
Abstract
Nonalcoholic fatty liver disease (NAFLD), now the leading cause of liver damage worldwide, is a potentially progressive condition to advanced hepatic fibrosis, the main risk factor for liver-related events and for hepatocellular carcinoma. In particular, about 10–30% of NAFLD patients can develop nonalcoholic steatohepatitis (NASH), which more frequently progresses to advanced liver disease. However, development of cardiovascular complications and of extra-hepatic neoplasm is a more frequent evolution of this condition, and there is a huge interindividual variability in liver disease susceptibility. Indeed, although the severity of metabolic alterations is the main risk factor for progressive NAFLD, qualitative components of the diet, physical activity, and inherited factors play also an important role. In particular, during the last years, common variants in PNPLA3, TM6SF2, MBOAT7 and GCKR have been shown to contribute to the full spectrum of NAFLD pathology by facilitating hepatic fat compartmentalization in the presence of environmental triggers. In the future, evaluation of genetic risk factors may help stratifying the risk of liver-related vs. extra-hepatic complications of the disease, and to guide pharmacological therapy.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Cirrhosis
- Fibrosis of the liver
- Genetics
- Hepatocellular carcinoma
- Natural history
- Nonalcoholic fatty liver disease
- Nonalcoholic steatohepatitis
- Steatosis
7.1 Features of NAFLD
Nonalcoholic fatty liver disease (NAFLD) is now the leading cause of liver disease worldwide [1], and prevalence is still on the rise [2]. The hallmark of NAFLD is represented by hepatic fat accumulation exceeding 5% of liver weight, which is not explained by at- risk alcohol intake, usually defined by a threshold of 30/20 g/day in males/females. The main risk factors for the disease are represented by obesity, the constellation of metabolic alterations associated with insulin resistance (the so called metabolic syndrome) and type 2 diabetes. In most of the cases, NAFLD represents the hepatic manifestation of insulin resistance [3]. Hepatic fat is mainly accumulated within intracellular lipid droplets in hepatocytes, under the form of triglycerides. Indeed, the esterification within triglycerides represents the safest way to store free fatty acids, which derive from the adipose tissue due to systemic insulin resistance and from de novo lipogenesis stimulated by hyperinsulinemia, and would otherwise cause severe lipotoxicity and activation of fibrogenesis [4]. However, lipid droplets formation, metabolism and catabolism are highly regulated, and several proteins involved in the pathogenesis of liver damage and potentially lipotoxic compounds are involved in this biological process [5].
The acronym NAFLD defines a wide spectrum of liver conditions, ranging from simple uncomplicated steatosis to forms of liver disease associated with hepatocellular damage (“ballooning”) and lobular inflammation, which is non-alcoholic steatohepatitis (NASH) [6]. NASH is more commonly associated with activation of hepatic fibrogenesis, initially at pericellular and perivenular level, which in susceptible individuals may lead to cirrhosis and advanced liver disease. The pathogenesis of the transition from simple steatosis to NASH or progressive disease is still not completely understood, and likely multifactorial [7]. Altered microbiota and gut permeability, the severity of metabolic alterations, oxidative stress and a proinflammatory imbalance in the release of mediators from the adipose tissue and the muscle are likely involved.
7.2 Natural History of NAFLD
The knowledge on the natural history of liver disease related to NAFLD is still limited by the relatively low number and the selection bias of patients with histological characterization of liver damage with available long-term follow-up, and conversely by the lack of detailed characterization of liver damage for most individuals included in prospective population studies. However, a few robust conclusions could be established. The first one is that a diagnosis of NAFLD seems to be associated with an increased mortality rate, the leading cause being cardiovascular disease, followed up by extra-hepatic cancer and liver disease (the latter with the higher relative risk as compared to the general population) [8,9,10,11]. Heightened cardiovascular risk seems to be related to accelerated atherogenesis, independently of classic risk factors [12, 13], but may also reflect more severe insulin resistance with increased susceptibility to develop type 2 diabetes [14].
