
Abstract
Purpose of Review
To present a method enabling in vivo quantification of tissue membrane potential (ΔΨT), a proxy of mitochondrial membrane potential (ΔΨm), to review the origin and role of ΔΨm, and to highlight potential applications of myocardial ΔΨT imaging.
Recent Findings
Radiolabelled lipophilic cations have been used for decades to measure ΔΨm in vitro. Using similar compounds labeled with positron emitters and appropriate compartment modeling, this technique now allows in vivo quantification of ΔΨT with positron emission tomography. Studies have confirmed the feasibility of measuring myocardial ΔΨT in both animals and humans. In addition, ΔΨT showed very low variability among healthy subjects, suggesting that this method could allow detection of relatively small pathological changes.
Summary
In vivo assessment of myocardial ΔΨT provides a new tool to study the pathophysiology of cardiovascular diseases and has the potential to serve as a new biomarker to assess disease stage, prognosis, and response to therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Mitochondria are double-membrane-bound cellular organelles found in almost all eukaryotic cells. They are involved in the production of the chemical energy required for cellular biomechanical reactions via the phosphorylation of adenosine diphosphate (ADP) into adenosine triphosphate (ATP). The mitochondrial membrane potential (ΔΨm), which represents the electric gradient across the mitochondrial inner membrane, is at the very core of mitochondrial energy production, fueling the conversion of ADP to ATP. Alteration of myocardial ΔΨm is present in a wide range of cardiac pathologies and typically appears early in the disease process, often preceding symptoms [1,2,3]. Until recently, assessment of myocardial ΔΨm was only possible in isolated mitochondria and explanted hearts [4, 5]. However, new methodological and technological advances enable in vivo quantification of membrane potential in absolute units of millivolts (mV). In this paper, the origin and role of myocardial ΔΨm will be overviewed. A method for membrane potential quantification with positron emission tomography (PET) will be presented. Finally, potential cardiovascular applications of this technique will be discussed.
Role of Mitochondria and Origin of ΔΨm
Mitochondria play several roles in cellular homeostasis and function, including hormonal, ion, and immune signaling; steroid synthesis; regulation of cellular metabolism; and apoptosis [6,7,8]. One of the principal function of mitochondria is the phosphorylation of ADP into ATP, providing the vast majority of the energy necessary for cell function, including cardiomyocytes’ contraction [8]. The first steps of this process involve glycolysis and oxidation of fatty acids to produce Acetyl-CoA. Acetyl-CoA is the common intermediate driving the production of NADH and FADH2 through the tricyclic (Krebs) cycle (Fig. 1). NADH and FADH2 then feed electrons to the electron transport chain (ETC). Redox reactions are taking place along the ETC, and protons are translocated across the inner membrane, from the mitochondrial matrix into the intermembrane space, establishing a proton-motive force (Δpm). This force is composed of the pH gradient (ΔpHm) and the mitochondrial membrane potential ΔΨm, with ΔΨm comprising the bulk of Δpm. Ultimately, the ATP synthase dissipates a small fraction of the energy stored in the Δpm to convert ADP into ATP by letting protons cross back in the mitochondrial matrix. Critical to this process is the impermeability of the mitochondrial inner membrane to most ions, allowing accumulation of protons in the intermembrane space.

Schematic representation of the electron transport chain and generation of the mitochondrial membrane potential (ΔΨm); ADP, adenosine diphosphate; ATP, adenosine triphosphate; ROS, reactive oxygen species; TCA, tricyclic cycle
When the ETC function is optimized, there is maximal coupling between proton pumping and phosphorylation of ADP to ATP. This occurs when there is minimal proton leakage across the inner membrane. When levels of ADP increase, as seen in states of high energy demand, there is increased phosphorylation and utilization (and thus dissipation) of the proton gradient, stimulating the ETC function and oxygen consumption. Conversely, low ADP levels slow phosphorylation rate, leading to inhibition of the ETC. [9] It is thought that ΔΨm has direct inhibitory effects on the rate of electron transport [10], and when there is significant reduction of ΔΨm, the ETC is free to run at maximal rate, and ΔΨm can be progressively restored. Collapse of ΔΨm via opening of the mitochondrial permeability transition pores (mPTP), and subsequent release of cytochrome C into the cytosol plays a central role in cellular apoptosis.
