
Abstract
Purpose of Review
To summarize the key factors contributing to the onset and progress of nonalcoholic fatty liver disease (NAFLD) and put them in a system genetics context. We particularly focus on how genetic regulation of hepatic lipids contributes to NAFLD.
Recent Findings
NAFLD is characterized by excessive accumulation of fat in the liver. This can progress to steatohepatitis (inflammation and hepatocyte injury) and eventually, cirrhosis. The severity of NAFLD is determined by a combination of factors including obesity, insulin resistance, and lipotoxic lipids, along with genetic susceptibility. Numerous studies have been conducted on large human cohorts and mouse panels, to identify key determinants in the genome, transcriptome, proteome, lipidome, microbiome and different environmental conditions contributing to NAFLD.
Summary
We review common factors contributing to NAFLD and put them in a systems genetics context. In particular, we describe how genetic regulation of liver lipids contributes to NAFLD. The combination of an unhealthy lifestyle and genetic predisposition increases the likelihood of accumulating lipotoxic specie lipids that may be one of the driving forces behind developing severe forms of NAFLD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nonalcoholic fatty liver disease (NAFLD) encompasses many abnormalities, ranging from liver steatosis to nonalcoholic steatohepatitis (NASH), and cirrhosis, in the absence of excessive consumption of alcohol and viral infection. Liver steatosis is defined by intrahepatic fat content exceeding 5% of liver weight, whereas NASH is defined as hepatic steatosis accompanied by inflammation and hepatocyte injury (ballooning) in the presence or absence of fibrosis [1, 2]. Cirrhosis is considered an advanced stage of NASH, with scarring/fibrosis of the liver caused by long-term tissue damage [2].
NAFLD is the most common liver disease in affluent societies [3]. Steatosis is usually not a major concern, but it increases the risk of developing the more severe inflammatory condition known as NASH, which is projected to increase by ~ 60% in the USA, from 17 million in 2015 to 27 million affected individuals in 2030 [4]. NASH is associated with increased mortality due to increased risk of liver-, cancer-, and cardiovascular-related mortality [5]. Progression of NASH can ultimately lead to end-stage liver diseases such as cirrhosis and hepatocellular carcinoma.
NAFLD is closely related to other metabolic abnormalities such as obesity, type 2 diabetes, and cardiovascular diseases (CVDs). It is linked to CVDs through dyslipidemia, insulin resistance, altered coagulation, and inflammation [6, 7]. Furthermore, many subjects with NAFLD have an increased risk of developing hypertension, coronary heart disease, cardiomyopathy, and cardiac arrhythmias [8]. However, studies conducted so far are insufficient in establishing NAFLD as an independent risk factor for CVDs, and reports suggesting increased risk for CVDs in individuals with NAFLD should be interpreted with caution. Several comprehensive studies involving large sample sizes and long-term follow-up suggest that NAFLD increases CVDs risk, but not necessarily CVD-related mortality in itself [9, 10]
Although the risk of developing NAFLD increases with obesity, insulin resistance, and unhealthy lifestyle, genetics play an important role in disease development [11, 12]. The importance of liver fat is underscored by a recent Mendelian randomization study suggesting a causal connection between long-term accumulation of hepatic fat in patients with NAFLD and development of fibrosis [13]. Although the presence of fat in the liver may play a direct role in the development of fibrosis in NAFLD patients, most patients with NAFLD do not develop NASH. Several factors seem to contribute to the progression of NAFLD into NASH, indicating a multi-factorial disease, where disease progression is influenced by a combined impact of multiple genetic and environmental factors. Identified factors at the cellular level include genetic variability, dysregulated lipid metabolism, oxidative stress, endoplasmic reticulum (ER) stress, mitochondrial dysfunction, abnormal secretion of adipokines and cytokines, lipotoxicity, and gut-derived endotoxins [14,15,16,17].
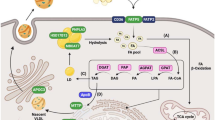
Recent technological and statistical advances have resulted in a paradigm shift in the studies of multi-factorial diseases. Integration of systems biology, multi-omics and computational modeling, offers a powerful approach to study metabolic diseases and is useful to identify factors contributing to disease development [18] (Fig. 1). We will describe some of the most common factors contributing to NAFLD and demonstrate how the combination of genetic variants and multi-omics approaches can be used to unravel new mechanisms in hepatic lipid metabolism and development of NAFLD.

Individuals exhibit different susceptibility to nonalcoholic fatty liver disease (NAFLD) due to genetic variation. For example, carriers of mutations in TM6SF2 and PNPLA3 genes have enhanced risk of developing NAFLD. Accumulation of lipotoxic lipids may contribute to the development of nonalcoholic steatohepatitis (NASH) [14, 19,20,21,22]. Obesity and type 2 diabetes exacerbate the risk of NAFLD, and lifestyle interventions can be particularly beneficial in certain patients, such as those harboring the PNPLA3-I148M variant [23]. Created with BioRender.com
Hepatic Fat Accumulation
Hepatic free fatty acids (FFAs) are derived from the hydrolysis of triacylglycerol (TAG) stored in the liver or adipose tissue, dietary lipids, or endogenously produced FFAs. FFAs produced in the liver by de novo lipogenesis are derived from either carbohydrates, amino acids, or alcohol. Regardless of their origin, all forms of hepatic FFAs can be incorporated into TAG and stored in hepatic lipid droplets (LDs), promoting hepatic steatosis. Alternatively, they may be exported as components of very low-density lipoprotein particles (VLDL) or used for ATP production [24]. Disruption of these pathways can lead to excessive hepatic fat accumulation, a hallmark of NAFLD [25].
Hepatic steatosis arises from an imbalance between lipid acquisition and disposal. This imbalance is mainly affected by four major routes: hepatic uptake of circulating lipids, de novo lipogenesis, fatty acid oxidation, and secretion of lipids in VLDL [26]. Thus, the precise molecular mechanisms driving pathological fat accumulation in the liver may differ and will often be obscure. We will explore pathological conditions that adversely impact lipid metabolism on a systemic level and thereby are likely to affect hepatic lipid metabolism and the progression of NAFLD into NASH.
