
Abstract
Purpose of Review
Cardiovascular disease is the leading contributor to mortality and morbidity. Many deaths of heart failure patients can be attributed to sudden cardiac death due primarily to ventricular arrhythmia. Currently, most anti-arrhythmics modulate ion channel conductivity or β-adrenergic signaling, but these drugs have limited efficacy for some indications, and can potentially be proarrhythmic.
Recent Findings
Recent studies have shown that mutations in proteins other than cardiac ion channels may confer susceptibility to congenital as well as acquired arrhythmias. Additionally, ion channels themselves are subject to regulation at the levels of channel expression, trafficking and post-translational modification; thus, research into the regulation of ion channels may elucidate disease mechanisms and potential therapeutic targets for future drug development.
Summary
This review summarizes the current knowledge of the molecular mechanisms of arrhythmia susceptibility and discusses technological advances such as induced pluripotent stem cell-derived cardiomyocytes, gene editing, functional genomics, and physiological screening platforms that provide a new paradigm for discovery of new therapeutic targets to treat congenital and acquired diseases of the heart rhythm.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cardiovascular disease (CVD) is the leading contributor to mortality and morbidity. However, development of drugs in this area of medicine is steadily declining [1]. CVD is among the indications with the highest attrition during drug development and the lowest rates of regulatory approval. Moreover, the complexities of discovering new therapeutic targets for heart disease have caused several major pharmaceutical companies to refocus their efforts away from the field. The underlying reasons are diverse and range from early scientific or clinical risk, to commercial risks when approved [2, 3•, 4]. To improve the success rate of new drugs for CVD, new modalities to identify and validate therapeutic agents are paramount for addressing the needs of the large and growing population of patients with CVD [5].
Induced pluripotent stem cell (iPSC) technology is an exciting new platform for modeling CVDs that provides a virtually limitless source of iPSC-derived cardiomyocytes (iPSC-CMs) from individual patients harboring congenital disease-causing mutations [6]. The technology provides a clear advantage over primary rodent cardiomyocytes in that the hiPSC-CMs retain human genetic and cellular context. Recent developments in high-throughput assays to rapidly quantify the kinetics of excitation-contraction coupling enable screening of chemical libraries for functional genomics and drug development applications. We anticipate that these technical advances will revolutionize disease modeling and drug discovery for CVD, particularly, diseases of heart rhythm. This review focuses on the molecular mechanisms of heart rhythm disorders and on the opportunity of using iPSC models to explore the influence of non-ion channel mechanisms to the development of cellular arrhythmia and the possibility to discover new classes of drug targets for clinical development.
Heart Rhythm Disorders and Current Therapeutic Strategies
Clinically, electrocardiography measures the instantaneous change in electrical field potential to infer the direction, magnitude, and duration of electrical activity during each cardiac cycle. Myocardial excitation is initiated by the sinoatrial (SA) pacemaker cells which activate the atria and then propagates through the cardiac conduction system to enable synchronized excitation and contraction of each ventricle. At the cellular level, each depolarization and repolarization cycle, known as an action potential (AP), initiates excitation-contraction (EC) coupling linking the electrical membrane potential to myosin-actin cross-bridge formation (Fig. 1). An AP is generated when the membrane potential reaches a supra-threshold voltage that activates voltage-gated sodium channels to rapidly depolarize the membrane, known as phase 0 of the AP. Phase 1 of the AP is a brief repolarization period mediated by two transient outward potassium currents, Ito,f and Ito,s. Phase 2 of the AP is maintained by the influx of Ca2+ ions through voltage-gated L-type calcium channels (LTCCs, ICa). Towards the end of phase 2, the efflux of K+ ions is conducted by voltage-gated potassium channels (IKr and IKs) to continue repolarization during phase 3 of the AP. The resting state, phase 4, is maintained by multiple mechanisms. The sodium-potassium ATPase utilizes ATP to actively extrude Na+ in exchange for influx of K+ that maintains the Na+ and K+ gradients. Additionally, the inwardly rectifying potassium current (IK1) is activated at hyperpolarized membrane potentials to maintain the resting potential.

Electrophysiological properties of cardiomyocytes are altered in congenital as well as acquired arrhythmic diseases and are responsible for increased susceptibility to arrhythmia and sudden cardiac death. Gene defects, differential expression from modulation at either the levels of gene expression or translation, aberrant signaling, and protein modifications can underpin the electrophysiological disease state
Dysfunction of impulse generation or aberrant conduction through the heart can cause abnormal heart rhythms. The SA node has a faster intrinsic rate than the atrioventricular node and therefore normally dictates heart rate. However, if other cardiac sites show enhanced automaticity, they may generate competing stimuli and induce arrhythmia. Such triggers are observed as early or delayed after depolarizations (EADs and DADs) at the cellular level (Fig. 1). Myocardial damage or abnormal electrophysiological properties may allow an electrical impulse to encounter conduction block to one region, propagate along an alternative path to then re-activate the original region, a condition known as reentry that can lead to premature and/or repeated reactivation of the atria or ventricles. Clinically, arrhythmias are managed pharmacologically with anti-arrhythmic drugs that either target ion channels directly or inhibit β-adrenergic signaling. However, current anti-arrhythmics have limited efficacy and can sometimes be proarrhythmic [7].
Genetic Basis for Arrhythmia Disorders
Unraveling the molecular mechanisms of human genetic disorders has yielded profound insights into normal as well as pathological human physiology. Many variants in predominantly ion channel proteins have been identified as being the causative or a modifying factor for arrhythmic disease (Table 1), including long QT syndrome (LQTS) [8], short QT syndrome (SQTS) [9], catecholaminergic polymorphic ventricular tachycardia (CPVT) [10], sick sinus syndrome (SSS) [11], atrial fibrillation (AFib) [12, 13], and Brugada syndrome (BrS) [14]. With a prevalence of 1% in the general population and 6% in people older than 65, AFib is the most common type of arrhythmia. However, the pathophysiology of AFib is complex as a patient’s genetic background can be both causative (familial AFib) and potentially act as a disease modifier (non-familial AFib) [13]. Similarly, many polymorphisms have been identified by genome-wide association studies (GWAS) that map to genes associated with arrhythmic diseases and may contribute to additional risk albeit with small effect sizes [15,16,17,18]. In contrast, LQTS is the most common type of congenital arrhythmia with a prevalence of 1:2000 [19]. Taken together, there are many genetic variants associated with distinct proarrhythmic diseases that can be mined to gain additional insights into molecular mechanisms.
