
Abstract
Over the past three decades, statins have become first-line treatment for reducing LDL cholesterol (LDL-C) and cardiovascular disease (CVD). They have provided a clear, robust, and reproducible relationship between the absolute LDL-C reduction and the decrease in CVD; every 1 mmol/L (∼40 mg/dL) in LDL-C reduction results in a 22 % decrease in CVD events. This relationship has recently been extended to reduction in LDL-C with a non-statin, ezetimibe, on top of statin therapy, further consolidating LDL-C as the cornerstone in CVD risk reduction. Despite these two effective and safe LDL-C-lowering drugs, there remains a need for additional drugs to reduce LDL-C, the focus of this review which covers agents which produce sufficient LDL-C reduction to potentially help address this unmet need and are either recently approved or currently in clinical trials.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Since the introduction of statins in the USA in 1987, they have become the cornerstone of efforts to reduce cardiovascular disease (CVD). Irrespective of the starting levels of LDL cholesterol (LDL-C) and the underlying absolute risk of CVD, large trials have clearly demonstrated that LDL-C reduction with statins significantly and substantially reduces morbidity and mortality [1–4]. Starting in the early twenty-first century, statins have been available as generic formulations with the most effective statins, atorvastatin and rosuvastatin, becoming generic in many countries in 2011 and 2013, respectively. Rosuvastatin is anticipated to become generically available in the USA in 2016 and Europe a year later. More recently, the cholesterol absorption transport inhibitor, ezetimibe, which lowers LDL-C only moderately but is well tolerated and has few side effects, has recently been shown to further reduce CVD when added to statin therapy compared to statin alone and that the relationship between LDL-C reduction and CVD reduction is consistent with that seen with statins [5••].
There are currently limited options for large LDL-C reductions for patients either unable to tolerate statins or effective doses of statins or who are unable to achieve specific LDL-C goals despite statins. Perhaps the best example of the latter is patients with the most common genetic disorder worldwide, heterozygous familial hypercholesterolemia (HeFH) and especially the rarer patients with homozygous (Ho) FH. Besides ezetimibe, older agents such as niacin, bile acid sequestrants, and fibrates, which have been used for decades, only reduce LDL-C by ~15 %, may be associated with significant adverse events, and in recent outcome trials have not been shown to further reduce CVD events when added to statins [6–9].
Therefore, there remains a pressing need for additional LDL-C-lowering agents as summarized in Table 1.
We review LDL-C-lowering agents currently in clinical development, or recently approved, in two categories; those intended for general or broad use in all patient populations and those for specific for orphan use in only HoFH (Table 2).
LDL-C-Lowering Agents Intended for General or Broad Use
At present, all these agents are still in clinical development. Two therapeutic classes are currently in phase 3 development with the most exciting agents, the monoclonal antibodies (mAbs) inhibiting proprotein convertase subtilisin/kexin type 9 (PCSK9), anticipating marketing approval in Europe and the USA before the end of 2015. These agents based on supporting genetics, mechanism of action, and large and predictable reductions in LDL-C combined with good safety and tolerability promise to be as revolutionary as the statins were a generation ago. The second class of agents is the cholesterol ester transfer protein (CETP) inhibitors which inhibit the exchange of cholesterol between HDL and Apo B-containing lipoproteins. The third class of agent, ETC-1002, targets early steps in the cholesterol synthetic pathway modulating adenosine triphosphate-citrate lyase and adenosine monophosphate-activated protein kinase and is entering phase 3.
PCSK9 Inhibitors
Background
There are a number of recent excellent reviews of what is an extremely interesting pathway from discovery of PCSK9 to development of clinical compounds; therefore, we provide just a brief overview [10, 11]. Over the last decade, understanding of the role of PCSK9 in LDL metabolism has advanced rapidly. Studies using a PCSK9 knockout mouse model which showed that LDLR activity was substantially increased with resultant LDL-C reduction were followed by studies in humans which confirmed that families with low plasma levels of LDL-C had “loss-of-function” (LOF) mutations in PCSK9 and a significant reduction in lifetime risk of CVD, with no apparent adverse health effects [12]. A number of LOF mutations have now been described associated with both reduced LDL-C and CVD risk. A key discovery was the demonstration by Legace and coworkers in 2006 that it was circulating or plasma PCSK9 that bound to the LDLR, was internalized along with the receptor and LDL-C, and then targeted the LDLR for degradation, thus preventing the LDLR from recycling [13••]. This finding was critical, leading to the rapid development of monoclonal antibodies which bind to PCSK9 and inhibit PCSK9 binding to the LDLR and restore, or increase, LDLR activity.
PCSK9 mAbs
Two fully human mAbs targeted to PCSK9, alirocumab and evolocumab, entered phase 1 trials in humans in 2010 with results published in early 2012 [14••, 15]. These early studies showed dramatic reductions in free PCSK9 and LDL-C in patients with both FH and non-FH. Over the last 3 years, clinical development has progressed very rapidly as shown in Fig. 1, with these two agents having completed phase 3 studies and under review by regulatory agencies in the USA and Europe for marketing approval. A third mAb, bococizumab, is in phase 3 clinical development.

PCSK9: rapid progress in humans from POC to CVD data [11]
Dosing and Dosing Intervals
The trials have demonstrated that the binding of PCSK9 is rapid and complete, exceeding 99 %, within hours after subcutaneous (SC) dosing and the resultant effect on LDL-C follows within days, reaching a maximum reduction of approximately 60 % within about 5 to 7 days after dosing of 140 to 150 mg, a dose which binds all available PCSK9. Increasing doses even three- or fourfold does not result in additional LDL-C reduction but does increase the duration of PCSK9 suppression, keeping LDL-C stable at the lower level and thus increasing the interval needed between SC injections of the mAb. The pharmacokinetics are not linear, and extensive studies have shown that the dose required to double the interval between dosing and keep LDL-C stable is threefold; for example, evolocumab 420 mg every 4 weeks is needed to achieve the same reduction in LDL-C as 140 mg given every 2 weeks [16•].