Oxidative Stress and ΔΨm
Reactive oxygen species (ROS) are normal by-products of the respiratory chain. Under physiological conditions, the ETC is efficient with minimal production of ROS [11]. At small concentrations, ROS play an important physiological role as signal transduction molecules within the mitochondria and cells. However, when ΔΨm falls outside its optimal physiological range, either above or below, there is a significant rise in the production of ROS by the ETC; increasing ΔΨm by only 10 mV leads to a 70–90% increase in ROS production [12, 13]. The respiratory chain complexes are very sensitive to oxidizing agents due to the vulnerability of their components to direct ROS attack. Their oxidative modifications manifest with a decreased enzymatic activity and dysfunction of the whole respiratory chain [14]. Oxidative damage to the respiratory chain complexes not only results in a decreased efficiency of ATP production but can also lead to further increase in ROS production, amplifying the ROS release [14]. Finally, oxidation of the inner membrane anion channels (IMACs) by ROS leads to their opening and partial dissipation of ΔΨm. Overall, in conditions associated with increased ROS, ΔΨm may be reduced, and because the ratio ATP:ADP decreases exponentially as a function of ΔΨm, relatively small mitochondrial depolarization elicits a large decrease in the ability to convert ADP to ATP [15].
Quantification of ΔΨ with PET Imaging
In vitro quantification of ΔΨm has been performed for decades by measuring the distribution of various lipophilic cations in isolated mitochondria [4, 5]. At equilibrium, the concentration of those compounds on each side of a membrane (\( \overline{C} \)) can be expressed by the Nernst equation (Eq. 1).
where ΔΨ is the membrane’s electric potential and \( \beta =\frac{zF}{RT} \) is the ratio of known physical parameters: z is the valence of the ionic probe, F denotes Faraday’s constant, R is the universal gas constant, and T is the temperature in Kelvin. Assessment of ΔΨm with PET imaging relies on the same methodology, using lipophilic cations labeled with positron emitters such as 18F-tetraphenylphosphonium (18F-TPP+). Those tracers cross freely and with minimal interaction the cellular and mitochondrial phospholipid bilayer membranes, and their distribution at equilibrium abides the Nernst equation. Radiotracer uptake is therefore exponentially proportional to ΔΨm; under physiological conditions, concentration of a monovalent lipophilic cation will be 3–10 times greater in the intracellular space compared to the extracellular space and 100–500 times greater in the mitochondrial matrix compared to the extracellular space [16].
An image voxel containing myocardium can be modeled into 4 compartments, which include the plasma, interstitial space, cytosol, and mitochondria (Fig. 2) [17••]. The plasma and interstitial space compose the extracellular space. Using this model, the tracer concentration in a voxel at equilibrium can be expressed as:

Compartmental model for a lipophilic cation in a positron emission tomography (PET) image voxel. Cp, Cinter, Ccyto, Cmito, and CECS represent the tracer concentration in the plasma, interstitial space, cytosol, mitochondria, and extracellular space respectively (adapted from [14])
where \( {\overline{C}}_{mito} \), \( {\overline{C}}_{cyto} \), and \( {\overline{C}}_{ECS} \) represent the probe concentration at equilibrium in the mitochondria, cytosol, and extracellular space, respectively, and fECS and fmito represent the extracellular space and mitochondrial volume fraction, respectively. Noting that plasma concentration (\( {\overline{C}}_p \)) is equal to \( {\overline{C}}_{ECS} \) at equilibrium, division of Eq. 2 by \( {\overline{C}}_p \) yields the following expression:
where VT, ∆Ψm, and ∆Ψc represent the volume of distribution of the tracer and the mitochondrial and cellular membrane potential, respectively. Because VT > > 1 and \( {e}^{-\beta \left(\Delta {\Psi}_m+\Delta {\Psi}_c\right)}\gg {e}^{-\beta \left(\Delta {\Psi}_c\right)} \), Eq. 3 can be approximated by:
where ΔΨT is the total tissue membrane potential, defined as ΔΨm + ΔΨc. Equation 4 can be rearranged as:
From Eq. 5, we note that 3 parameters are required to quantify ΔΨT: (1) the extracellular space fraction, (2) the mitochondrial fraction, and (3) the tracer volume of distribution. The fmito parameter is currently not measurable noninvasively. It has to be assumed constant, and a value of 0.25 can be used for human studies [18]. It is important to note that mitochondrial fraction may decrease in disease state, ultimately leading to an underestimation of membrane potential (less negative). The volume of distribution VT can be determined kinetically [14] or by taking the ratio between the voxel concentration measured by PET imaging and the plasma concentration at secular equilibrium [16, 19].