Congenital arrhythmia is typically caused by mutations affecting the biophysical properties of the pore-forming α-subunit of voltage-gated ion channels. However, biophysically normal α-subunit variants with hindered (a) synthesis or trafficking, (b) recycling and internalization, or (c) degradation pathways may also be disease causing, as ion channel density also determines current profiles [20]. Mutations affecting the pore-forming subunit, such as KCNQ1 (LQTS1, SQTS2), KCNH2 (LQTS2, SQTS1), and SCN5A (LQTS3, BrS), may cause heritable heart rhythm disorders. Importantly, it has become clear that variants in ancillary proteins, ranging from β-subunits to enzymes, may also cause or contribute to congenital arrhythmia. Both KCNE1 (LQTS5) and KCNE2 (LQTS6), which encode for β-subunits that modulate the conductance IKr and IKs, have been shown to be causative for LQTS by slowing repolarization [21,22,23]. Mutations in cytoskeletal and scaffolding proteins have also been linked to arrhythmia. LQTS4 is caused by a mutation in ankyrin-β (ANKB) [24]. ANKB mutations reduce the trafficking and expression of the Na+/K+ ATPase and Na+/Ca2+ exchanger as well as alter intracellular calcium handling that sensitizes the cells to arrhythmia [25]. Caveolae, which create small invaginations of the cell membrane, form micro-domains for ion channel expression and regulation [26, 27]. Mutations in CAV3 were shown to cause LQTS9 by increasing late sodium current [28] and modulating the pacemaker current If [29].
In addition to ion channels and ion channel-modifying proteins, intracellular calcium-handling proteins have profound effects on the electrophysiological properties of cardiomyocytes, and aberrant regulation of these genes can cause arrhythmia susceptibility. CPVT is an inherited arrhythmogenic disease caused by mutations in RYR2 [30, 31], calsequestrin (CASQ2) [32, 33], calmodulin (CALM) [34], TECRL [35], or triadin (TRDN) [36, 37]. RYR2 is the sarcoplasmic reticulum (SR)-anchored ligand-gated ion channel responsible for calcium flux from the SR to the cytosol to initiate contraction. SR calcium leak through RYR2 can induce arrhythmia when the leaked calcium is able to induce an ectopic action potential. Many other proteins, including TRDN, CALM, and CASQ2, associate with RYR2 and are involved in functional regulation of RYR2 or are responsible for calcium buffering in the SR. Hence, defects in the function of these proteins may adversely affect SR calcium physiology and render a patient susceptible to arrhythmia.
Mutations in the enzyme GPD1L were shown to cause BrS2 by modulating PKC-dependent phosphorylation of SCN5A resulting in reduced INa [38,39,40]. Interestingly, mutations in the enzyme TECRL were also linked to CPVT [35]. However, the precise function of this protein remains unknown, but a role in lipid metabolism is suspected. Moreover, variants in the hormone NPPA [41] and multiple transcription factors have also been implicated in AFib [42, 43]. Such variants likely act through modulation of ion channels transcription, atrial remodeling, pulmonary vein development, or development of cardiac conduction system [44, 45]. Taken together, defects not only in pore-forming α-subunits of ion channels, but also in a wide variety of other proteins have been shown to cause or modulate susceptibility to arrhythmia.
Ion Channel Regulation and Implications for Disease
Sudden cardiac death due to arrhythmia is the leading cause of mortality in patients with heart failure [46]. Altered ion channel expression and regulation as well as tissue damage are thought to underpin arrhythmic susceptibility in the failing heart. Cardiomyocytes in the failing heart have prolonged AP duration, decreased repolarization reserve, and a high rate of Ca2+-dependent arrhythmias resulting from electrical remodeling due to aberrant neurohormonal signaling. Understanding the regulatory mechanisms of ion channel function and how these are perturbed by CVDs may yield possibilities for drug development.
The electrophysiological properties of cardiomyocytes are heavily regulated by intrinsic signals, extrinsic signals, and mechanical stress in both healthy and diseased patients [47]. Several neurohormonal pathways are active in the heart and are responsible for adverse remodeling and progression of the disease [48, 49]. Ca2+-dependent signaling regulates transcriptional activity in the heart via CaMK2 [50,51,52]- and calcineurin [52, 53]-dependent pathways. In addition, LTCCs provide the cell with the ability to indirectly sense membrane voltage changes and alter gene expression as its C-terminal domain translocates to the nucleus to regulate gene expression [54]. miRNAs fine-tune expression of multiple genes by preventing translation of the target mRNA, and several miRNAs modulate cardiovascular physiology or are differentially expressed in heart disease [55,56,57,58,59,60]. Alternative splicing of SCN5A resulting in truncated non-functional channels is increased in heart failure [61]. In addition, multiple ion channels undergo differential expression in the failing human heart: KCNJ2, KCND3, CACNA1C, SCN5A, KCNH2, KCNQ1, RYR2, CASQ, SERCA2A, NCX, Na+/K+ ATPase, and calmodulin are reduced, whereas HCN4 (If) is increased [62], albeit with some heterogeneity and inconsistency across studies. These findings likely contribute to the AP prolongation, automaticity, and conduction disturbances observed in the failing heart.
Abnormal trafficking may be an important contributing factor to altered ion channel function in diseased hearts. However, the mechanisms of anterograde and retrograde trafficking of membrane proteins leading to the highly organized membrane structure of the cardiomyocyte remain poorly understood [63]. Drugs such as probucol and fluoxetine can cause drug-induced QT prolongation by blocking hERG channel trafficking [64]. In addition, hERG membrane expression is regulated by extracellular potassium concentration, as reduction of extracellular potassium leads to hERG internalization and degradation, and hence reduced IKr [65]. This mechanism may in part explain why hypokalemia is a risk factor for arrhythmia and sudden cardiac death in heart disease [66]. miR-1 is upregulated in patients with coronary artery disease and infarcted rat hearts [67], and overexpression of this miRNA induces arrhythmia in mice by downregulating trafficking genes [68]. Gap junctions, comprising connexin channels, play an important role in electrical conduction of the electrical excitation throughout the heart. In the healthy heart, gap junctions are localized at the intercalated discs for longitudinal spread of excitation. However, the expression of GJA1 is reduced at both the mRNA and protein level and improperly localized in heart failure [69, 70]. These deficits may contribute to conduction disturbances and proarrhythmia seen in heart failure.
Post-translational modifications (PTMs) enable transcription-independent modulation of cellular function, typically by inducing conformational changes. Phosphorylation is arguably the most studied reversible enzymatic PTM. Phosphate groups can be added to serine, threonine, and tyrosine residues by kinases, and removed by phosphatases. The human genome encodes for more than 500 kinases and many have been shown to play a critical role in heart disease [71]. Phosphorylation events orchestrated by PKA and CaMK2 participate in the excitation-contraction coupling as well as mediate the chronotropic, inotropic, and lusitropic effects of β-adrenergic stimulation on the heart [72]. PKA has many targets in the heart, including phospholamban, LTCCs, RYR, and troponin I [73, 74]. Similarly, CaMK2 has also been implicated in the phosphorylation of several ion channels in cardiomyocytes, such as sodium channels, LTCCs, RYR, and SERCA [75]. However, CaMK2 hyperactivity has been implicated in the pathogenesis of heart failure and arrhythmia [76]. Phosphorylation of sodium channels by CaMK2 increases late sodium current [77], while phosphorylation of RYR2 increases channel open probability leading to SR calcium leak and reduced SR calcium content [78]. As a result, inhibition of CaMK2 was recognized as a potential therapeutic strategy, for which industry has initiated a search for pharmaceutical grade compounds although none have yet reached clinical testing yet [76, 79].