Impact of Background Therapy
A number of larger trials have assessed the impact of background therapy on the LDL-C response when evolocumab or alirocumab was added to patients on diet alone, low- or high-dose statin therapy, and statin plus ezetimibe and found a consistent additional ∼60 % reduction [17, 18].
Effect in Patients with Statin-Associated Muscle Symptoms
Three trials, with nearly 800 patients in total, focusing on patients with a history of statin-associated muscle symptoms (SAMS) have been published or presented: two 12-week trials with evolocumab (GAUSS and GAUSS-2) and one 24-week trial with alirocumab (ODYSSEY Alternative) [19••, 20]. Based on intent-to-treat analysis, the two GAUSS trials reported a consistent 33–35 % additional reduction in LDL-C versus ezetimibe while ODYSSEY Alternative trial had a 30 % difference versus ezetimibe, reflecting in part that ∼50 % of patients remained on the 75 mg starting dose of alirocumab. Tolerability of the PCSK9 mAb was good for all three trials, adverse events overall, and muscle side effects in particular, were similar compared to ezetimibe and lower than the atorvastatin 20 mg group in ODYSSEY Alternative. These studies thus showed that PCSK9 inhibition can result in large LDL-C reductions, can achieve optimal LDL-C goals in many of these high-risk statin-intolerant patients, and confirm that it is not the reduction in LDL-C nor the LDL-C level achieved that is related to the muscle side effects reported with statins.
Effect in FH Patients
Large global trials in well over 1000 HeFH patients have shown that LDL-C reduction in FH with PCSK9 mAb therapy is the same ∼60 % seen in non-FH patients [21, 22, 23••]. These trials, all done on a background of high-dose efficacious statin ± ezetimibe, also demonstrated that for the first time nearly all HeFH patients could achieve LDL-C <100 mg/dL (2.6 mmol/L) and the majority LDL-C <70 mg/dL (1.8 mmol/L) with the addition of these novel agents. In the RUTHERFORD-2 trial, the potential role genetics plays in the LDL-C response in HeFH was evaluated [21]. As seen in Table 3, there was no difference between HeFH patients having a LDLR defect associated with no (“null”), some (“defective”), or unknown receptor activity, all experiencing a similar 60 % reduction in LDL-C. Thus, knowing the genetic abnormality in HeFH does not provide a guide to the use of PCSK9 mAb therapy in these patients. While there was general optimism that PCSK9 inhibitors would be effective in HeFH, there was significant skepticism that they would work in HoFH. However, following on a small proof-of-concept trial [24••], a larger, double-blind, randomized and placebo-controlled trial in 50 HoFH, the TESLA Part B trial, reported a 31 % decrease in LDL-C compared to placebo, half the reduction seen in HeFH or non-FH patients [25••]. However, due to very high baseline LDL-C levels in these patients, the absolute reduction in LDL-C of 93 mg/dL (2.4 mmol/L) was large. The study also confirmed that in HoFH, the LDL-C reduction is dependent on the underlying genetic defect(s) as reductions of 40 % were found in those with at least one defective LDLR and nearly 50 % in those with two defective LDLR genes. While very few of the patients were LDLR negative, it did appear that these, fortunately a small minority of HoFH patients, had no response.
Tolerability, Adherence, and Safety
Initial concern that the need for SC injections may lead to discontinuation and poor adherence to the therapy has not been substantiated. In fact, the number of patients withdrawing from trials has been no higher than trials with statins or other oral lipid-lowering drugs. Serious adverse events and treatment-related adverse events have also been no different from those reported in placebo, comparator, or control groups with no specific adverse events indicating any particular organ system. In terms of laboratory findings, no significant or sustained elevations of either liver function or muscle enzymes have been reported. The development of consistent or high titers of neutralizing antibodies with evolocumab has not been reported so far and very few with alirocumab, probably because these are fully humanized antibodies, which minimizes the potential for immune reactions. Concern regarding the very low levels of LDL-C that are achieved in some patients has been partially addressed in the short-term phase 3 trials by evaluating adverse events based on achieved LDL-C during the trials [26••, 27••]. From Tables 4 and 5, there appears to be no increased adverse events, clinically or in laboratory parameters, when LDL-C was as low as 25 mg/dL (0.65 mmol/L) compared to those with higher LDL-C or in placebo or control groups. While this data is reassuring and consistent with the epidemiological studies of patients with PCSK9 loss-of-function mutations, including the few patients with two such mutations leading to lifelong very low LDL-C, this question will only be definitively answered by the long-term currently ongoing clinical outcome trials the first of which are anticipated to conclude in 2017.
Effect on Cardiovascular Events
Two recent trials assessed CVD endpoints as exploratory or post hoc analysis as part of longer term safety trials [26••, 27••]. After a mean duration of 11 months, the OSLER trials showed a reduction in major adverse CVD events, 2.18 % with standard of care alone compared to 0.95 % in the group treated with evolocumab (HR 0.47, 95 % CI 0.28–0.78, P = 0.003), and in a post hoc analysis of the ODYSSEY Outcome trial, major adverse CVD events adjudicated in a blinded analysis were reduced with alirocumab compared to placebo (1.7 vs. 3.3 %; HR 0.52, 95 % CI 0.31–0.90, P = 0.02). While the total number of patients experiencing CVD events in both trials was relatively small, the uniformity of results from the two studies bodes well for the likely outcome of the large longer term trials in progress.
A number of additional mAbs to PCSK9 have entered clinical development including bococizumab (RN316) which is currently in early phase 3 [28], while mAbs developed by Eli Lilly, LY3015014, and Genentech/Roche, RG7652, have been in phase 2 with decision to progress to further development currently uncertain.
In summary, PCSK9 mAb therapy appears to be the most promising therapeutic class since statins and able to fill the current medical need outlined in Table 1. Despite the need for SC administration injection and the likely high cost associated with a biological agent, the large reduction in LDL-C and the ability to self-administer these drugs only every 2 or 4 weeks will likely result in PCSK9 monoclonal antibodies being widely accepted and used by patients. Based on the early CVD event data, it appears very likely that the additional LDL-C reductions attained with this therapy added to statin and ezetimibe will augment our ability to further reduce cardiovascular disease.