Obesity, Systemic Inflammation, and Insulin Resistance
Obesity is characterized by expansion of adipose tissue. The size of adipose tissue is positively associated with systemic low-grade inflammation where enlarged adipocytes and infiltrating immune cells secrete inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), interleukin-1 beta (IL-1β), monocyte chemoattractant protein-1 (MCP-1), plasminogen activator inhibitor-1 (PAI-1), and visfatin [27]. These mediators interfere with insulin-signaling pathways, causing insulin resistance, where cells in several organs fail to respond effectively to insulin. Thus, insulin fails to suppress the release of FFAs from adipose tissue promoting increased hepatic FFA uptake. Insulin resistance is also associated with failure to suppress hepatic gluconeogenesis and reduce insulin-mediated skeletal muscle glucose uptake. Instead, glucose diverted to the liver becomes substrate to hepatic de novo lipogenesis, driving lipid accumulation. This chronic inflammatory state and hyperinsulinemia work in a vicious cycle, each exacerbating the other, escalating insulin resistance and inflammation [28,29,30].
Altered Gut Microbiota
The gut microbiota is composed of 10–100 trillion of microorganisms [31]. These microorganisms generate metabolites and signal molecules affecting host metabolism including the immune system. The species composition of the gut microbiota seems to differ between healthy individuals and patients with chronic diseases like type 2 diabetes, obesity, CVDs, and NAFLD [32,33,34,35], suggesting a possible influence of altered gut microbiota in these metabolic diseases [36].
Dysbiosis refers to disturbed or abnormal gut microbiota with elevated intestinal permeability, change in bile acid metabolism, increased translocation of lipopolysaccharide (LPS) (from the outer membrane of mainly Gram-negative bacteria), and initiation of inflammatory pathways. Inflammation can also contribute to dysbiosis. The first reports linking gut dysbiosis to NAFLD emerged from descriptive human studies, suggesting an overrepresentation of small intestinal bacterial overgrowth in patients with NASH [37]. Animal experiments involving altered microbiome have later supported an association between dysbiosis, obesity, and NAFLD [38, 39]. Although the role of the microbiome is unclear, it is hypothesized that LPS from the gut may activate nuclear factor κB (NF-κB) in the liver of NAFLD patients [40]. This activation promotes increased levels in TNF-α and IL-1β, which in turn triggers recruitment of inflammatory cells, leading to elevated inflammation and eventually development of fibrosis [40]. Activation of the farnesoid X receptor (FXR), which is a bile acid receptor with roles in lipid, glucose, and energy metabolism, may suppress NF-κB activity and is able to mitigate hepatic inflammation [41, 42].
Investigation of the role of gut microbiota in NAFLD development is challenging, given the complexity of the microbiome. To improve studies, several key approaches can be employed. For instance, longitudinal design can be used to capture the dynamic changes in microbiota over time and trace such changes to manifestation of hepatic inflammation and fibrosis. Alternatively, large-scale cohort studies with diverse populations are expected to include individuals with various NASH severity. From such cohorts, parallel analyses of the microbiome and circulating or hepatic lipids can be used to identify factors contributing to NASH. In animal models, fecal microbiota transplantation experiments can be useful in demonstrating if a particular strain of bacteria plays a causal role. Standardized sampling and analytical methods would ensure comparability, including integration of clinical and microbiota data to uncover confounders and complex interactions.
Inflammation and Oxidative Stress
Oxidative stress is characterized by imbalance between generation of reactive oxygen species (ROS) and efficiency of the antioxidant defense system [43, 44]. This imbalance can be due to increased production of pro-oxidant substances or dysfunction of the antioxidant system. Even small increases in the levels of reactive species can cause oxidative stress, promoting DNA damage, ER stress, mitochondrial damage, endothelial dysfunction cytotoxicity, apoptosis, and fibrosis [45].
Oxidative stress [46] and mitochondrial dysfunction [47] are frequently observed in NAFLD. For instance, increased activity of endothelial nitric oxide synthase (eNOS) and inducible nitric oxide synthase (iNOS) may contribute to elevated oxidative damage in the liver vasculature, promoting inflammation and fibrosis [48]. Altered lipid metabolism, causing hepatic steatosis, often leads to the production of ROS in the mitochondria and the ER, and by extracellular NADPH oxidase. A disruption in the normal flow of electrons in the electron transport chain can cause a non-enzymatic reaction with oxygen. The outcome of such reaction is primarily two types of ROS called superoxide anion (O2•-) and hydrogen peroxide (H2O2). Additionally, the levels of antioxidant enzymes like glutathione (GSH) peroxidase, superoxide dismutase (SOD2) (converts two O2•- to H2O2), and different catalases that may neutralize ROS appear to be reduced in the liver of individuals with NASH, limiting mitochondrial ability to reduce levels of reactive ROS [47].
Subjects with NASH may exhibit elevated activity of cytochrome P450 2E1 (CYP2E1) [49], which generates ROS as a byproduct of fatty acid metabolism. When increased activity of CYP2E1 coincides with the presence of C47T polymorphisms in SOD2 (also known as MnSOD), the hepatic capacity to counteract oxidative stress can be compromised [50,51,52,53].
Genetic Determinants in the Development of NAFLD
Oligonucleotide scanning arrays used to conduct genome-wide association studies (GWAS) on large patient cohorts for nearly any disease have altered research in recent decades. GWAS has enabled identification of allelic gene variants significantly contributing to individual predisposition to NAFLD [54].
Despite the fact that surrogate markers for hepatic fat content (such as alanine transaminase (ALT) often has been used in human GWAS, allelic gene variants identified as important in the development of NAFLD are often linked to biological processes affecting hepatic fat accumulation. Allelic variants have been identified in genes encoding patatin-like phospholipase domain–containing protein 3 (PNPLA3), transmembrane 6 superfamily member 2 (TM6SF2), membrane-bound O-acyltransferase domain–containing 7 (MBOAT7), hydroxysteroid 17β-dehydrogenase (HSD17B13), and glucokinase regulator (GCKR) [55]. Genes involved in regulating oxidative stress and mitochondrial function also seem to play a significant role in NAFLD pathogenesis, such as SOD2, uncoupling protein 2 (UCP2), UCP3, and mitochondrial amidoxime reducing component 1 (MARC1) [22, 56]. We will briefly discuss the genetic contribution of PNPLA3 and TM6SF6 in NAFLD development [21].
PNPLA3 and NAFLD
PNPLA3 is localized to the LD surface and belongs to the patatin-like phospholipase domain–containing family [57, 58]. Whereas PNPLA3 exhibits lipase activity targeting TAGs in vitro [59], it mainly possesses acyltransferase activity on phospholipids in mouse livers [59, 60]. A specific variant of the PNPLA3 gene (isoleucine-to-methionine substitution), called rs738409[G] (I148M), leads to loss of TAG hydrolysis (PNPLA3 inactivation) [61] promoting elevated hepatic fat accumulation and inflammation [62]. However, whole body knock-out of Pnpla3 in mice did not result in hepatic steatosis [63].