Quantification of Extracellular Space
As stated above, measurement of extracellular volume (ECV) is critical to accurately quantify membrane potential. This is because there is a significant extracellular space variability among healthy individuals, and several diseases are associated with extracellular space expansion. Failure to account for ECV leads to significant underestimation of ΔΨ as well as increased variability between subjects [20]. ECV can be assessed with computed tomography (CT) or magnetic resonance imaging (MR). In both cases, images are acquired before and after administration of a contrast agent (iodine-based for CT and gadolinium-based for MR), which distributes in the extracellular space. At the present time, MR allows more robust and precise quantification of ECV without additional ionizing radiation. In that context, ΔΨT imaging represents an attractive application of hybrid PET/MR scanners. CT, on the other hand, is readily available, and acquisition can be performed on a conventional PET/CT system. Detailed methodology of ECV quantification is beyond the scope of this review and can be found elsewhere (see [21]).
In Vivo Quantification of Myocardial Membrane Potential
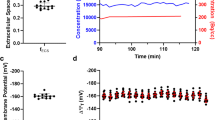
Fukuda et al. were the first to attempt in vivo quantification of ΔΨT in the 1980s [22]. Following a bolus injection of the lipophilic cation 11C-triphenylmethylphosphonium (11C-TPMP), they measured myocardial tracer concentration in dogs with PET imaging, as well as in mice and rats using autoradiography. Accounting for the extracellular volume, they calculated the ΔΨT using the Nernst equation with plasma and myocardial tracer concentrations. Nearly 25 years later, Gurm et al. reported an attempt to noninvasively quantify ΔΨm using PET imaging in a swine model of ischemic heart disease [23]. They used the monovalent lipophilic cation 18F-TPP+, which passively crosses membranes without significant interaction. Based on the Nernst equation, they reported an average ΔΨm of −91 mV (Table 1), which appears significantly discordant with previous measurements. This difference can be explained in part by the fact that ECV was not taken into account, leading to an underestimation of ΔΨm. Furthermore, both Gurm et al. and Fukuda et al. used bolus radiotracer injection but assumed that imaging was performed at secular equilibrium (i.e., constant tracer concentration in the different compartments), which was not the case. Such assumption may lead to erroneous quantification of membrane potential [20, 24]. More recently, Alpert et al. reported a successful method to quantify ΔΨT in swine [17••]. Using a bolus injection of 18F-TPP+ and a dynamic PET acquisition, they estimated VT in the myocardium using Logan analysis [25] and calculated ΔΨT using the Nernst equation, accounting for ECV. Subsequently, Alpert et al. reported that intracoronary infusion of BAM15, a proton gradient uncoupler affecting specifically the mitochondrial membrane without depolarizing the cellular membrane, leads to partial and reversible decreased in 18F-TPP+ concentrations (Fig. 3) [26•]. These results confirmed that myocardial 18F-TPP+ concentrations measured by PET are sensitive to temporal change in ΔΨm.

Effect of BAM15, a selective mitochondrial proton uncoupler, on tissue membrane potential (ΔΨT). Vertical long axis of a pig heart before (top) and after (bottom) intracoronary infusion of BAM15 in the left anterior descending artery demonstrating partial depolarization in the septal and apical segments (data from [16])
Pelletier-Galarneau et al. were the first to demonstrate the feasibility of ΔΨT in vivo quantification in humans [19••]. Using a bolus plus infusion of 18F-TPP+ and hybrid PET/MR imaging, they reported an average ΔΨT of −160.7 ± 3.7 mV among 13 healthy volunteers [19••]. The corresponding average ΔΨm was −123 mV, in excellent agreement with explanted heart measurements (−118 mV) [27]. Importantly, ΔΨT showed very low variability between subjects, suggesting that this method could prove useful to detect relatively small pathological changes and that small sample size could be sufficient to test hypothesis. In addition, high-quality parametric images can be generated with this method, allowing assessment of regional variability in ΔΨT (Fig. 4). Other radiotracers have been used in preclinical studies to assess mitochondrial membrane potential, including 18F-fluoropentyl-triphenylphosphonium (18F-FPTP), 18F-fluorobenzyl-triphenylphosphonium (18F-FBnTP), and 18F-MitoPhos, all of which share the property of being monovalent lipophilic cations [28,29,30]. So far, only 18F-TPP+ has been used in humans.