Non-monoclonal Antibody Inhibitors of PCSK9
Adnectins
There have been limited alternatives to immunoglobulin-based inhibitors of proteins or enzymes involved in disease processes. However, one alternative has been adnectins, which are small (≤12 kDa), compact proteins which, while not having sequence homology to immunoglobulins, still possess a β-sheet fold structure with diversified loops similar to the variable regions on antibodies. Adnectins have potential advantages compared to antibodies as they are not glycosylated, have no disulfides, have high thermal stability, and can be produced using bacterial expression systems. However, due to their size, adnectins are rapidly filtered by the kidney and thus require pharmacokinetic (PK) enhancement modification for longer duration in vivo therapeutics. While the highly stable adnectin polypeptide scaffold allows incorporation of various PK enhancement modalities, the first-generation PCSK9 adnectin, BMS-962476, employed polyethylene glycol [29]. In a proof-of-concept ascending single-dose study, the 0.3 mg/kg dose in statin-treated patients resulted in a LDL-C reduction of 48 % from baseline [30].
PCSK9 Synthesis Inhibitors
As it has been extremely difficult to develop a small molecule that can interfere with the binding of PCSK9 to the LDLR, the focus has been an alternative therapeutic approach, reducing intracellular PCSK9 production or release from the hepatocyte into the circulation. Initial studies in an animal model, using a second-generation antisense oligonucleotide (ASO) directed at murine PCKS9, reduced PCSK9 expression and decreased LDL-C by 38 % [31]. However, this approach has not yet entered human trials. Small interfering RNA (siRNA) which direct sequence-specific degradation of messenger RNA have been shown to suppress the synthesis of the corresponding proteins. A proof-of-concept single ascending-dose study with ALN-PCS, a first-generation siRNA that inhibits PCSK9 synthesis, formulated in a novel lipid nanoparticle for delivery, resulted in both plasma PCSK9 and LDL-C reductions [32]. The trial involved a single 1-h intravenous infusion of placebo or ALN-PCS in ascending doses with the highest dose, 0.4 mg/kg (Fig. 2) resulting in a mean 40 % reduction in LDL-C (P < 0.0001) relative to placebo. However, the formulation of ALN-PCS required that the evening prior to, and the morning of, dosing, participants received oral corticosteroids, histamine receptor blockers plus paracetamol to reduce the potential for infusion-related reactions Subsequent improvement in the formulation using a GalNAc-siRNA conjugate appears to hold greater promise of overcoming the potential infusion-related reactions and allowing for SC administration [33].

siRNA: plasma PCSK9 levels following 0.400 mg/kg of ALN-PCS. (Reprinted from Fitzgerald et al. [32] with permission from Elsevier)
Cholesterol Ester Transfer Protein Inhibitors
Cholesterol ester transfer protein (CETP) is a plasma protein that facilitates exchange of cholesteryl esters and triglyceride between HDL and Apo B-containing lipoproteins [34]. While the primary purpose was to increase HDL-C, early trials also noted a reduction in LDL-C, mechanistically consistent with the prevention of cholesterol transfer from HDL to LDL. The magnitude of LDL-C reduction has varied with different CETP inhibitors as well as with the dose of specific inhibitors [35, 36]. For example, minimal or no LDL-C reduction was seen with dalcetrapib, to reductions of ∼45 % recently reported with the most potent and efficacious agent, TA-8995. As increasing HDL-C is not recognized by any regulatory agency as a basis/surrogate for approving a drug to reduce CVD events, large CVD outcome trials are required. Two agents, torcetrapib and dalcetrapib, were terminated during development after their large CVD outcome trials were stopped, for toxicity and futility, respectively. However, three CETP inhibitors, TA-8995, evacetrapib, and anacetrapib, continue in development, the latter two now also in large CVD outcome trials [37, 38].
Anacetrapib, a potent, selective, but very long-acting CETP inhibitor, at doses of 10, 40, 150, and 300 mg daily reported LDL-C reductions ranging from 15 to 40 % [39]. In a large 76-week safety trial, The Determining the Efficacy and Tolerability of CETP Inhibition with Anacetrapib (DEFINE) study, LDL-C reduction with the 100 mg dose was originally reported as being 40 % from baseline [40]. However, a subsequent reanalysis of a large subset of the DEFINE study samples by ultracentrifugation rather than calculation by Friedewald formula indicated the LDL-C was more modest at 25 %. No significant difference in the primary end point of prespecified adjudicated CVD events was seen in the trial between those on anacetrapib (2.0 %) and placebo (2.6 %) (P = 0.40). More recently, the effects of 52 weeks of therapy with 100 mg daily of anacetrapib was published in subjects with HeFH [41]. The study randomized 306 subjects 2:1 to receive either anacetrapib or placebo. Anacetrapib reduced LDL-C by approximately 40 %, raised HDL-C >100 %, and decreased Lp(a) by 32 %. Of concern is while CVD events were very few, they occurred only in the anacetrapib group (2 %) with 0 % in the placebo group. The 100 mg dose is currently been evaluated in the large CVD outcome trial in the 30,624-patient REVEAL (Randomized EValuation of the Effects of Anacetrapib Through Lipid-modification) trial, with results anticipated in 2017 [37].
Evacetrapib, another potent inhibitor of CETP, showed increases in HDL-C of 54, 95, and 129 % and reductions in LDL-C of 14, 22, and 36 % with doses of 30, 100, and 500 mg per day [36]. A 12,000-patient CVD outcome trial with 130 mg of evacetrapib, ACCELERATE, is currently in progress with results also expected in 2017 [38].