The rs738409[G] variant also seems to cause a preferential accumulation of polyunsaturated (PUFA) and saturated fatty acids into liver LDs and VLDLs, respectively [64]. The preferential storage of PUFAs in LDs might cause a reduction of PUFA availability for phosphatidylcholine (PC) synthesis [64]. This alteration in PC composition might affect hepatic metabolism and inflammation, as ongoing research explores the potential of PC as an adjunct therapy in patients with NAFLD [65, 66].
Subjects with the rs738409[G] variant have increased risk of developing the full spectrum of NAFLD. They show on average twice the amount of hepatic fat as compared to individuals with other gene variants [62], an increased independent risk of developing fibrosis, and a ~ 12-fold increased risk of developing liver cancer [67]. However, it seems that individuals with NAFLD harboring the rs738409[G] variant exhibit less insulin resistance as compared to control subjects with NAFLD [68]. Intriguingly, weight loss decreased liver fat more effectively in subjects carrying the rs738409[G] variant as compared to controls with matched liver fat [23]. Another allelic variant of the PNPLA3 gene, rs6006460[T] or S453I (serine-to-isoleucine substitution), is associated with low hepatic fat content and reduced risk of developing NAFLD [62].
TM6SF2 and NAFLD
TM6SF2 is localized to ER and the Golgi apparatus of hepatocytes and enterocytes, where it is involved in synthesis and packaging of apolipoprotein B-containing lipoprotein particles [69]. In a comprehensive exome-wide association study including 80,000 individuals, a genetic variant in the TM6SF2 gene, rs58542926 (Glu167Lys), was associated with liver steatosis [21]. A similar association was observed in a meta-analysis involving 123,800 individuals across 44 studies [70], where the rs58542926 variant exhibited an increased risk of NAFLD progression and development of fibrosis, as well as elevated levels of alanine transaminase (ALT) and aspartate transaminase (AST). Similar to what has been found for the PNPLA3 genetic variants, this TM6SF2 allelic variant, rs58542926, was negatively associated with serum total cholesterol, low-density lipoprotein (LDL), and TAGs. Tm6sf2 knock-out mice showed a higher degree of hepatic steatosis, inflammation, combined with lowered plasma levels of both total and LDL cholesterol, mirroring the observed phenotype in humans [69, 71]. Furthermore, TM6SF2 siRNA inhibition in cultured human hepatocytes was associated with reduced secretion of TAG-rich lipoproteins and increased cellular TAG concentration [72]. TM6SF2 overexpression, on the other hand, reduced hepatic TAG accumulation [72]. Interestingly, using adeno-associated virus to overexpress Tm6sf2 in mice resulted in phenotypes previously observed in Tm6sf2-deficient mice including reduced plasma lipid levels, diminished hepatic TAGs secretion, and increased hepatic steatosis [73]. Taken together, the results demonstrate TM6SF2 as a causative gene in NAFLD.
Systems Biology to Study Liver Fat Metabolism
Due to recent methodological improvements in the detection of individual lipid species, there has been a shift from studying whole lipid classes towards targeting specific lipid species in the development of lipid-related diseases [74,75,76]. Lipids encompass a variety of hydrophobic molecules, such as fatty acids, mono-, di-, and triglycerides, phospholipids, sterols, sphingolipids, and other members of distinct lipid classes. Lipid species within the same class often have similar structures and functions as compared to species from different classes.
Whereas certain lipid species are associated with steatosis, the understanding of how these lipids contribute to NASH is limited. Synthesis of TAG and subsequent storage in stable intracellular LDs in hepatocytes seem to protect cells from accumulation of “lipotoxic” lipids [77]. When the intracellular LD level expands beyond its capacity, lipotoxic lipids may accumulate in the intracellular environment and contribute to inflammation, cell damage, and fibrosis by activating hepatic stellate cells to collagen-producing myofibroblasts [78]. Elevated levels of free cholesterol and saturated fatty acids, in addition to certain ceramides species, have all been proposed to increase the risk of NASH [79].
The use of inbred mice strain panels, like the Hybrid Mouse Diversity Panel (HMDP), has been particularly successful to study genetic regulation of hepatic lipids [80]. There are several advantages with the use of inbred mice cohorts to study genetic and environmental factors underlying complex traits, compared to, e.g., human GWAS [80]. First, all relevant tissues can be obtained. Second, the environment can be carefully controlled. Third, several individuals with identical genomes can be studied, which increases power substantially. Finally, results from independent studies can be integrated and reanalyzed. We will describe several studies on inbred mice cohorts to study liver lipid metabolism.
In a pioneering liver lipidomics study, Jha et al. conducted a comprehensive profiling of a subset of hepatic lipids related to storage, signaling, membrane, and mitochondrial functions, across 47 different BXD recombinant inbred mice strains fed either a chow or a high-fat/high-sucrose (HF/HS) diet [19]. The BXD lines represent a genetic reference population derived from a cross between the C57BL/6 J and DBA/2 J strains. The researchers integrated lipid profiling data with complementary multi-omics datasets, creating a comprehensive and integrated view of the genetic regulation of liver lipids in NAFLD. They identified several gene-lipid associations and lipid quantitative trait loci (lQTL) including genes relevant for energy metabolism like Tm6sf2, protein phosphatase 1 regulatory subunit 3B (Ppp1r3b), phospholipid transfer protein (Ptlp), cytochrome P450, family 26 (Cyp26a1), and pancreatic lipase related protein 2 (Pnliprp2). Of particular interest was the identification of certain cardiolipin (CL) species that seemed relevant for hepatic health. Out of 23 CL species, nine seemed particularly important. Two CL species, tetralinoleoyl-CL (CL(LLLL)) and its precursor/remodeling intermediate, trilinoleoyl-MLCL (MLCL(LLL)), exhibited a negative correlation with liver size, whereas seven CL species enriched in monounsaturated fatty acids (MUFAs), such as oleic acid and palmitoleic acid, all showed a positive correlation. These associations between specific CL species and NAFLD were confirmed in a follow-up study where C57BL/6 J mice were fed a HF/HS diet with or without nicotinamide riboside (NR), used to ameliorate the development of NAFLD induced by HF/HS feeding. Taken together, the “healthy” CLs exhibited a negative correlation with obesity and NAFLD traits, whereas the “unhealthy” CLs showed a positive correlation. This suggests that hepatic CL species might serve as markers of hepatic health, and even be important for the pathogenesis of NAFLD.