Secondly, the main prognostic determinant in patients with NAFLD is represented by the severity of liver fibrosis [15]. Overall evidence indicates that, compared to NAFLD patients with no fibrosis, NAFLD patients with fibrosis are at an increased risk for all-cause mortality, and this risk increased with increases in the stage of fibrosis. When NAFLD-related fibrosis was estimated non-invasively, this conclusion held true also in the general population, and the association was independent of several possible confounding factors [16]. Most importantly, the impact of fibrosis is more pronounced for liver-related mortality as the risk of liver-related mortality increased exponentially with each increase in the stage of fibrosis, even if these estimates could not be corrected for age [15]. For stage 1, mortality rate ratio was estimated at 1.41 (95% confidence interval (CI) 0.17–11.95); stage 2, 9.57 (95% CI 1.67–54.93); stage 3, 16.69 (95% CI 2.92–95.36); and stage 4 (cirrhosis), 42.30 (95% CI 3.51–510.34) [15]. In particular, in patients with cirrhosis, liver disease becomes the leading cause of death [17, 18]. Conversely, cardiovascular disease and extra-hepatic cancer predominate in those with lower fibrosis stages, but their incidence as liver function begins to deteriorate [17]. In contrast, although the presence of NASH overall is also associated with increased mortality as compared to simple mild steatosis [19], NASH without liver fibrosis does not seem to confer an increased risk of mortality [20, 21]. Indeed, fibrosis progression rate is influenced by basal fibrosis stage. Although there is a wide variability in the transition rates between different stages of fibrosis, estimates are consistent with lower rate transition between no to mild fibrosis (0.3–2.2%) than between intermediate to advanced fibrosis (2.8–13.3%) [2]. It should be taken into account that these estimates also account for disease regressors, about one-third of patients in prospective studies, mostly represented by individuals who lose weight or improved metabolic control during the follow-up [22].
However, the presence of histological NASH is likely associated with faster progression, on average, of liver fibrosis [23,24,25], in keeping with a role of oxidative stress and immune activation in determining the evolution of this liver disease. In particular, a meta-analysis of early studies led to an estimation that to progress of one fibrosis stage, it takes on average 14 years in patients with steatosis, while only 7 years in those with NASH, but fibrosis progression occurs in only about one-third of patients. However, there is a wide variability, and a significant proportion of patients without histological NASH show rapid progression of liver disease. Furthermore, even in patients without NASH, the presence of mild histological inflammation may be associated with a higher risk of disease progression [26].
The increasing prevalence of this condition, and especially due to the ageing of affected individuals and more and more frequent association with type 2 diabetes, is leading to a dramatic rise in the proportion of patients who may develop advanced fibrosis [2]. Indeed, NAFLD is already becoming a leading indication for liver transplantation in Western countries [27]. Hepatocellular carcinoma is also a rising cause of liver disease included in the disease spectrum of NAFLD [28, 29]. Although the risk of progression is higher with increasing severity of liver fibrosis, it should be noted that HCC may also develop in patients with NAFLD with significant liver disease, rendering surveillance, early diagnosis and application of curative treatments difficult tasks [30, 31].
7.3 Environmental Risk Factors for Disease Progression
The severity of the metabolic abnormalities, insulin resistance, and in particular the presence of type 2 diabetes represent the major risk factor associated with development of advanced liver disease, and with fibrosis progression in prospective studies in patients with NAFLD [23,24,25]. A key mediator of liver disease progression induced by metabolic risk factors may be represented by severity of hepatic fat accumulation, which has been linked with short- and long-term fibrosis progression independently of several confounders [32,33,34]. Cohort studies also highlighted a possible role of arterial hypertension as a risk factor for progressive worsening of fibrosis, possibly due to activation of the neurohormonal sympathetic system leading to stimulation of hepatic stellate cells. In keeping, variations in body weight and associated metabolic abnormalities represent the main clinical predictor of the liver disease evolution during the follow-up.
Cross-sectional studies also highlighted that independently of adiposity, physical exercise may have a protective role, while sarcopenia is associated with more severe liver damage. Furthermore, besides total caloric intake, the quality of diet also matters. Indeed, industrial fructose intake has been associated with higher risk of both development and progression of NAFLD, probably by inducing ATP depletion, stimulating lipogenesis and decreasing lipid oxidation [35], and an increase in the ratio of dietary saturated/unsaturated fat intake may also play a role. On the other hand, the role of red meat consumption is less established. Concerning beverages, besides a predisposing role of sodas containing fructose, a moderate intake of alcohol with wine, but not with beer, and not under the form of binge drinking, was associated with protection from fibrosis in cross-sectional epidemiological studies, but any alcohol intake was associated with increased risk of disease progression in those with clinically significant fibrosis [36]. Vice versa, coffee consumption may be protective by promoting the antioxidant response.
Lastly, some drugs active on cardiovascular risk factors may influence the risk of liver damage progression, e.g. statins and renin angiotensin aldosterone axis modulators may reduce fibrogenesis by reducing free cholesterol and altering activation of hepatic stellate cells. Exposure to environmental toxins may also play a role in disease susceptibility, but few data are available in the literature.