Parametric images of the volume of distribution (VT) and tissue membrane potential (ΔΨT). HLA, horizontal long axis; SA, short axis; VLA, vertical long axis (reproduced with permission from Springer Nature from Pelletier-Galarneau et al. Eur J Nucl Med Mol Imaging, 2020) [19••]
Potential Clinical Applications
Alterations in mitochondrial function, ROS production, and ΔΨm are seen in a broad range of pathologies, including diabetes, cancer, various degenerative diseases, and myopathy [29, 31, 32]. In cardiovascular diseases, mitochondrial dysfunction is thought to play a critical role in the development and progression of ventricular arrhythmias, heart failure, cardiotoxicity, and reperfusion injury just to name a few [33,34,35,36]. Noninvasive mapping of membrane potential could provide a new biomarker in mitochondrial-related diseases. In addition, it has the potential to play a role in the assessment of response to therapy, especially with the advent of new mitochondrial therapies.
Chemotherapy-Induced Cardiotoxicity
Chemotherapy-induced cardiotoxicity is a multifactorial complication of chemotherapy with interactions between the nature and dosage of the therapy, environmental factors, and genetic factors [37]. It is a relatively frequent side effect of many anticancer agents, including anthracyclines such as doxorubicin [38]. As the damages observed in chemotherapy-induced cardiotoxicity are often irreversible, it is imperative to detect cardiotoxicity early in order to minimize long-term morbidity and mortality, especially given the trend of improved cancer survival. The mechanisms underlying doxorubicin-induced cardiotoxicity (DIC) are incompletely understood. A key role has been attributed to mitochondrial dysfunction with mitochondria being the most injured intracellular organelle from exposure to doxorubicin [39]. While early research suggested that DIC was mediated through redox recycling of doxorubicin and subsequent mitochondrial and cellular damage, recent work has identified multiple alternative pathways of mitochondrial mediated DIC [40]. Doxorubicin has a high binding affinity with cardiolipin, a phospholipid present in the mitochondrial membrane, which is involved in maintaining mitochondrial structure and function as well as cell survival [41, 42]. Cardiolipin is particularly important for normal ETC activity, which is disrupted by doxorubicin binding to cardiolipin. The resulting increased electron transfer to oxygen molecules and generation of free radicals damage the mitochondrial membrane, leading to reduced ATP levels [39, 40, 42]. While the pathway of DIC through inhibition of the ETC can be observed as a result of acute mitochondrial exposure to doxorubicin [40], chronic effects of doxorubicin have been related to binding of doxorubicin to topoisomerase 2β, which is largely present in the mitochondria, resulting in DNA double-strand breaks and transcriptome changes leading to defective mitochondrial biogenesis and ROS formation [39, 43,44,45]. Further suggested DIC mechanisms include calcium dysregulation following increased mitochondrial ROS generation and failed energy production as well as activation of apoptotic pathways [1, 39]. The mitochondrial dysfunction underpinning DIC, particularly inhibition of ETC, can be associated with depolarization of ΔΨm. In fact, depolarization of ΔΨm following exposure to doxorubicin has been observed in in vitro studies in postnatal rat cardiac myocytes and human colon and breast cancer cell lines [1, 2]. Importantly, these molecular changes occur early in the disease stage before irreversible functional impairment can be observed.
At present, screening for cardiotoxicity is done mostly by serial assessment of left ventricular ejection fraction (LVEF) and echocardiographic strain imaging [46], both of which reflect changes that are occurring late in the disease process, after irreversible damages have occurred. In that context, noninvasive measurement of membrane potential could represent an interesting biomarker of cardiotoxicity, with preliminary data supporting this hypothesis. Indeed, McCluskey et al. showed that myocardial concentration of 18F-MitoPhos, a lipophilic cation, decreased significantly 48 h after a single infusion of doxorubicin, while other biomarkers (e.g., troponins) remained unchanged [47•]. Further studies are required to confirm whether change in membrane potential can predict cardiotoxicity and alter patient management.