Parker and colleagues studied genetic regulation of ~ 300 hepatic lipid species from 107 inbred mice strains developing fatty liver after 16 h of fasting [20]. The inbred mice strains were obtained from the HMDP [80]. Liver and plasma lipidomics were integrated with the liver proteome and genetic polymorphism data to identify high-confidence candidate genes that influence lipid accumulation. One of the candidate genes identified using lQTL as well as protein QTL, was proteasome 26S subunit, non-ATPase 9 (Psmd9). The effect of Psmd9 on the development of hepatic steatosis in mice fed a HF/HS diet was confirmed in DBA/2 J mice using antisense oligonucleotides (ASO) blocking Psmd9 expression. Finally, they combined hepatic and plasma lipid analysis and identified blood-based biomarkers for NAFLD using machine learning. The identified lipid signature was confirmed to predict hepatic lipid accumulation with high accuracy in a human cohort with varying degrees of hepatic steatosis.
In another HMDP study [81•], Norheim et al. investigated the genetic regulation of ~ 250 liver lipids obtained from 102 inbred strains fed a HF/HS diet for 8 weeks. First, they determined how chow and HF/HS diet affected the accumulated hepatic lipids in three specific mice strains (C57BL/6 J, DBA/2 J, and C3H/HeJ). Many lipids including ceramides, known to be involved in fatty liver development, increased significantly in response to the HF/HS diet as compared to the chow diet, regardless of the genetic background. However, certain lipids were differently altered in response to the two diets, depending on the mouse strain. For example, C3H/HeJ mice had lower phosphatidylcholine (PC) and higher phosphatidylethanolamine (PE) hepatic content compared to the other two strains (C57BL/6 J and DBA/2 J), suggesting that allelic gene variants contribute to hepatic accumulation of lipid species. Next, using lQTL analysis, gene-lipid correlations, and network modeling, biological pathways and genes underlying these interactions were examined and led to the identification of interferon-activated gene (Ifi203) and mitogen-activated protein kinase 6 (Map2k6) as regulators of hepatic phosphatidylcholine homeostasis and TAG accumulation, respectively. The effect of Map2k6 on hepatic steatosis was confirmed with hepatic overexpression of the gene in the liver using adeno-associated virus (AAV) [81•].
Although several researchers have reported genetic regulation of lipids accumulating in hepatic steatosis, none has investigated the genetic regulation of lipids that might contribute to the transition of hepatic steatosis into NASH. Future studies should focus on understanding this transition to identify new targets and mechanism that hopefully can result in important therapeutic breakthroughs in NASH treatment. Such studies can be performed in inbred mice strains that develop different degrees of NASH. Hepatic lipidomics data can then be integrated with clinical traits, different omics technologies, and gut microbiota.
NAFLD and Atherosclerosis
Considering the high prevalence of atherosclerosis in NAFLD patients [82,83,84], ongoing research aims to explore shared genetic and behavioral risk factors between NAFLD and atherosclerosis. Both NAFLD and atherosclerosis are classified as metabolic-associated disorders [85], and their shared risk factors include obesity, metabolic syndrome, hypertension, dyslipidemia, and type 2 diabetes. NAFLD involves excessive accumulation of lipids, mainly TAGs in hepatocytes [86]. Atherosclerosis is initiated by the accumulation of apoB lipoprotein–derived cholesterol and cholesteryl esters in the subendothelial space of middle-sized arteries [87, 88••].
There are complex genetic relationships between atherosclerosis and NAFLD. Whereas many genetic associations are positively correlated with both diseases, examples of opposing correlations between genetic associations for the two diseases are also known [89]. For instance, there is a discrepancy in findings that link NAFLD and atherosclerosis, even in well-documented allelic variants of the PNPLA3 gene; the rs738409[G] allelic variant is strongly associated with NAFLD development [62] but carriers of this allelic variant exhibit reduced lipid secretion into the circulation, promoting lower plasma cholesterol levels [90] and consequently reduced risk of developing atherosclerosis [91, 92]. On the other hand, some data suggest that the PNPLA3 allelic variant does not significantly affect the levels of TAG or cholesterol in plasma [64, 93, 94]. Moreover, data from 429 Italian NAFLD patients show that the PNPLA3-rs738409[G] allelic variant is associated with high severity of carotid atherosclerosis in individuals that are < 50 years old [95]. Other studies have linked the PNPLA-rs738409[G] allelic variant to chronic kidney disease, which is a well-known marker of increased cardiovascular risk in NAFLD patients [96]. Hence, the statement that the PNPLA3-rs738409[G] allelic variant promotes NAFLD while offering protective effects against atherosclerosis requires reexamination. New precise and comprehensive studies are required to tease out genetic interactions between NAFLD and atherosclerosis.
Concluding Remarks
The severity of NAFLD is determined by a combination of several factors such as obesity, insulin resistance, inflammation, and alterations in the microbiota, along with genetic susceptibility. Allelic gene variants identified in human GWAS are often linked to hepatic fat accumulation. Recently, several systems genetic studies using inbred mice panels have shown the importance of investigating the accumulation of specific lipids species. Future studies should investigate the genetic regulation of lipotoxic lipids in the development of NASH.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Roeb E, Steffen HM, Bantel H, Baumann U, Canbay A, Demir M, et al. S2k Guideline non-alcoholic fatty liver disease. Z Gastroenterol. 2015;53(7):668–723. https://doi.org/10.1055/s-0035-1553193.
Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, et al. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology. 2018;67(1):328–57. https://doi.org/10.1002/hep.29367.
Ratziu V, Bellentani S, Cortez-Pinto H, Day C, Marchesini G. A position statement on NAFLD/NASH based on the EASL 2009 special conference. J Hepatol. 2010;53(2):372–84. https://doi.org/10.1016/j.jhep.2010.04.008.
Estes C, Razavi H, Loomba R, Younossi Z, Sanyal AJ. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology. 2018;67(1):123–33. https://doi.org/10.1002/hep.29466.
Ahmed EA, El-Derany MO, Anwar AM, Saied EM, Magdeldin S. Metabolomics and lipidomics screening reveal reprogrammed signaling pathways toward cancer development in non-alcoholic steatohepatitis. Int J Mol Sci. 2022;24(1). https://doi.org/10.3390/ijms24010210.