Many other PTMs or amino acid modifications have been shown to modulate ion channel function and may have a role in disease. Glycosylation regulates multiple steps of protein biogenesis including protein folding, trafficking, and function [80]. Many voltage-gated channels are heavily glycosylated, and terminal sialic acid glycosylation plays an important role in voltage sensing and gating of ion channels [81]. Glycosylation-associated genes are differentially expressed in the heart with respect to chamber specification and developmental state [82]. Moreover, targeted deletion of such glycosylation-associated genes has profound effects on cardiac excitability. Aberrant glycosylation or sialylation of substrates, including calsequestrin [83], was also observed in CVD [84]. A missense mutation in calsequestrin (K206N) introduced an additional glycosylation site resulting in a larger molecular weight protein that reduced calcium-binding properties and caused arrhythmia by dysregulating calcium handling [85]. Protein methylation of sodium channels has also been observed on arginine residues implicated in Brugada syndrome and LQTS3 [86]. However, the mechanism by which methylation affects sodium channel function remains poorly understood. S-acylation is a reversible covalent fatty acid modification of cysteine residues and is enzymatically mediated by acyltransferases and is reversed by acyl-protein thioesterases. S-palmitoylation is the most common form of S-acylation [87, 88]. Palmitoylation motifs were shown to regulate late sodium current, and a mutation of a single cysteine (C981F) has been associated with susceptibility to arrhythmia [89]. Ubiquitinylation and SUMOylation are known to affect ion channel function by targeting the protein for degradation or manipulating activity [90]. SUMOylation was shown to modulate activation and inactivation kinetics of KCNQ1 (IKs) and KCNA5 (IKur), an atrial repolarizing potassium channel [91, 92]. In addition, a missense mutation attenuating deSUMOylation of TRPM4 associated with heart block resulted in impaired endocytosis and elevated cell membrane density [93]. Finally, oxidative stress is often associated with disease, including HF and AFib, and also enables protein modification and alteration of function [94, 95]. CaMK2 can be directly modified by reactive oxygen species leading to sustained activation and increased late sodium current, Na+/Ca2+ overload, and arrhythmogenesis [96, 97]. Oxidative stress has been linked to calcium channel [94] and potassium channel dysfunction [98]. In particular, KCNA5 is modified with sulfenic acid residues in response to oxidative stress in AFib leading to reduced surface expression and current density without altering the biophysical properties of the channel [99].
In summary, the biogenesis and function of cardiac ion channels is a tightly regulated process that is critical for the normal function of the heart. Ion channels are regulated at many levels from transcription and mRNA splicing to protein trafficking and post-translational modification. The results of genomic studies have identified many variants in regulatory genes of ion channels, and the emergence of new technologies to study the influence of these mutations in healthy and diseased states presents an exciting opportunity for the discovery of novel regulatory networks for future drug discovery.
iPSC-CMs for Arrhythmic Heart Disease Modeling
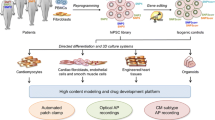
The ability to study the intricate mechanisms governing cardiomyocyte excitability in human cardiomyocytes has been extraordinarily difficult due to the scarce availability of human myocardial samples, and the inability to isolate and propagate primary human cardiomyocytes. Classical models to measure cardiovascular electrophysiology have been limited mostly to heterologous expression of α-subunits in cell lines, such as HEK293 and CHO that do not retain the cardiomyocytes context and rodent models that do not recapitulate the human cellular context. The development of human iPSCs as a stem cell source [100] and efficient protocols for deriving cardiomyocytes [101, 102] has enabled the routine production of virtually unlimited quantities of human iPSC-CMs. iPSC-CMs make it possible to assess the integrated effect of genetic variants or external stimuli on excitation-contraction coupling. Additionally, recent advances in gene-editing tools facilitate the generation of de novo mutations in a healthy patient background or correction of a putative disease-causing mutations in patient lines [103, 104]. As a result, iPSC-CMs have been utilized as a model to study monogenic CVDs and to address potential roles of putative at-risk alleles identified by GWAS [105]. Multiple familial arrhythmogenic disorders have been studied using iPSC-CMs, including LQTS [106, 107], CPVT [108], arrhythmogenic right ventricular dysplasia [109], and more recently BrS [110]. Initially, the focus was mainly channel α-subunit-dependent channelopathies, whereas more recently, the contribution of alternate mechanisms to arrhythmia has also been probed with iPSC-CMs, such as Cav3 in LQTS9 [111]. While this area remains understudied, we believe that iPSC-CMs can aid in deconvoluting complex mechanisms of arrhythmia, as these cells allow the investigation of the interaction between multiple molecular cascades in the context of a human cardiomyocyte.
Despite their key advantages, iPSC-CMs have limitations such as variability, impurity, physiological immaturity, and lack of chamber specificity, which we hope further research will resolve [112]. iPSC-CMs are considered immature counterparts for the human adult cardiomyocytes, with respect to their structure, metabolism, and electrophysiology [113]. For example, the slightly depolarized resting membrane potential and the lack of reliance on sodium current for AP generation may restrict the use of such immature cells to probe the contribution of late sodium current to arrhythmia [114•]. However, considerable progress has been made using culture media alteration and tissue engineering strategies to further purify and mature the physiology of the iPSC-CMs [115,116,117,118]. In addition, iPSC-CMs can be directed to ventricular or atrial fate by defined factors [119] and can hence be used to study cell-autonomous AFib mechanisms. Importantly, several companies have been founded to generate commercial grade human iPSC-CMs to satisfy the needs of industry.
High-Throughput iPSC-CM Models for Drug Discovery and Toxicity Screening
iPSC-CMs have considerable benefits for drug discovery as well, which remain to be explored, as these cells enable screening with physiological readouts compared to traditional target-based screens of ion channel α-subunits. This feature represents a significant advance for drug discovery by moving away from target-centric approaches and towards modulation of a physiological phenotype that is a result of many complex biological processes working in concert, such as APs or Ca2+ transients (Fig. 2). Physiological screens with iPSC-CMs have the following benefits: (1) the function of the channels can be probed in context with other ion channels and regulatory proteins that more closely resemble in vivo environment; (2) disease states can be recreated using iPSC-CMs derived from specific CVD patients; and (3) the contribution of non-ion channel proteins and cell signaling can be studied using powerful screening tools such as functional and chemical genomics approaches.