A third CETP inhibitor, TA-8995, is entering phase 3 development and recently published results from phase 2 in 364 patients with mild dyslipidemia [42]. The double-blind, placebo-controlled, parallel-group trial, randomized patients on diet only with fasting LDL-C between 2.5 and 4.5 mmol/L, HDL cholesterol levels 0.8 to 1.8 mmol/L, and triglyceride levels <4.5 mmol/L. Patients were allocated equally to receive one of the following nine treatments: TA-8995 1, 2.5, 5, or 10 mg daily or matching placebo; TA-8995 10 mg plus 20 mg atorvastatin; TA-8995 10 mg plus 10 mg rosuvastatin or 20 mg atorvastatin or 10 mg rosuvastatin alone. After 12 weeks, LDL-C was reduced by 27, 33, 45, and 45 % with the 1, 2.5, 5, and 10 mg doses of TA-8995, respectively. HDL-C increased by 76, 124, 157, and 179 % with the 1, 2.5, 5, and 10 mg, respectively. Combination with the statins was as anticipated, greater than the statin or TA-8995 alone for LDL-C but similar for HDL as TA-8995 alone. No serious clinical or laboratory adverse events were seen between treatment groups.
Depending on the results of the CVD outcome trials which if positive would likely result in their marketing approval, it may be that these agents would be used to reduce LDL-C further, although their effect on LDL-C is relatively small compared to that of PCSK9 inhibitors.
ACL and AMP-Kinase Modulator (ETC-1002)
8-Hydroxy-2,2,14,14-tetramethylpentadecanedioic acid (ETC-1002) is a dicarboxylic acid derivative with a dual action: activating adenosine monophosphate-activated protein kinase (AMPK) in its free form and, following rapid acetylation in the liver to form a thioester, inhibiting adenosine triphosphate-citrate lyase (ACL) [43]. These two enzymes play an important role in fatty acid and cholesterol metabolism with inhibition of ACL rapidly reducing acetyl-CoA, the final common substrate for both fatty acid and sterol synthesis, at a point in the cholesterol synthetic pathway prior to 3-hydroxy-3-methyl- glutaryl-CoA reductase, the enzyme inhibited by statins. Activation of AMP-activated protein kinase reduces lipid synthesis, regulates carbohydrate metabolism, and reduces inflammation and adiposity in non-clinical studies (Fig. 3). ETC-1002 is currently in phase 2 clinical development to reduce LDL-C, and a recent trial has also provided evidence for favorable effects on glycemic biomarkers in diabetics [44]. The initial dose-ranging phase 2 placebo-controlled trial in 177 patients, with baseline LDL-C 130 and 220 mg/dL, showed LDL-C levels of 18, 25, and 27 % with ETC-1002 40, 80, and 120 mg, respectively, compared to a reduction of 2 % with placebo (all, P < 0.0001). The drug was well tolerated and did not show clinically meaningful differences in adverse events or other safety assessments compared with placebo. A second phase 2 trial (ETC-1002-008) randomized ∼350 patients 4:4:4:1:1 to 5 treatment arms, ETC-1002 120 or 180 mg daily, ezetimibe 10 mg daily, or ETC1002 120 mg or 180 mg plus ezetimibe [45]. Baseline LDL-C across all groups was ∼160 mg/dL, and LDL-C decreased 27, 30, and 21 % with ETC-1002 120 mg, 180 mg and ezetimibe 10 mg monotherapies, respectively, and 43 and 48 % when ETC-1002 120 and 180 mg were combined with ezetimibe. More recently, ETC-1002 was investigated in a single-center phase 2 trial in 60 patients with type 2 diabetes mellitus and elevated LDL-C (>100 mg/dL) [46]. Patients stopped both their lipid-altering and hypoglycemic therapy ≥4 weeks prior to randomization to either ETC-1002 or placebo for 4 weeks. The dose of ETC-1002 was initiated at 80 mg daily and increased to 120 mg at week 2. At the end of 4 weeks, LDL-C decreased 43 % with ETC-1002 compared with a 4 % reduction with placebo (P < 0.0001). Significant improvements were seen in glycemic parameters with ETC-1002 compared to placebo, and adverse events were not significantly different. No data has yet been presented or published assessing the effect of ETC-1002 when added to statins, and this would be important given that it is likely to be added to patients already on statins, even if on low or intermittent dose. In addition, given that ETC-1002 works by reducing synthesis of precursors in the cholesterol pathway prior to HMG CoA, it will be important to determine if this results in added LDL-C reduction with statins. However, based on the efficacy and good initial safety finding with monotherapy or combined with ezetimibe, the drug is now entering phase 3 development.

ETC-1002: dual mechanism of action of on AMP-activated protein kinase (AMPK) activation and ATP-citrate lyase (ACL) inhibition (with permission from Gutierrez et al. [46])
LDL-C-Lowering Agents Intended for Specific or “Orphan” Use (HoFH Only)
Statins and PCSK9 inhibitors enhance LDL-C removal from the circulation and are thus dependent to a significant degree on existing LDLR activity. An alternative approach is to decrease production of the atherogenic, apolipoprotein (Apo) B-containing, lipoproteins. Two drugs approved in the last two years take this approach with selective inhibition of either hepatic Apo B100 synthesis with gene silencing antisense technology or non-selective inhibition of lipidation of Apo B100 and B48 lipoproteins by the enzyme microsomal triglyceride transport protein (MTP).
Apolipoprotein B Antisense (Mipomersen)
Antisense drugs have single-strand antisense nucleotide sequences complementary to mRNA, called antisense oligonucleotides or ASOs. They disappear from the bloodstream rapidly after SC injection but remain in tissue for prolonged periods, often months.
Mipomersen, the only Apo B antisense drug to have entered clinical development with progression to approval for any clinical use, is metabolized by nucleases, both endonucleases and exonucleases, and is broken down to fragments excreted in the urine.