Targher G, Byrne CD, Tilg H. NAFLD and increased risk of cardiovascular disease: clinical associations, pathophysiological mechanisms and pharmacological implications. Gut. 2020;69(9):1691–705. https://doi.org/10.1136/gutjnl-2020-320622.
Przybyszewski EM, Targher G, Roden M, Corey KE. Nonalcoholic fatty liver disease and cardiovascular disease. Clin Liver Dis (Hoboken). 2021;17(1):19–22. https://doi.org/10.1002/cld.1017.
Kasper P, Martin A, Lang S, Kutting F, Goeser T, Demir M, et al. NAFLD and cardiovascular diseases: a clinical review. Clin Res Cardiol. 2021;110(7):921–37. https://doi.org/10.1007/s00392-020-01709-7.
Stepanova M, Younossi ZM. Independent association between nonalcoholic fatty liver disease and cardiovascular disease in the US population. Clin Gastroenterol Hepatol. 2012;10(6):646–50. https://doi.org/10.1016/j.cgh.2011.12.039.
Lazo M, Hernaez R, Bonekamp S, Kamel IR, Brancati FL, Guallar E, et al. Non-alcoholic fatty liver disease and mortality among US adults: prospective cohort study. BMJ. 2011;343:d6891. https://doi.org/10.1136/bmj.d6891.
Weston SR, Leyden W, Murphy R, Bass NM, Bell BP, Manos MM, et al. Racial and ethnic distribution of nonalcoholic fatty liver in persons with newly diagnosed chronic liver disease. Hepatology. 2005;41(2):372–9. https://doi.org/10.1002/hep.20554.
Anstee QM, Daly AK, Day CP. Genetic modifiers of non-alcoholic fatty liver disease progression. Biochim Biophys Acta. 2011;1812(11):1557–66. https://doi.org/10.1016/j.bbadis.2011.07.017.
Cai B, Dongiovanni P, Corey KE, Wang X, Shmarakov IO, Zheng Z, et al. Macrophage MerTK promotes liver fibrosis in nonalcoholic steatohepatitis. Cell Metab. 2020;31(2):406-21 e7. https://doi.org/10.1016/j.cmet.2019.11.013.
Dongiovanni P, Stender S, Pietrelli A, Mancina RM, Cespiati A, Petta S, et al. Causal relationship of hepatic fat with liver damage and insulin resistance in nonalcoholic fatty liver. J Intern Med. 2018;283(4):356–70. https://doi.org/10.1111/joim.12719.
Mota M, Banini BA, Cazanave SC, Sanyal AJ. Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metabolism. 2016;65(8):1049–61. https://doi.org/10.1016/j.metabol.2016.02.014.
Lonardo A, Nascimbeni F, Maurantonio M, Marrazzo A, Rinaldi L, Adinolfi LE. Nonalcoholic fatty liver disease: evolving paradigms. World J Gastroenterol. 2017;23(36):6571–92. https://doi.org/10.3748/wjg.v23.i36.6571.
Musso G, Cassader M, Paschetta E, Gambino R. Bioactive lipid species and metabolic pathways in progression and resolution of nonalcoholic steatohepatitis. Gastroenterology. 2018;155(2):282-302 e8. https://doi.org/10.1053/j.gastro.2018.06.031.
Seldin M, Yang X, Lusis AJ. Systems genetics applications in metabolism research. Nat Metab. 2019;1(11):1038–50. https://doi.org/10.1038/s42255-019-0132-x.
Jha P, McDevitt MT, Gupta R, Quiros PM, Williams EG, Gariani K, et al. Systems analyses reveal physiological roles and genetic regulators of liver lipid species. Cell Syst. 2018;6(6):722-33 e6. https://doi.org/10.1016/j.cels.2018.05.016.
Parker BL, Calkin AC, Seldin MM, Keating MF, Tarling EJ, Yang P, et al. An integrative systems genetic analysis of mammalian lipid metabolism. Nature. 2019;567(7747):187–93. https://doi.org/10.1038/s41586-019-0984-y.
Kozlitina J, Smagris E, Stender S, Nordestgaard BG, Zhou HH, Tybjærg-Hansen A, et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2014;46(4):352–6. https://doi.org/10.1038/ng.2901.
Taliento AE, Dallio M, Federico A, Prati D, Valenti L. Novel insights into the genetic landscape of nonalcoholic fatty liver disease. Int J Environ Res Public Health. 2019;16(15). https://doi.org/10.3390/ijerph16152755.
Sevastianova K, Kotronen A, Gastaldelli A, Perttila J, Hakkarainen A, Lundbom J, et al. Genetic variation in PNPLA3 (adiponutrin) confers sensitivity to weight loss-induced decrease in liver fat in humans. Am J Clin Nutr. 2011;94(1):104–11. https://doi.org/10.3945/ajcn.111.012369.
Norum KR, Berg T, Helgerud P, Drevon CA. Transport of cholesterol. Physiol Rev. 1983;63(4):1343–419. https://doi.org/10.1152/physrev.1983.63.4.1343.
Saponaro C, Gaggini M, Carli F, Gastaldelli A. The subtle balance between lipolysis and lipogenesis: a critical point in metabolic homeostasis. Nutrients. 2015;7(11):9453–74. https://doi.org/10.3390/nu7115475.
Liu Q, Bengmark S, Qu S. The role of hepatic fat accumulation in pathogenesis of non-alcoholic fatty liver disease (NAFLD). Lipids Health Dis. 2010;9:42. https://doi.org/10.1186/1476-511X-9-42.
Fain JN. Release of inflammatory mediators by human adipose tissue is enhanced in obesity and primarily by the nonfat cells: a review. Mediators Inflamm. 2010;2010:513948. https://doi.org/10.1155/2010/513948.
Fabbrini E, Sullivan S, Klein S. Obesity and nonalcoholic fatty liver disease: biochemical, metabolic, and clinical implications. Hepatology. 2010;51(2):679–89. https://doi.org/10.1002/hep.23280.
Chitturi S, Abeygunasekera S, Farrell GC, Holmes-Walker J, Hui JM, Fung C, et al. NASH and insulin resistance: insulin hypersecretion and specific association with the insulin resistance syndrome. Hepatology. 2002;35(2):373–9. https://doi.org/10.1053/jhep.2002.30692.
Utzschneider KM, Kahn SE. Review: The role of insulin resistance in nonalcoholic fatty liver disease. J Clin Endocrinol Metab. 2006;91(12):4753–61. https://doi.org/10.1210/jc.2006-0587.