Induced pluripotent stem cells can be derived with high efficiency from patient tissue samples and efficiently differentiated to virtually all cell types of the body, including cardiomyocytes (iPSC-CMs). iPSC-CMs can be utilized to study mechanisms of arrhythmia dependent on extrinsic (pharmacological drugs and chemical environment) as well as intrinsic factors (genetics). While traditional techniques for quantifying electrophysiological parameters and excitation-contraction coupling are low throughput, recent developments in both screening technology and high-throughput assays enable phenotypical screens of moderate sized chemical libraries (small molecules, nucleic acids, or CRISPR/Cas9) in parallel. We believe these technologies will drive our current understanding of molecular arrhythmia mechanisms further and will revolutionize target identification and drug discovery for anti-arrhythmics in addition to determining of cardiotoxicity liabilities of drug candidates
Currently, mainly low-throughput assays have been used to probe iPSC-CMs as well as animal-derived cardiomyocytes. Recent developments in high-throughput optical platforms to record AP kinetics enable the ability to screen chemical libraries and quantify physiological parameters [120]. These methods may truly revolutionize cardiovascular drug discovery. Optically, recording of APs overcomes the inherent throughput limitations of single-cell patch clamping electrophysiology assays, as cells can be imaged in multi-well plates rather than as single cells. Additionally, the reduced cost of using voltage-sensitive dyes provides an advantage over more expensive approaches such as microelectrode arrays that may be prohibitively costly for functional genomics applications. Organic small molecule calcium and voltage dyes are available with appropriate fluorescent properties to enable high-throughput screening [114•, 121, 122]. The high-throughput quantification of cardiomyocyte contractility is more challenging. Many assays use cellular displacement as a surrogate for contractility, since the direct measurement of force is more difficult to accomplish at a micrometer scale [123, 124]. For example, traction force microscopy (TFM) on single cells and on micro-patterned gels [115], micropost deflection assays [125], or engineered heart tissues [126] can be used to measure force, but difficulties in fabrication of suitable devices as well as development of instruments to measure force in a high-throughput fashion have limited the widespread adoption of these tools.
There is also considerable interest to use iPSC-CMs to study the cardiovascular safety of drug candidates [127,128,129]. A traditional approach to determine the proarrhythmic liability of a given drug candidate has been to measure inhibition of hERG (IKr) channels. Although measuring IKr inhibition has kept potentially toxic drugs off the market, the technique does not predict Torsade de Points (TdP) as some drugs inhibit IKr without causing TdP [130, 131] and other drugs cause TdP without inhibiting IKr [132,133,134]. Given the phenotypical screening potential of iPSC-CMs, these cells can be utilized for toxicity screening without bias towards the underlying mechanisms. The utility of iPSC-CMs for prediction of cardiotoxicity is currently being investigated by the FDA under the Comprehensive in vitro Proarrhythmia Assay (CiPA) initiative. The project aims to develop a more predictive model for TdP by bringing together academia, pharmaceutical industry, and regulatory agencies to facilitate the adoption of a new paradigm for assessment of drug toxicity. Current CiPA studies support the utility of iPSC-CMs for proarrhythmic testing; however, further development and validation are likely warranted before adoption [135•, 136•, 137•]. Additionally, proarrhythmia is not the only potential liability of drug candidates as some drugs have been shown to induce cardiomyopathy and heart failure [138]. iPSC-CMs can be similarly applied for such toxicities using other phenotypic readouts. Sharma et al. [128] recently analyzed the cardiotoxic propensity of 21 anti-cancer kinase inhibitors by developing a risk index based on the relationship between plasma Cmax and the dose that affected both contractility and viability in vitro in iPSC-CMs. The results correlated well with clinically reported adverse events in patients.
iPSC-CMs represent a flexible platform for screening of diverse chemical collections that probe the contribution of both coding and non-coding mechanisms to arrhythmia. By combining genetic, pharmacological, or environmentally induced iPSC-CMs models of disease with physiological screens using libraries of small molecules [139], (anti-) miRNAs, siRNAs, and/or Cas9 guide RNAs [140, 141], the role of individual proteins or signaling pathways on the cardiomyocyte excitability can be rapidly tested (Fig. 2). For example, by knocking down individual kinases, the impact of signaling cascades on arrhythmia propensity can be probed directly under normal or diseased conditions. With CRISPR gene-editing technology, individual PTM sites, such as for phosphorylation or ubiquitinylation, can be mutated at the genomic level to study the role of specific PTM events on arrhythmia propensity. As these edited proteins remain under endogenous promotor regulation, this methodology is superior to traditional viral overexpression. Taken together, iPSC-CMs offer the ability to conduct phenotypical screening assays on human cardiomyocytes, which provides relevant new avenues for drug discovery, both for conducting screens for rescuing disease phenotypes and for quantifying cardiotoxic liabilities.
Conclusion
Arrhythmia is a significant disease burden and the development of novel drugs for this indication has stagnated in recent years. It is becoming increasingly clear that multiple proteins in addition to ion channels may confer susceptibility to arrhythmia. Additional research into the regulation of ion channel expression, trafficking, and regulation is needed and may provide novel directions for drug development. New technologies such as iPSC-CMs, gene editing, functional genomics, and high-throughput screening platforms provide new approaches to our search for novel arrhythmia therapies and, hopefully, will lead to the discovery of new drugs to alleviate arrhythmogenesis in patients with CVDs.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Hay M, Thomas DW, Craighead JL, Economides C, Rosenthal J. Clinical development success rates for investigational drugs. Nat Biotechnol. 2014;32:40–51.
Packer M. The imminent demise of cardiovascular drug development. JAMA Cardiol. 2017;2:1293–4.
• Fordyce CB, Roe MT, Ahmad T, Libby P, Borer JS, Hiatt WR, et al. Cardiovascular drug development: is it dead or just hibernating? J Am Coll Cardiol. 2015;65:1567–82. This paper describes the diminished efforts to develop new cardiac drugs and the underlying reasons.
Hwang TJ, Lauffenburger JC, Franklin JM, Kesselheim AS. Temporal trends and factors associated with cardiovascular drug development, 1990 to 2012. JACC Basic Transl Sci. 2016;1:301–8.
Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, et al. Heart disease and stroke statistics—2017 update: a report from the American Heart Association. Circulation. 2017;135:e146–603.
Karakikes I, Ameen M, Termglinchan V, Wu JC. Human induced pluripotent stem cell-derived cardiomyocytes: insights into molecular, cellular, and functional phenotypes. Circ Res. 2015;117:80–8.
Chaudhry GM, Haffajee CI. Antiarrhythmic agents and proarrhythmia. Crit Care Med. 2000;28:N158–64.
Nakano Y, Shimizu W. Genetics of long-QT syndrome. J Hum Genet. 2016;61:51–5.
Gollob MH, Redpath CJ, Roberts JD. The short QT syndrome: proposed diagnostic criteria. J Am Coll Cardiol. 2011;57:802–12.
Priori SG, Chen SRW. Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ Res. 2011;108:871–83.
Anderson JB, Benson DW. Genetics of sick sinus syndrome. Cardiac Electrophysiol Clin. 2010;2:499–507.