Heart Failure
Mitochondrial dysfunction is a hallmark of heart failure (HF), with both preserved and reduced ejection fraction [48]. Several alterations in mitochondrial function have been reported in heart failure, including ATP production, glucose metabolism, increased oxidative stress, dysregulation of calcium metabolism, protein modifications, and apoptosis [49, 50]. In addition, therapies improving mitochondrial function have been shown to improve outcomes of patients with HF. For instance, the Q-SYMBIO trial showed that coenzyme Q10 (CoQ10), a molecule targeting oxidative stress and mitochondrial dysfunction, improves symptoms and reduces major adverse cardiovascular events in patients with HF [51]. Other molecules such as ACE inhibitors have also been shown to improve mitochondrial function [52], reinforcing the importance of mitochondrial dysfunction in HF. In the failing heart, there is increased production of ROS by the mitochondria [53, 54]. Excess ROS is associated to cellular dysfunction and DNA damage and can lead to cell death. In the myocardium, ROS can directly impair contractile function and activate pathways leading to hypertrophy, necrosis, and apoptosis [55]. Excess ROS was also shown to dissipate the ΔΨm via different mechanisms [56]. ΔΨm is directly related to the capacity of the ETC to pump protons across the inner mitochondrial membrane and is therefore necessary for ATP generation [56, 57]. The decrease of ΔΨm in HF has been confirmed in animal models of HF with decreased activity of the ETC complexes leading to decreased ΔΨm, which in turn resulted in reduced mitochondrial Ca2+ concentration and impaired myocyte contractility [34]. Imaging of membrane potential could provide insight into the pathophysiological processes of the disease, help with development of new therapeutic interventions, and allow for therapy optimization.
Ischemia-Reperfusion Injury
Acute myocardial infarction is one of the leading causes of death and disability worldwide. Treatment of acute ischemia relies on timely reperfusion. However, myocardial reperfusion itself may further damage the affected cardiomyocytes, a phenomenon known as ischemia-reperfusion injury (IRI), and it has been estimated that approximately 50% of the final infarcted area is related to IRI [58]. The complex mechanisms of IRI involve different biological pathways and are not yet fully understood. However, a key role has been attributed to opening of the mPTP during reperfusion [33]. This results in immediate depolarization of ΔΨm, disruption of ATP production, production of ROS, and the release or activation of several pro-apoptotic proteins like cytochrome C [3, 59]. Furthermore, uncoupling of the ETC results in reversal of ATPase, which switches from synthesis of ATP to active hydrolysis. The resulting rapid decline of intracellular ATP concentration leads to disruption of ionic and metabolic homeostasis and the activation of degradative enzymes, in turn, resulting in irreversible damage to the cell [60]. Triggers for mPTP opening include Ca2+ overload, rapid normalization of pH, oxidative stress, and mitochondrial depolarization, all conditions that occur during post-ischemic reperfusion [60, 61]. Noninvasive assessment of membrane potential could provide a new tool to study the pathophysiology of IRI and assess the effect of new therapies.
Conclusion
In vivo quantification of ΔΨm with PET imaging relies on the extension of well-established chemistry principles and benchtop in vitro methods. With radiolabelled lipophilic cations, such as 18F-TPP+, and appropriate compartment modeling, in vivo quantification of the tissue membrane potential (ΔΨT), a proxy of ΔΨm, in absolute units of millivolts can be achieved. Studies have demonstrated the feasibility of measuring myocardial ΔΨT in both animals and humans and that myocardial 18F-TPP+ concentration measured by PET is sensitive to temporal changes in ΔΨm. In addition, ΔΨT showed very low variability between subjects, suggesting that this method could be used to detect relatively small pathological changes. As alteration in mitochondrial function and membrane potential is ubiquitous in cardiovascular diseases, this imaging technique could provide a new tool to better understand the pathophysiology of those diseases and has the potential to serve as a new biomarker to assess disease stage, prognosis, and response to therapy. Further studies are needed to establish these potential roles.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Kuznetsov AV, Margreiter R, Amberger A, Saks V, Grimm M. Changes in mitochondrial redox state, membrane potential and calcium precede mitochondrial dysfunction in doxorubicin-induced cell death. Biochim Biophys Acta BBA - Mol Cell Res. 2011;1813:1144–52.
Dhingra R, Margulets V, Chowdhury SR, Thliveris J, Jassal D, Fernyhough P, et al. Bnip3 mediates doxorubicin-induced cardiac myocyte necrosis and mortality through changes in mitochondrial signaling. Proc Natl Acad Sci Natl Acad Sci. 2014;111:E5537–44.
Honda HM, Korge P, Weiss JN. Mitochondria and ischemia/reperfusion injury. Ann N Y Acad Sci. 2005;1047:248–58.
Kamo N, Muratsugu M, Hongoh R, Kobatake Y. Membrane potential of mitochondria measured with an electrode sensitive to tetraphenyl phosphonium and relationship between proton electrochemical potential and phosphorylation potential in steady state. J Membr Biol. 1979;49:105–21.