Parks BW, Nam E, Org E, Kostem E, Norheim F, Hui ST, et al. Genetic control of obesity and gut microbiota composition in response to high-fat, high-sucrose diet in mice. Cell Metab. 2013;17(1):141–52. https://doi.org/10.1016/j.cmet.2012.12.007.
Brahe LK, Astrup A, Larsen LH. Can we prevent obesity-related metabolic diseases by dietary modulation of the gut microbiota? Adv Nutr. 2016;7(1):90–101. https://doi.org/10.3945/an.115.010587.
He M, Shi B. Gut microbiota as a potential target of metabolic syndrome: the role of probiotics and prebiotics. Cell Biosci. 2017;7:54. https://doi.org/10.1186/s13578-017-0183-1.
Wortelboer K, Nieuwdorp M, Herrema H. Fecal microbiota transplantation beyond Clostridioides difficile infections. EBioMedicine. 2019;44:716–29. https://doi.org/10.1016/j.ebiom.2019.05.066.
Hui ST, Kurt Z, Tuominen I, Norheim F, Davis RC, Pan C, et al. The genetic architecture of diet-induced hepatic fibrosis in mice. Hepatology. 2018;68(6):2182–96. https://doi.org/10.1002/hep.30113.
Leung C, Rivera L, Furness JB, Angus PW. The role of the gut microbiota in NAFLD. Nat Rev Gastroenterol Hepatol. 2016;13(7):412–25. https://doi.org/10.1038/nrgastro.2016.85.
Wigg AJ, Roberts-Thomson IC, Dymock RB, McCarthy PJ, Grose RH, Cummins AG. The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut. 2001;48(2):206–11. https://doi.org/10.1136/gut.48.2.206.
Cho I, Yamanishi S, Cox L, Methe BA, Zavadil J, Li K, et al. Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature. 2012;488(7413):621–6. https://doi.org/10.1038/nature11400.
Mazagova M, Wang L, Anfora AT, Wissmueller M, Lesley SA, Miyamoto Y, et al. Commensal microbiota is hepatoprotective and prevents liver fibrosis in mice. FASEB J. 2015;29(3):1043–55. https://doi.org/10.1096/fj.14-259515.
Fang J, Yu CH, Li XJ, Yao JM, Fang ZY, Yoon SH, et al. Gut dysbiosis in nonalcoholic fatty liver disease: pathogenesis, diagnosis, and therapeutic implications. Front Cell Infect Microbiol. 2022;12:997018. https://doi.org/10.3389/fcimb.2022.997018.
Carr RM, Reid AE. FXR agonists as therapeutic agents for non-alcoholic fatty liver disease. Curr Atheroscler Rep. 2015;17(4):500. https://doi.org/10.1007/s11883-015-0500-2.
Khalid Q, Bailey I, Patel VB. Non-alcoholic fatty liver disease: the effect of bile acids and farnesoid X receptor agonists on pathophysiology and treatment. Liver Res - Open J. 2015;1(2):32–40. https://doi.org/10.17140/lroj-1-106.
Hikida RS, Staron RS, Hagerman FC, Sherman WM, Costill DL. Muscle fiber necrosis associated with human marathon runners. J Neurol Sci. 1983;59(2):185–203.
Nagashimada M, Ota T. Role of vitamin E in nonalcoholic fatty liver disease. IUBMB Life. 2019;71(4):516–22. https://doi.org/10.1002/iub.1991.
Masarone M, Rosato V, Dallio M, Gravina AG, Aglitti A, Loguercio C, et al. Role of oxidative stress in pathophysiology of nonalcoholic fatty liver disease. Oxid Med Cell Longev. 2018;2018:9547613. https://doi.org/10.1155/2018/9547613.
DelliBovi AP, Marciano F, Mandato C, Siano MA, Savoia M, Vajro P. Oxidative stress in non-alcoholic fatty liver disease. An updated mini review. Front Med (Lausanne). 2021;8:595371. https://doi.org/10.3389/fmed.2021.595371.
Kawahara H, Fukura M, Tsuchishima M, Takase S. Mutation of mitochondrial DNA in livers from patients with alcoholic hepatitis and nonalcoholic steatohepatitis. Alcohol Clin Exp Res. 2007;31(1 Suppl):S54-60. https://doi.org/10.1111/j.1530-0277.2006.00287.x.
Persico M, Masarone M, Damato A, Ambrosio M, Federico A, Rosato V, et al. Non alcoholic fatty liver disease and eNOS dysfunction in humans. BMC Gastroenterol. 2017;17(1):35. https://doi.org/10.1186/s12876-017-0592-y.
Chalasani N, Gorski JC, Asghar MS, Asghar A, Foresman B, Hall SD, et al. Hepatic cytochrome P450 2E1 activity in nondiabetic patients with nonalcoholic steatohepatitis. Hepatology. 2003;37(3):544–50. https://doi.org/10.1053/jhep.2003.50095.
El-Koofy NM, El-Karaksy HM, Mandour IM, Anwar GM, El-Raziky MS, El-Hennawy AM. Genetic polymorphisms in non-alcoholic fatty liver disease in obese Egyptian children. Saudi J Gastroenterol. 2011;17(4):265–70. https://doi.org/10.4103/1319-3767.82582.
Namikawa C, Shu-Ping Z, Vyselaar JR, Nozaki Y, Nemoto Y, Ono M, et al. Polymorphisms of microsomal triglyceride transfer protein gene and manganese superoxide dismutase gene in non-alcoholic steatohepatitis. J Hepatol. 2004;40(5):781–6. https://doi.org/10.1016/j.jhep.2004.01.028.
Nobili V, Donati B, Panera N, Vongsakulyanon A, Alisi A, Dallapiccola B, et al. A 4-polymorphism risk score predicts steatohepatitis in children with nonalcoholic fatty liver disease. J Pediatr Gastroenterol Nutr. 2014;58(5):632–6. https://doi.org/10.1097/MPG.0000000000000279.
Varela NM, Quinones LA, Orellana M, Poniachik J, Csendes A, Smok G, et al. Study of cytochrome P450 2E1 and its allele variants in liver injury of nondiabetic, nonalcoholic steatohepatitis obese women. Biol Res. 2008;41(1):81–92.
Krawczyk M, Liebe R, Lammert F. Toward genetic prediction of nonalcoholic fatty liver disease trajectories: PNPLA3 and beyond. Gastroenterology. 2020;158(7):1865-80 e1. https://doi.org/10.1053/j.gastro.2020.01.053.
Eslam M, Sanyal AJ, George J, International Consensus P. MAFLD: A consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology. 2020;158(7):1999-2014 e1. https://doi.org/10.1053/j.gastro.2019.11.312.