Christophersen IE, Ellinor PT. Genetics of atrial fibrillation: from families to genomes. J Hum Genet. 2016;61:61–70.
Campuzano O, Perez-Serra A, Iglesias A, Brugada R. Genetic basis of atrial fibrillation. Genes Dis. 2016;3:257–62.
Nielsen MW, Holst AG, Olesen S-P, Olesen MS. The genetic component of Brugada syndrome. Front Physiol. 2013;4:179.
Marsman RF, Tan HL, Bezzina CR. Genetics of sudden cardiac death caused by ventricular arrhythmias. Nat Rev Cardiol. 2014;11:96–111.
Milan DJ, Lubitz SA, Kääb S, Ellinor PT. Genome-wide association studies in cardiac electrophysiology: recent discoveries and implications for clinical practice. Heart Rhythm. 2010;7:1141–8.
Sinner MF, Ellinor PT, Meitinger T, Benjamin EJ, Kaab S. Genome-wide association studies of atrial fibrillation: past, present, and future. Cardiovasc Res. 2011;89:701–9.
Behr ER, Ritchie MD, Tanaka T, Kääb S, Crawford DC, Nicoletti P, et al. Genome wide analysis of drug-induced torsades de pointes: lack of common variants with large effect sizes. PLoS One. 2013;8:e78511.
Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, et al. Prevalence of the congenital long-QT syndrome. Circulation. 2009;120:1761–7.
Curran J, Mohler PJ. Alternative paradigms for ion channelopathies: disorders of ion channel membrane trafficking and posttranslational modification. Annu Rev Physiol. 2015;77:505–24.
Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, et al. MiRP1 forms I Kr potassium channels with HERG and is associated with cardiac arrhythmia. Cell. 1999;97:175–87.
Brenyo AJ, Huang DT, Aktas MK. Congenital long and short QT syndromes. Cardiology. 2012;122:237–47.
Splawski I, Tristani-Firouzi M, Lehmann MH, Sanguinetti MC, Keating MT. Mutations in the hminK gene cause long QT syndrome and suppress lKs function. Nat Gen. 1997;17:338–40.
Mohler PJ, Splawski I, Napolitano C, Bottelli G, Sharpe L, Timothy K, et al. A cardiac arrhythmia syndrome caused by loss of ankyrin-B function. Proc Natl Acad Sci. 2004;101:9137–42.
Mohler PJ, Schott J-J, Gramolini AO, Dilly KW, Guatimosim S, WH DB, et al. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature. 2003;421:634–9.
Yarbrough TL. Localization of cardiac sodium channels in caveolin-rich membrane domains: regulation of sodium current amplitude. Circ Res. 2002;90:443–9.
Balijepalli RC, Foell JD, Hall DD, Hell JW, Kamp TJ. Localization of cardiac L-type ca(2+) channels to a caveolar macromolecular signaling complex is required for beta(2)-adrenergic regulation. Proc Natl Acad Sci. 2006;103:7500–5.
Vatta M, Ackerman MJ, Ye B, Makielski JC, Ughanze EE, Taylor EW, et al. Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome. Circulation. 2006;114:2104–12.
Motloch LJ, Larbig R, Darabi T, Reda S, Motloch KA, Wernly B, et al. Long-QT syndrome-associated caveolin-3 mutations differentially regulate the hyperpolarization-activated cyclic nucleotide gated channel 4. Physiol Int. 2017;104:130–8.
Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001;103:196–200.
Laitinen PJ, Brown KM, Piippo K, Swan H, Devaney JM, Brahmbhatt B, et al. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation. 2001;103:485–90.
Lahat H, Pras E, Olender T, Avidan N, Ben-Asher E, Man O, et al. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am J Hum Genet. 2001;69:1378–84.
Postma AV, Denjoy I, Hoorntje TM, Lupoglazoff J-M, Da Costa A, Sebillon P, et al. Absence of calsequestrin 2 causes severe forms of catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2002;91:e21–6.
Gomez-Hurtado N, Boczek NJ, Kryshtal DO, Johnson CN, Sun J, Nitu FR, et al. Novel CPVT-associated calmodulin mutation in CALM3 (CALM3-A103V) activates arrhythmogenic ca waves and sparks. Circ Arrhythm Electrophysiol. 2016;9
Devalla HD, Gélinas R, Aburawi EH, Beqqali A, Goyette P, Freund C, et al. TECRL, a new life-threatening inherited arrhythmia gene associated with overlapping clinical features of both LQTS and CPVT. EMBO Mol Med. 2016:e201505719.
Roux-Buisson N, Cacheux M, Fourest-Lieuvin A, Fauconnier J, Brocard J, Denjoy I, et al. Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human. Hum Mol Genet. 2012;21:2759–67.
Rooryck C, Kyndt F, Bozon D, Roux-Buisson N, Sacher F, Probst V, et al. New family with catecholaminergic polymorphic ventricular tachycardia linked to the triadin gene. J Cardiovasc Electrophysiol. 2015;26:1146–50.
Van Norstrand DW, Valdivia CR, Tester DJ, Ueda K, London B, Makielski JC, et al. Molecular and functional characterization of novel glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) mutations in sudden infant death syndrome. Circulation. 2007;116:2253–9.
London B, Michalec M, Mehdi H, Zhu X, Kerchner L, Sanyal S, et al. Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias. Circulation. 2007;116:2260–8.
Valdivia CR, Ueda K, Ackerman MJ, Makielski JC. GPD1L links redox state to cardiac excitability by PKC-dependent phosphorylation of the sodium channel SCN5A. Am J Physiol Heart Circ Physiol. 2009;297:H1446–52.
Hodgson-Zingman DM, Karst ML, Zingman LV, Heublein DM, Darbar D, Herron KJ, et al. Atrial natriuretic peptide frameshift mutation in familial atrial fibrillation. N Engl J Med. 2008;359:158–65.
Huang R-T, Xue S, Xu Y-J, Zhou M, Yang Y-Q. A novel NKX2.5 loss-of-function mutation responsible for familial atrial fibrillation. Int J Mol Med. 2013;31:1119–26.
Yang Y-Q, Wang M-Y, Zhang X-L, Tan H-W, Shi H-F, Jiang W-F, et al. GATA4 loss-of-function mutations in familial atrial fibrillation. Clin Chim Acta. 2011;412:1825–30.
Zhou M, Liao Y, Tu X. The role of transcription factors in atrial fibrillation. J Thorac Dis. 2015;7:152–8.
Mahida S. Transcription factors and atrial fibrillation. Cardiovasc Res. 2014;101:194–202.
Klein L, Hsia H. Sudden cardiac death in heart failure. Cardiol Clin. 2014;32:135–44.
Rosati B. Regulation of ion channel expression. Circ Res. 2004;94:874–83.
Lefkowitz RJ, Rockman HA, Koch WJ. Catecholamines, cardiac -adrenergic receptors, and heart failure. Circulation. 2000;101:1634–7.