Kauppinen RA, Hassinen IE. Monitoring of mitochondrial membrane potential in isolated perfused rat heart. Am J Phys. 1984;247:H508–16.
Angajala A, Lim S, Phillips JB, Kim J-H, Yates C, You Z, et al. Diverse roles of mitochondria in immune responses: novel insights into immuno-metabolism. Front Immunol [Internet]. Frontiers; 2018 [cited 2020 Oct 23];9. Available from: https://www.frontiersin.org/articles/10.3389/fimmu.2018.01605/full
O’Rourke B. Metabolism: beyond the power of mitochondria. Nat Rev Cardiol. 2016;13:386–8.
O’Rourke B, Cortassa S, Aon MA. Mitochondrial ion channels: gatekeepers of life and death. Physiol Bethesda Md. 2005;20:303–15.
Lesnefsky EJ, Moghaddas S, Tandler B, Kerner J, Hoppel CL. Mitochondrial dysfunction in cardiac disease: ischemia–reperfusion, aging, and heart failure. J Mol Cell Cardiol. 2001;33:1065–89.
Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. Electron-transport chains and their proton pumps. 2002;
Sanderson TH, Reynolds CA, Kumar R, Przyklenk K, Hüttemann M. Molecular mechanisms of ischemia-reperfusion injury in brain: pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol Neurobiol. 2013;47:9–23.
Korshunov SS, Skulachev VP, Starkov AA. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997;416:15–8.
Liu SS. Cooperation of a “reactive oxygen cycle” with the Q cycle and the proton cycle in the respiratory chain--superoxide generating and cycling mechanisms in mitochondria. J Bioenerg Biomembr. 1999;31:367–76.
Marchi S, Giorgi C, Suski JM, Agnoletto C, Bononi A, Bonora M, et al. Mitochondria-ROS crosstalk in the control of cell death and aging. J Signal Transduct. 2012;329635:2012.
Chinopoulos C. Mitochondrial consumption of cytosolic ATP: Not so fast. FEBS Lett. 2011;585:1255–9.
Murphy MP. Targeting lipophilic cations to mitochondria. Biochim Biophys Acta BBA - Bioenerg. 1777;2008:1028–31.
•• Alpert NM, Guehl N, Ptaszek L, Pelletier-Galarneau M, Ruskin J, Mansour MC, et al. Quantitative in vivo mapping of myocardial mitochondrial membrane potential. PLoS One. 2018;13:e0190968. This study presents the first successful method for in vivo assessment of ΔΨT in swine, accounting for extracellular space and employing kinetic analysis to estimate tracer volume of distribution.
Barth E, Stämmler G, Speiser B, Schaper J. Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. J Mol Cell Cardiol. 1992;24:669–81.
•• Pelletier-Galarneau M, Petibon Y, Ma C, Han P, Kim SJW, Detmer FJ, et al. In vivo quantitative mapping of human mitochondrial cardiac membrane potential: a feasibility study. Eur J Nucl Med Mol Imaging. 2020. First-in-human study demonstrating the feasibility of in vivo ΔΨT quantification. The observed ΔΨT had very low inter-subject variability among healthy volunteers, suggesting that relatively small pathological changes could be detected.
Alpert NM, Pelletier-Galarneau M, Petibon Y, Normandin MD, El Fakhri G. In vivo quantification of mitochondrial membrane potential. Nature. 2020;583:E17–8.
Scully PR, Bastarrika G, Moon JC, Treibel TA. Myocardial extracellular volume quantification by cardiovascular magnetic resonance and computed tomography. Curr Cardiol Rep [Internet]. 2018 [cited 2020 Nov 18];20. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5840231/
Fukuda H, Syrota A, Charbonneau P, Vallois J, Crouzel M, Prenant C, et al. Use of 11C-triphenylmethylphosphonium for the evaluation of membrane potential in the heart by positron-emission tomography. Eur J Nucl Med. 1986;11:478–83.
Gurm GS, Danik SB, Shoup TM, Weise S, Takahashi K, Laferrier S, et al. 4-[18F]-Tetraphenylphosphonium as a PET tracer for myocardial mitochondrial membrane potential. JACC Cardiovasc Imaging. 2012;5:285–92.
Dedkova EN, Blatter LA. Measuring mitochondrial function in intact cardiac myocytes. J Mol Cell Cardiol. 2012;52:48–61.
Logan J. Graphical analysis of PET data applied to reversible and irreversible tracers. Nucl Med Biol. 2000;27:661–70.
