac regeneration after myocardial infarction. Clin Sci (Lond). 2017;131(24):2919–32.
Saraswati S, Alfaro MP, Thorne CA, Atkinson J, Lee E, Young PP. Pyrvinium, a potent small molecule Wnt inhibitor, promotes wound repair and post-MI cardiac remodeling. PLoS ONE. 2010;5(11): e15521.
Xie S, Fu W, Yu G, Hu X, Lai KS, Peng X, et al. Discovering small molecules as Wnt inhibitors that promote heart regeneration and injury repair. J Mol Cell Biol. 2020;12(1):42–54.
Zhao L, Ben-Yair R, Burns CE, Burns CG. Endocardial notch signaling promotes cardiomyocyte proliferation in the regenerating zebrafish heart through wnt pathway antagonism. Cell Rep. 2019;26(3):546–54 e5.
•• Peng X, Lai KS, She P, Kang J, Wang T, Li G, et al. Induction of Wnt signaling antagonists and p21-activated kinase enhances cardiomyocyte proliferation during zebrafish heart regeneration. J Mol Cell Biol. 2021;13(1):41–58. This study finds that Wnt signaling is dampened upon cardiac injury in zebrafish adult heart. Coordination of Wnt signaling inhibition and Pak2/pS675-β-catenin signaling enhances heart regeneration by supporting CM dedifferentiation and proliferation.
Peng X, Fan S, Tan J, Zeng Z, Su M, Zhang Y, et al. Wnt2bb induces cardiomyocyte proliferation in zebrafish hearts via the jnk1/c-jun/creb1 pathway. Front Cell Dev Biol. 2020;25(8):323.
Bertozzi A, Wu CC, Hans S, Brand M, Weidinger G. Wnt/beta-catenin signaling acts cell-autonomously to promote cardiomyocyte regeneration in the zebrafish heart. Dev Biol. 2022;481:226–37.
Mallick A, Taylor SKB, Ranawade A, Gupta BP. Axin family of scaffolding proteins in development: lessons from C. elegans. J Dev Biol. 2019;7(4):20.
Lin SC, Li Q. Axin bridges Daxx to p53. Cell Res. 2007;17(4):301–2.
Cheedipudi SM, Fan S, Rouhi L, Marian AJ. Pharmacological suppression of the WNT signaling pathway attenuates age-dependent expression of the phenotype in a mouse model of arrhythmogenic cardiomyopathy. J Cardiovasc Aging. 2021;1(3). https://doi.org/10.20517/jca.2021.04.
Zhao Z, Liu H, Li Y, Tian J, Deng S. Wnt-C59 attenuates pressure overload-induced cardiac hypertrophy via interruption of Wnt pathway. Med Sci Monit. 2020;26: e923025.
Halloin C, Schwanke K, Lobel W, Franke A, Szepes M, Biswanath S, et al. Continuous WNT control enables advanced hPSC cardiac processing and prognostic surface marker identification in chemically defined suspension culture. Stem Cell Reports. 2019;13(2):366–79.
Tao J, Wei X, Huang Y, Liu F, Wu Y, Adi D, et al. Sfrp1 protects against acute myocardial ischemia (AMI) injury in aged mice by inhibiting the Wnt/beta-catenin signaling pathway. J Cardiothorac Surg. 2021;16(1):12.
Martin S, Lin H, Ejimadu C, Lee T. Tissue-nonspecific alkaline phosphatase as a target of sFRP2 in cardiac fibroblasts. Am J Physiol Cell Physiol. 2015;309(3):C139–47.
Cao Q, Zhang J, Gao L, Zhang Y, Dai M, Bao M. Dickkopf3 upregulation mediates the cardioprotective effects of curcumin on chronic heart failure. Mol Med Rep. 2018;17(5):7249–57.
Laeremans H, Hackeng TM, van Zandvoort MA, Thijssen VL, Janssen BJ, Ottenheijm HC, et al. Blocking of frizzled signaling with a homologous peptide fragment of wnt3a/wnt5a reduces infarct expansion and prevents the development of heart failure after myocardial infarction. Circulation. 2011;124(15):1626–35.
Uitterdijk A, Hermans KC, de Wijs-Meijler DP, Daskalopoulos EP, Reiss IK, Duncker DJ, et al. UM206, a selective Frizzled antagonist, attenuates adverse remodeling after myocardial infarction in swine. Lab Invest. 2016;96(2):168–76.
Liu J, Zheng X, Zhang C, Zhang C, Bu P. Lcz696 Alleviates myocardial fibrosis after myocardial infarction through the sFRP-1/Wnt/beta-catenin signaling pathway. Front Pharmacol. 2021;12: 724147.
Burridge PW, Matsa E, Shukla P, Lin ZC, Churko JM, Ebert AD, et al. Chemically defined generation of human cardiomyocytes. Nat Methods. 2014;11(8):855–60.
Sassi Y, Avramopoulos P, Ramanujam D, Gruter L, Werfel S, Giosele S, et al. Cardiac myocyte miR-29 promotes pathological remodeling of the heart by activating Wnt signaling. Nat Commun. 2017;8(1):1614.
Badimon L, Casani L, Camino-Lopez S, Juan-Babot O, Borrell-Pages M. GSK3beta inhibition and canonical Wnt signaling in mice hearts after myocardial ischemic damage. PLoS ONE. 2019;14(6): e0218098.
Willems L, Daniels A, Fanton Y, Linsen L, Evens L, Bito V, et al. Differentiation of human cardiac atrial appendage stem cells into adult cardiomyocytes: a role for the Wnt pathway? Int J Mol Sci. 2020;21(11):3931.
Sun HY, Wang XL, Ma LC, Yang M, Yang HJ, Huang HW, et al. Influence of MiR-154 on myocardial apoptosis in rats with acute myocardial infarction through Wnt/beta-catenin signaling pathway. Eur Rev Med Pharmacol Sci. 2019;23(2):818–25.
Acknowledgements
We thank Mengke Qiang and Meng Wan for drawing schematic model pictures and T.P.Z. laboratory members for comments on the manuscript.
Funding
This work was supported by grants from the Ministry of Science and Technology of China (2018YFA0800103 and 2018YFA0801004) and the National Natural Science Foundation of China (31970780).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Regenerative Medicine
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Li, D., Sun, J. & Zhong, T.P. Wnt Signaling in Heart Development and Regeneration. Curr Cardiol Rep 24, 1425–1438 (2022). https://doi.org/10.1007/s11886-022-01756-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11886-022-01756-8