Al-Serri A, Anstee QM, Valenti L, Nobili V, Leathart JB, Dongiovanni P, et al. The SOD2 C47T polymorphism influences NAFLD fibrosis severity: evidence from case-control and intra-familial allele association studies. J Hepatol. 2012;56(2):448–54. https://doi.org/10.1016/j.jhep.2011.05.029.
Smagris E, BasuRay S, Li J, Huang Y, Lai KM, Gromada J, et al. Pnpla3I148M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology. 2015;61(1):108–18. https://doi.org/10.1002/hep.27242.
BasuRay S, Wang Y, Smagris E, Cohen JC, Hobbs HH. Accumulation of PNPLA3 on lipid droplets is the basis of associated hepatic steatosis. Proc Natl Acad Sci U S A. 2019;116(19):9521–6. https://doi.org/10.1073/pnas.1901974116.
Pingitore P, Pirazzi C, Mancina RM, Motta BM, Indiveri C, Pujia A, et al. Recombinant PNPLA3 protein shows triglyceride hydrolase activity and its I148M mutation results in loss of function. Biochim Biophys Acta. 2014;1841(4):574–80. https://doi.org/10.1016/j.bbalip.2013.12.006.
Kumari M, Schoiswohl G, Chitraju C, Paar M, Cornaciu I, Rangrez AY, et al. Adiponutrin functions as a nutritionally regulated lysophosphatidic acid acyltransferase. Cell Metab. 2012;15(5):691–702. https://doi.org/10.1016/j.cmet.2012.04.008.
He S, McPhaul C, Li JZ, Garuti R, Kinch L, Grishin NV, et al. A sequence variation (I148M) in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis. J Biol Chem. 2010;285(9):6706–15. https://doi.org/10.1074/jbc.M109.064501.
Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40(12):1461–5. https://doi.org/10.1038/ng.257.
Basantani MK, Sitnick MT, Cai L, Brenner DS, Gardner NP, Li JZ, et al. Pnpla3/Adiponutrin deficiency in mice does not contribute to fatty liver disease or metabolic syndrome. J Lipid Res. 2011;52(2):318–29. https://doi.org/10.1194/jlr.M011205.
Luukkonen PK, Nick A, Holtta-Vuori M, Thiele C, Isokuortti E, Lallukka-Bruck S, et al. Human PNPLA3-I148M variant increases hepatic retention of polyunsaturated fatty acids. JCI Insight. 2019;4(16). https://doi.org/10.1172/jci.insight.127902.
Lu Y, Feng T, Zhao J, Jiang P, Xu D, Zhou M, et al. Polyene phosphatidylcholine ameliorates high fat diet-induced non-alcoholic fatty liver disease via remodeling metabolism and inflammation. Front Physiol. 2022;13:810143. https://doi.org/10.3389/fphys.2022.810143.
Maev IV, Samsonov AA, Palgova LK, Pavlov CS, Shirokova EN, Vovk EI, et al. Effectiveness of phosphatidylcholine as adjunctive therapy in improving liver function tests in patients with non-alcoholic fatty liver disease and metabolic comorbidities: real-life observational study from Russia. BMJ Open Gastroenterol. 2020;7(1):e000368. https://doi.org/10.1136/bmjgast-2019-000368.
Liu YL, Patman GL, Leathart JB, Piguet AC, Burt AD, Dufour JF, et al. Carriage of the PNPLA3 rs738409 C >G polymorphism confers an increased risk of non-alcoholic fatty liver disease associated hepatocellular carcinoma. J Hepatol. 2014;61(1):75–81. https://doi.org/10.1016/j.jhep.2014.02.030.
Sookoian S, Pirola CJ. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology. 2011;53(6):1883–94. https://doi.org/10.1002/hep.24283.
Smagris E, Gilyard S, BasuRay S, Cohen JC, Hobbs HH. Inactivation of Tm6sf2, a gene defective in fatty liver disease, impairs lipidation but not secretion of very low density lipoproteins. J Biol Chem. 2016;291(20):10659–76. https://doi.org/10.1074/jbc.M116.719955.
Li XY, Liu Z, Li L, Wang HJ, Wang H. TM6SF2 rs58542926 is related to hepatic steatosis, fibrosis and serum lipids both in adults and children: a meta-analysis. Front Endocrinol (Lausanne). 2022;13:1026901. https://doi.org/10.3389/fendo.2022.1026901.
Fan Y, Lu H, Guo Y, Zhu T, Garcia-Barrio MT, Jiang Z, et al. Hepatic transmembrane 6 superfamily member 2 regulates cholesterol metabolism in mice. Gastroenterology. 2016;150(5):1208–18. https://doi.org/10.1053/j.gastro.2016.01.005.
Mahdessian H, Taxiarchis A, Popov S, Silveira A, Franco-Cereceda A, Hamsten A, et al. TM6SF2 is a regulator of liver fat metabolism influencing triglyceride secretion and hepatic lipid droplet content. Proc Natl Acad Sci U S A. 2014;111(24):8913–8. https://doi.org/10.1073/pnas.1323785111.
Ehrhardt N, Doche ME, Chen S, Mao HZ, Walsh MT, Bedoya C, et al. Hepatic Tm6sf2 overexpression affects cellular ApoB-trafficking, plasma lipid levels, hepatic steatosis and atherosclerosis. Hum Mol Genet. 2017;26(14):2719–31. https://doi.org/10.1093/hmg/ddx159.
Gronert K, Kantarci A, Levy BD, Clish CB, Odparlik S, Hasturk H, et al. A molecular defect in intracellular lipid signaling in human neutrophils in localized aggressive periodontal tissue damage. J Immunol. 2004;172(3):1856–61. https://doi.org/10.4049/jimmunol.172.3.1856.
Koybasi S, Senkal CE, Sundararaj K, Spassieva S, Bielawski J, Osta W, et al. Defects in cell growth regulation by C18:0-ceramide and longevity assurance gene 1 in human head and neck squamous cell carcinomas. J Biol Chem. 2004;279(43):44311–9. https://doi.org/10.1074/jbc.M406920200.
Kroesen BJ, Pettus B, Luberto C, Busman M, Sietsma H, de Leij L, et al. Induction of apoptosis through B-cell receptor cross-linking occurs via de novo generated C16-ceramide and involves mitochondria. J Biol Chem. 2001;276(17):13606–14. https://doi.org/10.1074/jbc.M009517200.