Side effects in clinical trials appear to be related to the ASOs, injection site reactions (ISRs), and mechanism of action, namely hepatic fat accumulation and liver enzyme elevation [47]. Mipomersen takes up to 6 months to achieve steady state and showed a dose response-related reduction in LDL-C and Apo B reaching nearly 70 % at the 400-mg-per-week dose, in phase 2 [48]. However, the dose selected for final development was 200 mg which provides substantially less LDL-C reduction. As the drug is only approved for use in HoFH in the USA, it is relevant only to review the data pertinent to this population. The definitive trial for regulatory approval randomized 51 HoFH patients, 2:1 to mipomersen 200 mg SC weekly or matching placebo for 26 weeks [49]. Mipomersen resulted in a significant (P < 0.001) mean reduction in LDL-C compared to placebo of 21.4 % (24.7 % mipomersen and 3.3 % with placebo). The most common, and troubling, adverse event were ISRs which were three times as frequent in mipomersen-treated HoFH patients. Additional clinically important side effects included flu-like symptoms. The most common laboratory findings in HoFH related to liver function tests with increases of ≥3 × ULN in hepatic transaminases, in 12 % of mipomersen-treated patients versus none in the placebo group. Longer term and larger studies are needed in order to determine whether the steatosis shown on hepatic imaging will become self-limiting or clinically significant and result in long-term hepatic damage. Mipomersen has therefore only been approved in the USA and is confined to use in HoFH under a strict REMS program [16•].
Microsomal Triglyceride Transfer Protein Inhibitors
Microsomal triglyceride transport protein (MTP) is a transfer protein localized in the endoplasmic reticulum (ER) of hepatocytes and enterocytes that plays a critical role in lipidation of both hepatic Apo B100 and intestinal Apo B48 to form lipoproteins which are then released into the circulation. Lack of MTP results in the inability to form chylomicrons, VLDL, and their downstream lipoproteins including remnants, IDL and LDL.
Pharmacological inhibition of MTP followed the discovery in 1992 by Wetterau and others that MTP deficiency was the cause of a rare inherited disorder, abetalipoproteinemia, characterized by very low levels of LDL-C, fat malabsorption, steatorrhea, hepatic steatosis, and neurological disorders due to the inability to transport vitamin E and other lipid-soluble vitamins in apo B-containing lipoproteins [50].
By the early 2000s, two compounds were in development, BAY 13-9952 (implitapide) from Bayer and BMS-201038 (lomitapide) from Bristol Myers Squibb. Implitapide demonstrated significant LDL-C reductions ranging from 8.2 % with 20 mg to 55.1 % with 160 mg daily. However, side effects, especially gastrointestinal and elevations in hepatic transaminases >3 times ULN in over 25 % in those receiving the 160 mg/day dose resulted in Bayer abandoning further development of the compound [51, 52].
BMS-201038 (lomitapide) was reported in a 7-day multiple ascending-dose, phase 1 study to produce large reductions in LDL-C ranging from 54 to 86 % with doses of 25 to 100 mg/day. However, a similar high rate of hepatosteatosis and gastrointestinal adverse experiences was encountered and Bristol Myers Squibb terminated further drug development. Both implitapide and lomitapide were investigated further by individual academic investigators, and small studies to develop them in HoFH and severe HeFH were continued [53]. Based on the results of the proof-of-concept study with lomitapide in HoFH, a larger study, partially funded by the FDA orphan drug program, enrolled 29 adult HoFH patients [54]. Patients on maximally tolerated lipid-lowering drug therapy, including 16 on LDL apheresis, participated in an open-label phase 3 study where lomitapide was started at a dose of 5 mg per day and titrated at approximately 4-week intervals to a maximum of 60 mg daily. Six patients terminated and 23 patients completed the 26-week efficacy phase. Based on an intention-to-treat analysis of all 29 patients, the mean LDL-C reduction was 40 % from baseline (P < 0.001), with patients receiving a median dose of 40 mg per day. Based on this open-label trial, lomitapide was approved by the FDA as an orphan drug for the treatment of adults with HoFH in December 2012 and by the European Medicines Agency (EMA) in early 2013. The FDA approval carried a black-box warning regarding the potential for hepatic toxicity as well as the REMS program similar to that of mipomersen. FDA also required an additional trial in pediatric HoFH patients which is still ongoing. A phase 3, open-label trial in Japan to meet requirement by the Japanese regulatory authorities has recently been completed [55]. Since approval in the USA, use of mipomersen has been very limited while lomitapide has been more widely used with 400 to 500 patients reported by the manufacturer to be currently receiving the drug. However, recently, the company reported a cumulative 46 % of patients prescribed lomitapide had stopped taking it, presumably due mostly to side effects. Thus, while effective in reducing LDL-C, lomitapide is likely to find a limited role in the treatment of HoFH due to its tolerability and side effects.
Peroxisome Proliferator Activated Receptor Delta Agonist
Peroxisome proliferator activated receptor (PPAR)-α and PPAR-γ agonists have been approved and used for dyslipidemia and diabetes mellitus, respectively, although humans are relatively resistant to proliferation of peroxisomes with PPAR agonists. The PPAR-δ receptor has represented a potential therapeutic target for many years due to widespread expression in metabolically active tissues and its role in lipid and glucose metabolism. As yet, no PPAR-δ agonist has been approved despite studies exploring potential metabolic benefits such as a reduction in Apo B-100, VLDL, and LDL-C. MBX-8025, an orally active, potent, PPAR-δ specific agonist with >750-fold and >2500-fold activity compared with PPAR-α and PPAR-γ on receptors, respectively, originally entered development as a lipid-altering agent intended for widespread use [56]. In a phase 2 trial at doses of 50 and 100 mg daily, MBX-8025 reduced LDL-C 18 and 22 %, respectively, versus placebo and, due to its robust effect on VLDL and triglycerides, non-HDL-C by 20 to 24 %. In the subgroup with the highest baseline LDL-C levels of >185 mg/dL, the reductions in LDL-C were more pronounced and up to 35 %. The drug was not continued further for broad development but is now being focused as an orphan drug for HoFH. Supportive studies in an animal model for human HoFH, the Watanabe-heritable hyperlipidemic (WHHL) rabbit, characterized by low (<5 %) hepatic LDL receptor activity and extremely elevated LDL-C, demonstrated that MBX-8025 produced a rapid and sustained reduction in LDL-C of −33, −45, and −42 % at weeks 1, 2, and 3, respectively [57] (Fig. 4).

Effect of MBX-8025 in WHHL rabbit (% change in LDL-C from baseline N = 5) (reprinted by permission, CymaBay Therapeutics Inc., unpublished results) [57]
Based on the good prior human tolerability and safety, the apparent greater LDL-C decrease with high baseline LDL-C, and the good efficacy in the WHHL rabbit, MBX-8025 is now conducting phase 2 proof-of-concept trials in HoFH which if successful will progress to a larger phase trial.