• Alpert NM, Pelletier-Galarneau M, Kim SJW, Petibon Y, Sun T, Ramos-Torres KM, et al. In-vivo imaging of mitochondrial depolarization of myocardium with positron emission tomography and a proton gradient uncoupler. Front Physiol. 2020;11:491. A study demonstrating that intracoronary infusion of a mitochondrial proton uncoupler leads to decreased 18F-TPP+ concentration in the corresponding territory, confirming that PET imaging with 18F-TPP+ is sensitive to temporal changes in ΔΨm.
Wan B, Doumen C, Duszynski J, Salama G, Vary TC, LaNoue KF. Effects of cardiac work on electrical potential gradient across mitochondrial membrane in perfused rat hearts. Am J Phys. 1993;265:H453–60.
Kim D-Y, Kim H-S, Le UN, Jiang SN, Kim H-J, Lee K-C, et al. Evaluation of a mitochondrial voltage sensor, (18F-fluoropentyl) triphenylphosphonium cation, in a rat myocardial infarction model. J Nucl Med. 2012;53:1779–85.
Momcilovic M, Jones A, Bailey ST, Waldmann CM, Li R, Lee JT, et al. In vivo imaging of mitochondrial membrane potential in non-small-cell lung cancer. Nature. 2019;575:380–4.
Madar I, Huang Y, Ravert H, Dalrymple SL, Davidson NE, Isaacs JT, et al. Detection and quantification of the evolution dynamics of apoptosis using the PET voltage sensor 18F-fluorobenzyl triphenyl phosphonium. J Nucl Med. 2009;50:774–80.
Widlansky ME, Wang J, Shenouda SM, Hagen TM, Smith AR, Kizhakekuttu TJ, et al. Altered mitochondrial membrane potential, mass, and morphology in the mononuclear cells of humans with type 2 diabetes. Transl Res J Lab Clin Med. 2010;156:15–25.
De Felice FG, Ferreira ST. Inflammation, defective insulin signaling, and mitochondrial dysfunction as common molecular denominators connecting type 2 diabetes to Alzheimer disease. Diabetes. 2014;63:2262–72.
Turer AT, Hill JA. Pathogenesis of myocardial ischemia-reperfusion injury and rationale for therapy. Am J Cardiol. 2010;106:360–8.
Lin L, Sharma VK, Sheu S-S. Mechanisms of reduced mitochondrial Ca2+ accumulation in failing hamster heart. Pflugers Arch. 2007;454:395–402.
Rasola A, Bernardi P. Mitochondrial permeability transition in Ca(2+)-dependent apoptosis and necrosis. Cell Calcium. 2011;50:222–33.
Rutledge C, Dudley S. Mitochondria and arrhythmias. Expert Rev Cardiovasc Ther. 2013;11:799–801.
Berardi R, Caramanti M, Savini A, Chiorrini S, Pierantoni C, Onofri A, et al. State of the art for cardiotoxicity due to chemotherapy and to targeted therapies: a literature review. Crit Rev Oncol Hematol. 2013;88:75–86.
Witteles RM, Bosch X. Myocardial protection during cardiotoxic chemotherapy. Circulation. 2015;132:1835–45.
Wenningmann N, Knapp M, Ande A, Vaidya TR, Ait-Oudhia S. Insights into doxorubicin-induced cardiotoxicity: molecular mechanisms, preventive strategies, and early monitoring. Mol Pharmacol. 2019;96:219.
Pointon AV, Walker TM, Phillips KM, Luo J, Riley J, Zhang S-D, et al. Doxorubicin in vivo rapidly alters expression and translation of myocardial electron transport chain genes, leads to ATP loss and caspase 3 activation. Melov S, editor. PLoS One. 2010;5:e12733.
de Wolf FA. Binding of doxorubicin to cardiolipin as compared to other anionic phospholipids—an evaluation of electrostatic effects. Biosci Rep. 1991;11:275–84.
Schlame M, Rua D, Greenberg ML. The biosynthesis and functional role of cardiolipin. Prog Lipid Res. 2000;39:257–88.
Zhang S, Liu X, Bawa-Khalfe T, Lu L-S, Lyu YL, Liu LF, et al. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med. 2012;18:1639–42.
Lyu YL, Kerrigan JE, Lin C-P, Azarova AM, Tsai Y-C, Ban Y, et al. Topoisomerase II mediated DNA double-strand breaks: implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res. 2007;67:8839–46.