Yamaguchi K, Yang L, McCall S, Huang J, Yu XX, Pandey SK, et al. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology (Baltimore, MD). 2007;45(6):1366–74. https://doi.org/10.1002/hep.21655.
Marra F, Svegliati-Baroni G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J Hepatol. 2018;68(2):280–95. https://doi.org/10.1016/j.jhep.2017.11.014.
Brankovic M, Jovanovic I, Dukic M, Radonjic T, Opric S, Klasnja S, et al. Lipotoxicity as the leading cause of non-alcoholic steatohepatitis. Int J Mol Sci. 2022;23(9). https://doi.org/10.3390/ijms23095146.
Lusis AJ, Seldin MM, Allayee H, Bennett BJ, Civelek M, Davis RC, et al. The Hybrid Mouse Diversity Panel: a resource for systems genetics analyses of metabolic and cardiovascular traits. J Lipid Res. 2016;57(6):925–42. https://doi.org/10.1194/jlr.R066944.
• Norheim F, Chella Krishnan K, Bjellaas T, Vergnes L, Pan C, Parks BW, et al. Genetic regulation of liver lipids in a mouse model of insulin resistance and hepatic steatosis. Mol Syst Biol. 2021;17(1):e9684. https://doi.org/10.15252/msb.20209684. (This study examined the role of lipid species’ role in NAFLD, using various analytical methods on data from 100 inbred mice strains fed a high-fat/high-sucrose diet. They discovered two genes, Ifi203 and Map2k6, that control phosphatidylcholine homeostasis and triacylglycerol accumulation in the liver, respectively.)
Drozdz K, Nabrdalik K, Kwiendacz H, Hendel M, Olejarz A, Tomasik A, et al. Risk factors for cardiovascular disease in patients with metabolic-associated fatty liver disease: a machine learning approach. Cardiovasc Diabetol. 2022;21(1):240. https://doi.org/10.1186/s12933-022-01672-9.
Duell PB, Welty FK, Miller M, Chait A, Hammond G, Ahmad Z, et al. Nonalcoholic fatty liver disease and cardiovascular risk: a scientific statement from the American Heart Association. Arterioscler Thromb Vasc Biol. 2022;42(6):e168–85. https://doi.org/10.1161/ATV.0000000000000153.
Friedrich-Rust M, Schoelzel F, Maier S, Seeger F, Rey J, Fichtlscherer S, et al. Severity of coronary artery disease is associated with non-alcoholic fatty liver disease: a single-blinded prospective mono-center study. PLoS One. 2017;12(10):e0186720. https://doi.org/10.1371/journal.pone.0186720.
Zhang Z, Zheng M, Lei H, Jiang Z, Chen Y, He H, et al. A clinical study of the correlation between metabolic-associated fatty liver disease and coronary plaque pattern. Sci Rep. 2023;13(1):7224. https://doi.org/10.1038/s41598-023-34462-8.
Loomba R, Friedman SL, Shulman GI. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell. 2021;184(10):2537–64. https://doi.org/10.1016/j.cell.2021.04.015.
Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. 2018;24(7):908–22. https://doi.org/10.1038/s41591-018-0104-9.
•• Finney AC, Das S, Kumar D, McKinney MP, Cai B, Yurdagul A Jr, et al. The interplay between nonalcoholic fatty liver disease and atherosclerotic cardiovascular disease. Front Cardiovasc Med. 2023;10:1116861. https://doi.org/10.3389/fcvm.2023.1116861. (This review is focused on the intricate connections between nonalcoholic fatty liver disease and cardiovascular disease. It also ventures into the exploration of potential therapeutic strategies targeting both ailments simultaneously)
Chew NWS, Chong B, Ng CH, Kong G, Chin YH, Xiao W, et al. The genetic interactions between non-alcoholic fatty liver disease and cardiovascular diseases. Front Genet. 2022;13:971484. https://doi.org/10.3389/fgene.2022.971484.
Diogo D, Tian C, Franklin CS, Alanne-Kinnunen M, March M, Spencer CCA, et al. Phenome-wide association studies across large population cohorts support drug target validation. Nat Commun. 2018;9(1):4285. https://doi.org/10.1038/s41467-018-06540-3.
Ruschenbaum S, Schwarzkopf K, Friedrich-Rust M, Seeger F, Schoelzel F, Martinez Y, et al. Patatin-like phospholipase domain containing 3 variants differentially impact metabolic traits in individuals at high risk for cardiovascular events. Hepatol Commun. 2018;2(7):798–806. https://doi.org/10.1002/hep4.1183.
Wu JT, Liu SS, Xie XJ, Liu Q, Xin YN, Xuan SY. Independent and joint correlation of PNPLA3 I148M and TM6SF2 E167K variants with the risk of coronary heart disease in patients with non-alcoholic fatty liver disease. Lipids Health Dis. 2020;19(1):29. https://doi.org/10.1186/s12944-020-01207-9.
BasuRay S, Smagris E, Cohen JC, Hobbs HH. The PNPLA3 variant associated with fatty liver disease (I148M) accumulates on lipid droplets by evading ubiquitylation. Hepatology. 2017;66(4):1111–24. https://doi.org/10.1002/hep.29273.
Linden D, Ahnmark A, Pingitore P, Ciociola E, Ahlstedt I, Andreasson AC, et al. Pnpla3 silencing with antisense oligonucleotides ameliorates nonalcoholic steatohepatitis and fibrosis in Pnpla3 I148M knock-in mice. Mol Metab. 2019;22:49–61. https://doi.org/10.1016/j.molmet.2019.01.013.
Petta S, Valenti L, Marchesini G, Di Marco V, Licata A, Camma C, et al. PNPLA3 GG genotype and carotid atherosclerosis in patients with non-alcoholic fatty liver disease. PLoS One. 2013;8(9):e74089. https://doi.org/10.1371/journal.pone.0074089.
Musso G, Cassader M, Gambino R. PNPLA3 rs738409 and TM6SF2 rs58542926 gene variants affect renal disease and function in nonalcoholic fatty liver disease. Hepatology. 2015;62(2):658–9. https://doi.org/10.1002/hep.27643.
Acknowledgements
The figure is created with BioRender.com.
Funding
Shirin Pourteymour has received funding from the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No. 801133.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Pourteymour, S., Drevon, C.A., Dalen, K.T. et al. Mechanisms Behind NAFLD: a System Genetics Perspective. Curr Atheroscler Rep 25, 869–878 (2023). https://doi.org/10.1007/s11883-023-01158-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11883-023-01158-3