Acetyl Coenzyme A Carboxylase Inhibitor
Gemcabene (CI-1027) is the monocalcium salt of a dialkyl ether dicarboxylic acid, and represents a new class of lipid-regulating compounds. Gemcabene has structural features of an acetyl coenzyme A (CoA) carboxylase inhibitor, a rate-limiting enzyme in fatty acid synthesis and inhibitor of cholesterol synthesis inhibiting the production of lipids needed for assembly of VLDL and its subsequently remodeled lipoproteins including LDL [58]. It also has weak, indirect PPARα activity and direct PPARγ activity. Because gemcabene acts early in this process, lipid precursors are able to be utilized in other metabolic processes thereby avoiding subsequent accumulation of intracellular hepatic fat and triglycerides, which is a significant limitation of other currently available orphan therapies for HoFH. Initially under development by Pfizer as a LDL-C-lowering agent for broad use, gemcabene doses ranging from 150 to 900 mg daily were studied in approximately 900 subjects in phase 1 and 2 studies. These studies evaluated safety, dose regimen, pharmacokinetics, pharmacodynamics, potential drug–drug interactions, and LDL-C-lowering efficacy. An 8-week, double-blind, randomized, placebo- and active-controlled, dose-ranging study of gemcabene 300, 600, and 900 mg daily reported LDL-C reductions compared to placebo of 17.0, 25.5, and 28.7 %, respectively, but drug development was terminated for commercial reasons [59]. Recently, the drug was acquired by Gemphire and in studies in LDL receptor-deficient mice, a preclinical model for HoFH, gemcabene significantly reduced LDL-C both alone and in combination with atorvastatin. The drug has been granted an orphan designation by FDA allowing it to enter clinical development for HoFH, starting with a phase 2 proof-of-concept trial which, if successful, will progress to a larger phase 3 definitive trial.
Conclusion
The last 30 years has seen the LDL hypothesis completely validated and become perhaps the major modifiable risk factor for both primary and secondary prevention of CVD. This has been achieved most convincingly with statins, due to the large and robust LDL-C reductions produced by the class, but is also consistent with CVD risk reductions achieved by a bile acid sequestrant and a cholesterol absorption inhibitor. However, cardiovascular disease still remains the major cause of mortality and morbidity in industrialized societies, and is rapidly becoming the major cause of morbidity and mortality in recently industrializing countries like India and China which together constitute nearly half the world’s population. Of major importance is the availability of effective statins as generic and very inexpensive formulations. However, there remains a large unmet medical need for new, effective, well-tolerated, and safe agents, especially for patients unable to either tolerate statins or achieve optimal LDL-C on current therapies. It is likely that many, if not all, the agents discussed in this review will fill this unmet need.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 1994; 344(8934):1383-1389.
Sacks FM, Pfeffer MA, Moye LA, et al. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and recurrent events trial investigators. N Engl J Med. 1996;335:1001–9.
Shepherd J, Cobbe SM, Ford I, Isles CG, Lorimer AR, MacFarlane PW, et al. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. N Engl J Med. 1995;333:1301–7.
Heart Protection Study Collaborative Group. MRC/BHF heart protection study of cholesterol lowering with simvastatin in 20 536 high-risk individuals: a randomised placebo-controlled trial. Lancet. 2002;360:7–22.
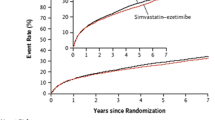
Cannon CP, Blazing MA, Giugliano RP, et al. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med. 2015. doi:10.1056/NEJMa1410489. Important trial which is first to demonstrate additional LDL-C lowering with non-statin added to statin resulted in further CVD risk reduction compared to statin alone.
Davidson MH, Dillon MA, Gordon B, et al. Colesevelam hydrochloride (cholestagel): a new, potent bile acid sequestrant associated with a low incidence of gastrointestinal side effects. Arch Intern Med. 1999;159(16):1893–900.
Ginsberg HN, Elam MB, Lovato LC, et al. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med. 2010;362:1563–74.
Boden WE, Probstfield JL, Anderson T, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365:2255–67. HPS2-THRIVE.
Landray MJ, Haynes R, Hopewell JC, et al. Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med. 2014;371:203–12.
Lambert G, Sjouke B, Choque B, et al. The PCSK9 decade. J Lipid Res. 2012;53:2515–24.
Stein EA, Raal FJ. Reduction of low density lipoprotein cholesterol by monoclonal antibody inhibition of PCSK9. Ann Rev Med. 2014;65:417–31.
Cohen JC, Boerwinkle E, Mosley Jr TH, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354:1264–72.
Lagace TA, Curtis DE, Garuti R, et al. Secreted PCSK9 decreases the number of LDL receptors in hepatocytes and in livers of parabiotic mice. J Clin Invest. 2006;116:2995–3005. Key finding in the PCSK9 relationship to high LDL-C in that it was plasma PCSK9 that resulted in LDL-C elevation thus allowing development of monoclonal antibodies to PCSK9.
Stein EA, Mellis S, Yancopoulos GD, et al. Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. N Engl J Med. 2012;366:1108–83. First proof of concept in man that inhibition of PCSK9 would lower LDL-C in non-FH and FH patients.
Dias CS, Shaywitz AJ, Wasserman SM, et al. Effects of AMG 145 on low-density lipoprotein cholesterol levels: results from 2 randomized, double-blind, placebo controlled, ascending-dose phase 1 studies in healthy volunteers and hypercholesterolemic subjects on statins. J Am Coll Cardiol. 2012;60:1888–98.
Stein EA, Raal FJ. New therapies for reducing low-density lipoprotein cholesterol. Endocrinol Metab Clin N Am. 2014;43:1007–33. Good review of drugs in development that lower LDL-C.
McKenney JM, Koren MJ, Kereiakes DJ, et al. Safety and efficacy of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease, SAR236553/REGN727, in patients with primary hypercholesterolemia receiving ongoing stable atorvastatin therapy. J Am Coll Cardiol. 2012;59:2344–53.