Schrier RW, Abraham WT. Hormones and hemodynamics in heart failure. N Engl J Med. 1999;341:577–85.
Backs J, Song K, Bezprozvannaya S, Chang S, Olson EN. CaM kinase II selectively signals to histone deacetylase 4 during cardiomyocyte hypertrophy. J Clin Invest. 2006;116:1853–64.
Li C, Cai X, Sun H, Bai T, Zheng X, Zhou XW, et al. The δA isoform of calmodulin kinase II mediates pathological cardiac hypertrophy by interfering with the HDAC4-MEF2 signaling pathway. Biochem Biophys Res Commun. 2011;409:125–30.
Zhu W, Zou Y, Shiojima I, Kudoh S, Aikawa R, Hayashi D, et al. Ca 2+/calmodulin-dependent kinase II and calcineurin play critical roles in endothelin-1-induced cardiomyocyte hypertrophy. J Biol Chem. 2000;275:15239–45.
Zhang Z-Y, Liu X-H, Hu W-C, Rong F, Wu X-D. The calcineurin-myocyte enhancer factor 2c pathway mediates cardiac hypertrophy induced by endoplasmic reticulum stress in neonatal rat cardiomyocytes. Am J Physiol Heart Circ Physiol. 2010;298:H1499–509.
Lu L, Sirish P, Zhang Z, Woltz RL, Li N, Timofeyev V, et al. Regulation of gene transcription by voltage-gated L-type calcium channel, Ca v1.3. J Biol Chem. 2015;290:4663–76.
Liu X, Xiao J, Zhu H, Wei X, Platt C, Damilano F, et al. miR-222 is necessary for exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell Metabolism. 2015;21:584–95.
Wahlquist C, Jeong D, Rojas-Muñoz A, Kho C, Lee A, Mitsuyama S, et al. Inhibition of miR-25 improves cardiac contractility in the failing heart. Nature. 2014;508:531–5.
Lu Y, Zhang Y, Wang N, Pan Z, Gao X, Zhang F, et al. MicroRNA-328 contributes to adverse electrical remodeling in atrial fibrillation. Circulation. 2010;122:2378–87.
Wang J, Song Y, Zhang Y, Xiao H, Sun Q, Hou N, et al. Cardiomyocyte overexpression of miR-27b induces cardiac hypertrophy and dysfunction in mice. 2012;22:516–27.
Huang Z-P, Wang D-Z. miR-22 in cardiac remodeling and disease. Trends Cardiovasc Med. 2014;24:267–72.
Kim GH. MicroRNA regulation of cardiac conduction and arrhythmias. Transl Res. 2013;161:381–92.
Shang LL, Pfahnl AE, Sanyal S, Jiao Z, Allen J, Banach K, et al. Human heart failure is associated with abnormal C-terminal splicing variants in the cardiac sodium channel. Circ Res. 2007;101:1146–54.
Nattel S, Frelin Y, Gaborit N, Louault C, Demolombe S. Ion-channel mRNA-expression profiling: insights into cardiac remodeling and arrhythmic substrates. J Mol Cell Cardiol. 2010;48:96–105.
Balse E, Boycott HE. Ion channel trafficking: control of ion channel density as a target for arrhythmias? Front Physiol. 2017;8:H1851.
Dennis A, Wang L, Wan X, Ficker E. hERG channel trafficking: novel targets in drug-induced long QT syndrome. Biochem Soc Trans. 2007;35:1060–3.
Guo J, Massaeli H, Xu J, Jia Z, Wigle JT, Mesaeli N, et al. Extracellular K+ concentration controls cell surface density of IKr in rabbit hearts and of the HERG channel in human cell lines. J Clin Invest. 2009;119:2745–57.
Kjeldsen K. Hypokalemia and sudden cardiac death. Exp Clin Cardiol. 2010;15:e96–9.
Yang B, Lin H, Xiao J, Lu Y, Luo X, Li B, et al. The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat Med. 2007;13:486–91.
Su X, Liang H, Wang H, Chen G, Jiang H, Wu Q, et al. Over-expression of microRNA-1 causes arrhythmia by disturbing intracellular trafficking system. Sci Rep. 2017;7:46259.
Smyth JW, Hong T-T, Gao D, Vogan JM, Jensen BC, Fong TS, et al. Limited forward trafficking of connexin 43 reduces cell-cell coupling in stressed human and mouse myocardium. J Clin Investig. 2010;120:266–79.
Dupont E, Matsushita T, Kaba RA, Vozzi C, Coppen SR, Khan N, et al. Altered connexin expression in human congestive heart failure. J Mol Cell Cardiol. 2001;33:359–71.
Fuller SJ, Osborne SA, Leonard SJ, Hardyman MA, Vaniotis G, Allen BG, et al. Cardiac protein kinases: the cardiomyocyte kinome and differential kinase expression in human failing hearts. Cardiovasc Res. 2015;108:87–98.
Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205.
Scheuer T. Regulation of sodium channel activity by phosphorylation. Semin Cell Dev Biol. 2011;22:160–5.
Davis MJ, Wu X, Nurkiewicz TR, Kawasaki J, Gui P, Hill MA, et al. Regulation of ion channels by protein tyrosine phosphorylation. Am J Physiol Heart Circ Physiol. 2001;281:H1835–62.
Bers DM, Grandi E. Calcium/calmodulin-dependent kinase II regulation of cardiac ion channels. J Cardiovasc Pharmacol. 2009;54:180–7.
Pellicena P, Schulman H. CaMKII inhibitors: from research tools to therapeutic agents. Front Pharmacol. 2014;5:21.
Wagner S, Dybkova N, Rasenack ECL, Jacobshagen C, Fabritz L, Kirchhof P, et al. Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J Clin Invest. 2006;116:3127–38.
Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res. 2005;97:1314–22.
Neef S, Steffens A, Pellicena P, Mustroph J, Lebek S, Ort KR, et al. Improvement of cardiomyocyte function by a novel pyrimidine-based CaMKII-inhibitor. J Mol Cell Cardiol. 2018;115:73–81.
Fozzard HA, Kyle JW. Do defects in ion channel glycosylation set the stage for lethal cardiac arrhythmias? Sci STKE. 2002;2002:pe19.
Baycin-Hizal D, Gottschalk A, Jacobson E, Mai S, Wolozny D, Zhang H, et al. Physiologic and pathophysiologic consequences of altered sialylation and glycosylation on ion channel function. Biochem Biophys Res Commun. 2014;453:243–53.
Montpetit ML, Stocker PJ, Schwetz TA, Harper JM, Norring SA, Schaffer L, et al. Regulated and aberrant glycosylation modulate cardiac electrical signaling. Proc Natl Acad Sci. 2009;106:16517–22.
Kiarash A, Kelly CE, Phinney BS, Valdivia HH, Abrams J, Cala SE. Defective glycosylation of calsequestrin in heart failure. Cardiovasc Res. 2004;63:264–72.