Zhu H, Sarkar S, Scott L, Danelisen I, Trush MA, Jia Z, et al. Doxorubicin redox biology: redox cycling, topoisomerase inhibition, and oxidative stress. React Oxyg Species Apex NC. 2016;1:189–98.
Juan Carlos Plana, Maurizio Galderisi, Ana Barac, Michael S Ewer, Bonnie Ky, Marielle Scherrer-Crosbie, Javier Ganame, Igal A Sebag, Deborah A Agler, Luigi P Badano, Jose Banchs, Daniela Cardinale, et al. Expert consensus for multimodality imaging evaluation of adult patients during and after cancer therapy: a report from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J. Am. Soc. Echocardiogr. Off. Publ. Am. Soc. Echocardiogr. J Am Soc Echocardiogr; 2014 [cited 2020 Nov 12]. Available from: https://pubmed.ncbi.nlm.nih.gov/25172399/
• McCluskey S, Haslop A, Coello C, Gunn R, Tate E, Southworth R, et al. Imaging chemotherapy induced acute cardiotoxicity with 18F-labelled lipophilic cations. J Nucl Med. 2019;jnumed.119.226787. This study demonstrates that myocardial concentration of a lipophilic cation radiotracer is significantly reduced following doxorubicin administration, indicating that PET assessment of ΔΨ has potential applications in chemotherapy-induced cardiotoxicity.
Kumar AA, Kelly DP, Chirinos JA. Mitochondrial dysfunction in heart failure with preserved ejection fraction. Circulation. 2019;139:1435–50.
Zhou B, Tian R. Mitochondrial dysfunction in pathophysiology of heart failure. J Clin Invest Am Soc Clin Investig. 2018;128:3716–26.
Bayeva M, Gheorghiade M, Ardehali H. Mitochondria as a therapeutic target in heart failure. J Am Coll Cardiol. 2013;61:599–610.
Mortensen SA, Rosenfeldt F, Kumar A, Dolliner P, Filipiak KJ, Pella D, et al. The effect of coenzyme Q10 on morbidity and mortality in chronic heart failure: results from Q-SYMBIO: a randomized double-blind trial. JACC Heart Fail. 2014;2:641–9.
Sanbe A, Tanonaka K, Kobayasi R, Takeo S. Effects of long-term therapy with ACE inhibitors, captopril, enalapril and trandolapril, on myocardial energy metabolism in rats with heart failure following myocardial infarction. J Mol Cell Cardiol. 1995;27:2209–22.
Ide T, Tsutsui H, Kinugawa S, Utsumi H, Kang D, Hattori N, et al. Mitochondrial electron transport complex I is a potential source of oxygen free radicals in the failing myocardium. Circ Res. 1999;85:357–63.
Odagiri K, Katoh H, Kawashima H, Tanaka T, Ohtani H, Saotome M, et al. Local control of mitochondrial membrane potential, permeability transition pore and reactive oxygen species by calcium and calmodulin in rat ventricular myocytes. J Mol Cell Cardiol. 2009;46:989–97.
Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol. 2011;301:H2181–90.
Gustafsson AB, Gottlieb RA. Heart mitochondria: gates of life and death. Cardiovasc Res. 2008;77:334–43.
O’Rourke B. Mitochondrial ion channels. Annu Rev Physiol. 2007;69:19–49.
Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. Massachusetts Med Soc. 2007;357:1121–35.
Halestrap AP. Calcium, mitochondria and reperfusion injury: a pore way to die. Biochem Soc Trans. 2006;34:232–7.
Halestrap A. Mitochondrial permeability transition pore opening during myocardial reperfusion—a target for cardioprotection. Cardiovasc Res. 2004;61:372–85.
Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J. 1999;341(Pt 2):233–49.
Funding
This work was funded in part by the National Institute of Health under P41EB022544 and R01HL137230.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Conflict of Interest
Dr. Normandin has a patent US20190125281A1 issued. Dr. Alpert has a patent US020190125281A120190502 issued. Dr. El Fakhri has a patent 16/092,650 pending. The other authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Nuclear Cardiology
Rights and permissions
About this article

Cite this article
Pelletier-Galarneau, M., Detmer, F.J., Petibon, Y. et al. Quantification of Myocardial Mitochondrial Membrane Potential Using PET. Curr Cardiol Rep 23, 70 (2021). https://doi.org/10.1007/s11886-021-01500-8
Accepted:
Published:
DOI: https://doi.org/10.1007/s11886-021-01500-8