Roth EM, McKenney JM, Hanotin C, et al. Atorvastatin with or without an antibody to PCSK9 in primary hypercholesterolemia. N Engl J Med. 2012;367:1891–900.
Stroes E, Colquhoun D, Sullivan D, et al. Anti-PCSK9 antibody effectively lowers cholesterol in patients with statin intolerance: the gauss-2 randomized, placebo-controlled phase 3 clinical trial of evolocumab. J Am Coll Cardiol. 2014;63(23):2541–8. Large trial of statin adverse patients unable to tolerate at least 2 statins, or minimum doses of statins.
Moriarty PM, Jacobson TA, Bruckert E, et al. Efficacy and safety of alirocumab, a monoclonal antibody to PCSK9, in statin-intolerant patients: design and rationale of ODYSSEY ALTERNATIVE, a randomized phase 3 trial. J Clin Lipidol. 2014;8(6):554–61.
Stein EA, Gipe D, Bergeron J, et al. Effect of a monoclonal antibody to PCSK9, REGN727/SAR236553, to reduce low-density lipoprotein cholesterol in patients with heterozygous familial hypercholesterolaemia on stable statin dose with or without ezetimibe therapy: a phase 2 randomised controlled trial. Lancet. 2012;380:29–36.
Raal FJ, Scott R, Somaratne R, et al. Low-density lipoprotein cholesterol-lowering effects of AMG 145, a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease in patients with heterozygous familial hypercholesterolemia. The reduction of LDL-C with PSCK9 inhibition in heterozygous familial hypercholesterolemia disorder (RUTHERFORD) randomized trial. Circulation. 2012;126:2408–17.
Raal FJ, Stein EA, Dufour R, et al. PCSK9 inhibition with evolocumab (AMG 145) in heterozygous familial hypercholesterolaemia (RUTHERFORD-2): results of a randomised double-blind, placebo controlled trial. Lancet. 2015;385:331–40. First trial in HeFH to assess genetic abnormalities causing FH and relationship to LDL-C response to evolocumab and demonstrating underlying defect was not related to response.
Stein EA, Honarpour N, Wasserman SM, Zu F, Scott R, Raal FJ. Effect of the proprotein convertase subtilisin/kexin 9 monoclonal antibody, AMG 145, in homozygous familial hypercholesterolemia. Circulation. 2013;128:2113–20. Proof-of-concept trial demonstrating PCSK9 inhibition would be effective in reducing LDL-C in HoFH.
Raal FJ, Honarpour N, Blom D, et al. Inhibition of PCSK9 with evolocumab in homozygous familial hypercholesterolaemia (TESLA Part B): results of a randomised, double-blind, placebo-controlled trial. Lancet. 2015;385:341–50. Critical phase 3 trial of evolocumab in HoFH demonstrating response was related to underlying LDL receptor activity.
Sabatine MC, Giugliano RP, Wiviott SD, et al. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372:1500–9. First trial with PCSK9 monoclonal antibody to show in predefined exploratory analysis that CVD events are reduced.
Robinson JG, Farnier M, Krempf M, et al. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372:1489–99. Confirmatory post hoc analysis with alirocumab to show reduction in CVD events with PCSK9 monoclonal antibody.
Ballantyne CM, Neutel J, Cropp A, et al. Results of bococizumab, a monoclonal antibody against proprotein convertase subtilisin/kexin type 9, from a randomised, placebo-controlled, dose-ranging study in statin treated subjects with hypercholesterolemia. Am J Cardiol. 2015;115:1212–21.
Mitchell T, Chao G, Sitkoffv D. Pharmacologic profile of the adnectin BMS-962476, a small protein biologic alternative to PCSK9 antibodies for LDL lowering. J Pharmacol Exp Ther. 2014;350(2):412–24.
Stein EA, Kasichayanula S, Turner T, et al. LDL cholesterol reduction with BMS-962476, an adnectin inhibitor of PCSK9: results of a single ascending dose study. J Am Coll Cardiol. 2014;63(12 Suppl):A172. doi:10.1016/S0735-1097(14)61372-3.
Graham MJ, Lemonidis KM, Whipple CP, et al. Antisense inhibition of proprotein convertase subtilisin/kexin type 9 reduces serum LDL in hyperlipidemic mice. J Lipid Res. 2007;48:763–7.
Fitzgerald K, Frank-Kamenetsky M, Shulga-Morskaya S, et al. Effect of an RNA interference drug on the synthesis of proprotein convertase subtilisin/kexin type 9 (PCSK9) and the concentration of serum LDL cholesterol in healthy volunteers: a randomised, single-blind, placebo-controlled, phase 1 trial. Lancet. 2014;383:60–8.
http://www.alnylam.com/web/assets/ALNY-ESC-GalNAc-siRNA-TIDES-May2014-Capella.pdf [accessed 9 June 2015]
Thompson A, Di Angelantonio E, Sarwar N, et al. Association of cholesteryl ester transfer protein genotypes with CETP mass and activity, lipid levels, and coronary risk. JAMA. 2008;299:2777–88.
Krishna R, Anderson MS, Bergman AJ, et al. Effect of the cholesteryl ester transfer protein inhibitor, anacetrapib, on lipoproteins in patients with dyslipidaemia and on 24-h ambulatory blood pressure in healthy individuals: two double-blind, randomised placebo-controlled phase I studies. Lancet. 2007;370:1907–14.
Nicholls SJ, Brewer HB, Kastelein JJP, et al. Effects of the CETP inhibitor evacetrapib administered as monotherapy or in combination with statins on HDL and LDL cholesterol: a randomized controlled trial. JAMA. 2011;306:2099–109.
A study of evacetrapib in high-risk vascular disease (ACCELERATE) http://clinicaltrials.gov/show/NCT01687998
REVEAL: Randomized EValuation of the Effects of Anacetrapib Through Lipid-modification. http://clinicaltrials.gov/show/NCT01252953
Bloomfield D, Carlson GL, Sapre A, et al. Efficacy and safety of the cholesteryl ester transfer protein inhibitor anacetrapib as monotherapy and coadministered with atorvastatin in dyslipidemic patients. Am Heart J. 2009;157(2):352–60.