Rong J, Han J, Dong L, Tan Y, Yang H, Feng L, et al. Glycan imaging in intact rat hearts and glycoproteomic analysis reveal the upregulation of sialylation during cardiac hypertrophy. J Am Chem Soc. 2014;136:17468–76.
Kirchhefer U, Wehrmeister D, Postma AV, Pohlentz G, Mormann M, Kucerova D, et al. The human CASQ2 mutation K206N is associated with hyperglycosylation and altered cellular calcium handling. J Mol Cell Cardiol. 2010;49:95–105.
Beltran-Alvarez P, Pagans S, Brugada R. The cardiac sodium channel is post-translationally modified by arginine methylation. J Proteome Res. 2011;10:3712–9.
Shipston MJ. Ion channel regulation by protein palmitoylation. J Biol Chem. 2011;286:8709–16.
Shipston MJ. Ion channel regulation by protein S-acylation. J Gen Physiol. 2014;143:659–78.
Pei Z, Xiao Y, Meng J, Hudmon A, Cummins TR. Cardiac sodium channel palmitoylation regulates channel availability and myocyte excitability with implications for arrhythmia generation. Nat Commun. 2016;7:12035.
Rougier J-S, Albesa M, Abriel H. Ubiquitylation and SUMOylation of cardiac ion channels. J Cardiovasc Pharmacol. 2010;56:22–8.
Benson MD, Li Q-J, Kieckhafer K, Dudek D, Whorton MR, Sunahara RK, et al. SUMO modification regulates inactivation of the voltage-gated potassium channel Kv1.5. Proc Natl Acad Sci. 2007;104:1805–10.
Xiong D, Li T, Dai H, Arena AF, Plant LD, Goldstein SAN. SUMOylation determines the voltage required to activate cardiac IKschannels. Proc Natl Acad Sci. 2017;114:E6686–94.
Kruse M, Schulze-Bahr E, Corfield V, Beckmann A, Stallmeyer B, Kurtbay G, et al. Impaired endocytosis of the ion channel TRPM4 is associated with human progressive familial heart block type I. J Clin Invest. 2009;119:2737–44.
Hool LC. Evidence for the regulation of L-type Ca2+ channels in the heart by reactive oxygen species: mechanism for mediating pathology. Clin Exp Pharmacol Physiol. 2008;35:229–34.
Seddon M, Looi YH, Shah AM. Oxidative stress and redox signalling in cardiac hypertrophy and heart failure. Heart. 2007;93:903–7.
Erickson JR, Joiner M-LA, Guan X, Kutschke W, Yang J, Oddis CV, et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. 2008;133:462–74.
Wagner S, Ruff HM, Weber SL, Bellmann S, Sowa T, Schulte T, et al. Reactive oxygen species-activated ca/calmodulin kinase II is required for late INa augmentation leading to cellular Na and Ca overload. Circ Res. 2011;108:555–65.
Zhang Y, Xiao J, Wang H, Luo X, Wang J, Villeneuve LR, et al. Restoring depressed HERG K +channel function as a mechanism for insulin treatment of abnormal QT prolongation and associated arrhythmias in diabetic rabbits. Am J Physiol Heart Circ Physiol. 2006;291:H1446–55.
Svoboda LK, Reddie KG, Zhang L, Vesely ED, Williams ES, Schumacher SM, et al. Redox-sensitive sulfenic acid modification regulates surface expression of the cardiovascular voltage-gated potassium channel Kv1.5. Circ Res. 2012;111:842–53.
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–72.
Burridge PW, Matsa E, Shukla P, Lin ZC, Churko JM, Ebert AD, et al. Chemically defined generation of human cardiomyocytes. Nat Meth. 2014;11:855–60.
Lian X, Hsiao C, Wilson G, Zhu K, Hazeltine LB, Azarin SM, et al. Cozzarelli prize winner: robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc Natl Acad Sci. 2012;109:E1848–57.
Karakikes I, Termglinchan V, Cepeda DA, Lee J, Diecke S, Hendel A, et al. A comprehensive TALEN-based knockout library for generating human-induced pluripotent stem cell–based models for cardiovascular diseases novelty and significance. Circ Res. 2017;120:1561–71.
Seeger T, Porteus M, Wu JC. Genome editing in cardiovascular biology. Circ Res. 2017;120:778–80.
Panopoulos AD, D'Antonio M, Benaglio P, Williams R, Hashem SI, Schuldt BM, et al. iPSCORE: a resource of 222 iPSC lines enabling functional characterization of genetic variation across a variety of cell types. Stem Cell Rep. 2017;8:1086–100.
Moretti A, Bellin M, Welling A, Jung CB, Lam JT, Bott-Flügel L, et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N Engl J Med. 2010;363:1397–409.
Matsa E, Rajamohan D, Dick E, Young L, Mellor I, Staniforth A, et al. Drug evaluation in cardiomyocytes derived from human induced pluripotent stem cells carrying a long QT syndrome type 2 mutation. Eur Heart J. 2011;32:952–62.
Itzhaki I, Maizels L, Huber I, Gepstein A, Arbel G, Caspi O, et al. Modeling of catecholaminergic polymorphic ventricular tachycardia with patient-specific human-induced pluripotent stem cells. J Am Coll Cardiol. 2012;60:990–1000.
Kim C, Wong J, Wen J, Wang S, Wang C, Spiering S, et al. Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature. 2013;494:105–10.
Liang P, Sallam K, Wu H, Li Y, Itzhaki I, Garg P, et al. Patient-specific and genome-edited induced pluripotent stem cell-derived cardiomyocytes elucidate single-cell phenotype of Brugada syndrome. J Am Coll Cardiol. 2016;68:2086–96.
Vaidyanathan R, Markandeya YS, Kamp TJ, Makielski JC, January CT, Eckhardt LL. IK1-enhanced human-induced pluripotent stem cell-derived cardiomyocytes: an improved cardiomyocyte model to investigate inherited arrhythmia syndromes. Am J Physiol Heart Circ Physiol. 1st ed. 2016;310:H1611–21.
Kolanowski TJ, Antos CL, Guan K. Making human cardiomyocytes up to date: derivation, maturation state and perspectives. Int J Cardiol. 2017;241:379–86.
Robertson C, Tran DD, George SC. Concise review: maturation phases of human pluripotent stem cell-derived cardiomyocytes. Stem Cells. 2013;31:829–37.
• McKeithan WL, Savchenko A, Yu MS, Cerignoli F, Bruyneel AAN, Price JH, et al. An automated platform for assessment of congenital and drug-induced arrhythmia with hiPSC-derived cardiomyocytes. Front Physiol. 2017;8:766. Describes methods for a high-throughput assay to measure action potential kinetics in 384-well plate format for congenital and pharmacological arrhythmia disease modeling.