Cannon CP, Shah S, Dansky HM, et al. Safety of anacetrapib in patients with or at high risk for coronary heart disease. N Engl J Med. 2010;363:2406–15.
Kastelein JJP, Besseling J, Shah S, et al. Anacetrapib as lipid-modifying therapy in patients with heterozygous familial hypercholesterolaemia (REALIZE): a randomised, double-blind, placebo-controlled, phase 3 study. Lancet. 2015;385:2153–61.
Hovingh GK, Kastelein JJP, van Deventer SJH, et al. Cholesterol ester transfer protein inhibition by TA-8995 in patients with mild dyslipidaemia (TULIP): a randomised, double-blind, placebo-controlled phase 2 trial. Lancet. 2015;386:452–60.
Pinkosky SL, Filippov S, Srivastava RA, et al. AMP-activated protein kinase and ATP-citrate lyase are two distinct molecular targets for ETC-1002, a novel small molecule regulator of lipid and carbohydrate metabolism. J Lipid Res. 2013;54:134–51.
Goldberg R. Targeting low-density lipoprotein and dysmetabolism in type 2 diabetes mellitus. Arterioscler Thromb Vasc Biol. 2014;34:477–8.
Filippov S, Pinkosky SL, Newton RS. LDL-cholesterol reduction in patients with hypercholesterolemia by modulation of adenosine triphosphate-citrate lyase and adenosine monophosphate-activated protein kinase. Curr Opin Lipidol. 2014;25(4):309–15.
Gutierrez MJ, Rosenberg NL, MacDougall DE, et al. Efficacy and safety of ETC-1002, a novel investigational low-density lipoprotein-cholesterol-lowering therapy for the treatment of patients with hypercholesterolemia and type 2 diabetes mellitus. Arterioscler Thromb Vasc Biol. 2014;34:676–83.
Rader DJ, Kastelein JJP. Lomitapide and mipomersen: two first-in-class drugs for reducing low-density lipoprotein cholesterol in patients with homozygous familial hypercholesterolemia. Circulation. 2014;129:1022–32.
Kastelein JJP, Wedel MK, Baker BF, et al. Potent reduction of apolipoprotein B and low-density lipoprotein cholesterol by short-term administration of an antisense inhibitor of apolipoprotein B. Circulation. 2006;114:1729–35.
Raal FJ, Santos RD, Blom DJ, et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double-blind, placebo-controlled trial. Lancet. 2010;375:998–1006.
Wetterau JR, Aggerbeck LP, Bouma ME, et al. Absence of microsomal triglyceride transfer protein in individuals with abetalipoproteinemia. Science. 1992;258:999–1001.
Stein EA, Isaacsohn JL, Mazzu A, Ziegler R. Effect of BAY 13-9952, a microsomal triglyceride transfer protein inhibitor on lipids and lipoproteins in dyslipoproteinemic patients. Circulation 1999;100(18,Suppl 1) :Abst 1342
52.Farnier, M., E. Stein, S. Megnien, L. et al Efficacy and safety of implitapide, a microsomal triglyceride transfer protein inhibitor in patients with primary hypercholesterolemia. Abstract Book of the XIV International Symposium on Drugs Affecting Lipid Metabolism in New York, 2001
Implitapide in patients with homozygous familial hypercholesterolemia (HoFH) on maximal concurrent lipid-lowering therapy https://clinicaltrials.gov/ct2/show/NCT00079846 [accessed June 9, 2015]
Cuchel M, Meagher EA, du Toit TH, et al. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: a single-arm, open-label, phase 3 study. Lancet. 2013;381:40–6.
Efficacy and safety of lomitapide in Japanese patients with HoFH on concurrent lipid-lowering therapy http://clinicaltrials.gov/ct2/show/record/NCT02173158 [accessed June 9, 2015]
Bays HE, Schwartz S, Littlejohn TIII, et al. MBX-8025, a novel peroxisome proliferator receptor-6 agonist: lipid and other metabolic effects in dyslipidemic overweight patients treated with and without atorvastatin. J Clin Endocrinol Metab. 2011;96:2889–97.
http://ir.cymabay.com/press-releases/detail/244/cymabay-therapeutics-announces-preclinical-data-demonstrating-the-potential-of-mbx-8025-to-treat-homozygous-familial-hypercholesterolemia [accessed June 9, 2015]
Ellis JM, Frahm JL, Li LO, Coleman RA. Acyl-coenzyme a synthetases in metabolic control. Curr Opin Lipidol. 2010;21:212–7.
Bisgaier CL, Essenburg AD, Barnett BC, et al. A novel compound that elevates high density lipoprotein and activates the peroxisome proliferator activated receptor. J Lipid Res. 1998;39:17–30.
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
Evan A. Stein has received honoraria for consulting on the development of PCSK9 inhibitors from Amgen, Regeneron/Sanofi, Genentech/Roche, and BMS/Adnectin. He is a co-inventor of a use patent owned by Amgen for the use of PCSK9 monoclonal antibodies in homozygous familial hypercholesterolemia for which he receives no financial compensation. He also sits on the scientific advisory board for Catabasis, CymaBay, and Gemphire and has been a consultant for AstraZeneca.
Frederick J. Raal reports grants from the University of Witwatersrand, has received consulting fees from Amgen and Sanofi related to PCSK9 inhibitors and from Genzyme related to apolipoprotein B inhibitors, and has received personal fees and non-financial support from AstraZeneca, Pfizer, and Merck.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Lipid Abnormalities and Cardiovascular Prevention
Rights and permissions
About this article

Cite this article
Stein, E.A., Raal, F.J. Lipid-Lowering Drug Therapy for CVD Prevention: Looking into the Future. Curr Cardiol Rep 17, 104 (2015). https://doi.org/10.1007/s11886-015-0659-8
Published:
DOI: https://doi.org/10.1007/s11886-015-0659-8