Ribeiro AJS, Ang Y-S, Fu J-D, Rivas RN, Mohamed TMA, Higgs GC, et al. Contractility of single cardiomyocytes differentiated from pluripotent stem cells depends on physiological shape and substrate stiffness. Proc Natl Acad Sci. 2015;112:12705–10.
Tohyama S, Hattori F, Sano M, Hishiki T, Nagahata Y, Matsuura T, et al. Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell. 2013;12:127–37.
Yang X, Rodriguez M, Pabon L, Fischer KA, Reinecke H, Regnier M, et al. Tri-iodo-l-thyronine promotes the maturation of human cardiomyocytes-derived from induced pluripotent stem cells. J Mol Cell Cardiol. 2014;72:296–304.
Liaw NY, Zimmermann W-HH. Mechanical stimulation in the engineering of heart muscle. Adv Drug Deliv Rev. 2016;96:156–60.
Zhang Q, Jiang J, Han P, Yuan Q, Zhang J, Zhang X, et al. Direct differentiation of atrial and ventricular myocytes from human embryonic stem cells by alternating retinoid signals. Cell Res. 2010;21:579–87.
del Álamo JC, Lemons D, Serrano R, Savchenko A, Cerignoli F, Bodmer R, et al. High throughput physiological screening of iPSC-derived cardiomyocytes for drug development. Biochim Biophys Acta. 1863;2016:1717–27.
Sirenko O, Crittenden C, Callamaras N, Hesley J, Chen Y-W, Funes C, et al. Multiparameter in vitro assessment of compound effects on cardiomyocyte physiology using iPSC cells. J Biomol Screen. 2013;18:39–53.
Bedut S, Seminatore-Nole C, Lamamy V, Caignard S, Boutin JA, Nosjean O, et al. High-throughput drug profiling with voltage- and calcium-sensitive fluorescent probes in human iPSC-derived cardiomyocytes. AJP Heart Circ Physiol. 2016;311:H44–53.
Hayakawa T, Kunihiro T, Ando T, Kobayashi S, Matsui E, Yada H, et al. Image-based evaluation of contraction–relaxation kinetics of human-induced pluripotent stem cell-derived cardiomyocytes: correlation and complementarity with extracellular electrophysiology. J Mol Cell Cardiol. 2014;77:178–91.
Maddah M, Heidmann JD, Mandegar MA, Walker CD, Bolouki S, Conklin BR, et al. A non-invasive platform for functional characterization of stem-cell-derived cardiomyocytes with applications in cardiotoxicity testing. Stem Cell Rep. 2015;4:621–31.
Beussman KM, Rodriguez ML, Leonard A, Taparia N, Thompson CR, Sniadecki NJ. Micropost arrays for measuring stem cell-derived cardiomyocyte contractility. Methods. 2016;94:43–50.
Breckwoldt K, Letuffe-Brenière D, Mannhardt I, Schulze T, Ulmer B, Werner T, et al. Differentiation of cardiomyocytes and generation of human engineered heart tissue. Nat Protoc. 2017;12:1177–97.
Matsa E, Burridge PW, Yu K-H, Ahrens JH, Termglinchan V, Wu H, et al. Transcriptome profiling of patient-specific human iPSC-cardiomyocytes predicts individual drug safety and efficacy responses in vitro. Cell Stem Cell. 2016;19:311–25.
Sharma A, Burridge PW, McKeithan WL, Serrano R, Shukla P, Sayed N, et al. High-throughput screening of tyrosine kinase inhibitor cardiotoxicity with human induced pluripotent stem cells. Sci Transl Med. 2017;9:eaaf2584.
Liang P, Lan F, Lee AS, Gong T, Sanchez-Freire V, Wang Y, et al. Drug screening using a library of human induced pluripotent stem cell-derived cardiomyocytes reveals disease-specific patterns of cardiotoxicity. Circulation. 2013;127:1677–91.
Redfern WS, Carlsson L, Davis AS, Lynch WG, MacKenzie I, Palethorpe S, et al. Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc Res. 2003;58:32–45.
Kramer J, Obejero-Paz CA, Myatt G, Kuryshev YA, Bruening-Wright A, Verducci JS, et al. MICE models: superior to the HERG model in predicting torsade de pointes. Sci Rep. 2013;3:2100.
Shah RR, Hondeghem LM. Refining detection of drug-induced proarrhythmia: QT interval and TRIaD. Heart Rhythm. 2005;2:758–72.
Roden DM. Mechanisms and management of proarrhythmia. Am J Cardiol. 1998;82:49I–57I.
Lacerda AE, Kuryshev YA, Chen Y, Renganathan M, Eng H, Danthi SJ, et al. Alfuzosin delays cardiac repolarization by a novel mechanism. J Pharmacol Exp Ther. 2008;324:427–33.
• Blinova K, Stohlman J, Vicente J, Chan D, Johannesen L, Hortigon-Vinagre MP, et al. Comprehensive translational assessment of human-induced pluripotent stem cell derived cardiomyocytes for evaluating drug-induced arrhythmias. Toxicol Sci. 2017;155:234–47. CiPA initiative paper.
• Ando H, Yoshinaga T, Yamamoto W, Asakura K, Uda T, Taniguchi T, et al. A new paradigm for drug-induced torsadogenic risk assessment using human iPS cell-derived cardiomyocytes. J Pharmacol Toxicol Methods. 2017;84:111–27. CiPA initiative paper.
• Huang H, Pugsley MK, Fermini B, Curtis MJ, Koerner J, Accardi M, et al. Cardiac voltage-gated ion channels in safety pharmacology: review of the landscape leading to the CiPA initiative. J Pharmacol Toxicol Methods. 2017;87:11–23. CiPA initiative paper.
Yeh ETH, Bickford CL. Cardiovascular complications of cancer therapy: incidence, pathogenesis, diagnosis, and management. J Am Coll Cardiol. 2009;53:2231–47.
Jones LH, Bunnage ME. Applications of chemogenomic library screening in drug discovery. Nat Rev Drug Discov. 2017;16:285–96.
Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–4.
Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–7.
Acknowledgments
We acknowledge Drs Sanjiv M Narayan and Ioannis Karakikes for providing helpful suggestions and feedback on the manuscript.
Funding
We gratefully acknowledge grant support from the NIH (1R01HL132225, 1R01HL130840, 1R01HL128072, 1R01HL113006, and HHSN268200900044C), Stanford University, the Fondation Leducq (Shapeheart), to MM. DF is funded by the European Union’s Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No 708459.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Arne A. N. Bruyneel, Wesley L. McKeithan, Dries A. M. Feyen, and Mark Mercola declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Regenerative Medicine
Rights and permissions
About this article

Cite this article
Bruyneel, A.A.N., McKeithan, W.L., Feyen, D.A.M. et al. Using iPSC Models to Probe Regulation of Cardiac Ion Channel Function. Curr Cardiol Rep 20, 57 (2018). https://doi.org/10.1007/s11886-018-1000-0
Published:
DOI: https://doi.org/10.1007/s11886-018-1000-0