A cartoon depicting the natural history of liver disease in NAFLD is presented in Fig. 7.1. Given the wide uncertainty concerning the progression (and possible regression) rates across the different stages of the disease, which may vary according to the specific populations, wide confidence intervals are indicated.

Natural history of NAFLD. The majority of individuals with environmental, metabolic and genetic risk factors develop some form of NAFLD, and 10–30% of them NASH. With time and ageing, NASH can progress to advanced fibrosis at variable rates (although progression from simple steatosis cannot be excluded) and then to liver failure or hepatocellular carcinoma (HCC). Yr per year
7.4 Role of Heritable Factors in NAFLD
Accumulating evidence indicate that hepatic fat and NAFLD are strongly heritable conditions [37]. First, twin studies led to the estimation that in the general population more than half of the variability of aminotransferases levels in individuals without viral hepatitis or alcohol abuse and of hepatic fat content are accounted for by heritable factors [38, 39]. Interestingly, they also suggested that heritability of liver fat and fibrosis share are shared traits, in line with a causal role of hepatic fat accumulation in triggering progressive liver disease [38, 39]. Second, multi-ethnic cohort studies demonstrated that there is a strong interethnic variability in the susceptibility towards NAFLD development, being higher in Hispanics, intermediate in Europeans and lower in African Americans, independently of adiposity, type 2 diabetes and socioeconomic factors [40]. Lastly, family studies showed that cases of NAFLD progressing to advanced fibrosis tend to cluster in specific families. Indeed, the risk of progressive NAFLD is higher in first-degree relatives of patients with NAFLD cirrhosis as compared to the general population, independently of several confounders [41].
7.5 Genetic Determinants of NAFLD Development and Progression: The PNPLA3 I148M Variant
The most important common genetic determinants of hepatic fat variability and the susceptibility to develop NAFLD have been uncovered thanks to the advent of genome-wide association studies in the last years. The major one is the rs738409 C>G encoding for the I148M protein variant of Patatin-like phospholipase domain-containing 3 (PNPLA3), accounting for a large fraction of the increased risk of this condition in Hispanics [42]. The I148M variant is specifically associated with increased hepatic fat, without major influences on adiposity, circulating lipids and risk of type 2 diabetes [42]. Importantly, the I148M variant increases susceptibility to the whole spectrum of liver damage related to NAFLD, from simple steatosis to NASH, fibrosis and cirrhosis, thereby representing a general modifier of liver disease progression [43]. Furthermore, the I148M variant heightens the risk of progression to hepatocellular carcinoma development independently of the effect on fibrosis. In Europeans, homozygosity for the mutation is enriched almost ninefold in patients who develop NAFLD-HCC as compared to the general population, while absence of the variant can rule out HCC risk with a high specificity in the general population [43,44,45]. Homozygosity of the mutation is also associated with increased risk of hepatic decompensation in patients with fatty liver and portal hypertension [46].
Carriage of this variant impacts on the risk of liver disease particularly during the developmental age [47, 48], interacting with dietary factors such as intake of fructose-enriched drinks and lack of physical activity [49]. However, the major environmental trigger of the phenotypic expression of the variant in those who do not drink excess alcohol is represented by excess adiposity [50]. Indeed, during obesity and insulin resistance, the PNPLA3 protein is induced by insulin and expressed at the surface of lipid droplets, where it mediates the remodelling of triglycerides and phospholipids, in particular by mediating the hydrolysis of oleic acid [51]. The mechanism behind the association with steatosis is related to accumulation of the mutated I148M protein on the surface of lipid droplets altering lipid remodelling and turnover [51,52,53]. Furthermore, the I148M alters retinol release from hepatic stellate cells directly favouring inflammation and fibrogenesis [54,55,56].
7.6 Other Common Genetic Determinants of NAFLD
Other common genetic mutations regulating hepatocellular lipid handling contribute to the risk of NAFLD. The rs58542926 C>T encoding for the E167K variant in Transmembrane 6 superfamily member 2 (TM6SF2) favours hepatic fat accumulation by decreasing lipid secretion in very-low-density lipoproteins (VLDL), also leading to increased susceptibility to liver damage. At the same time, this genetic factor protects from cardiovascular disease by reducing circulating lipids [57,58,59]. Variants in Glucokinase regulator (GCKR) [60, 61] and in membrane bound O-acyl transferase 7 (MBOAT7) [62] also contribute to the risk, by increasing de novo lipogenesis and altering the remodelling of phospholipids, respectively. All these factors result in fat accumulation and higher risk of liver disease. Conversely, a variant in protein phosphatase 1 regulatory subunit 3B (PPP1R3B) has recently been demonstrated to protect against hepatic fat accumulation, possibly by shunting the excessive energy supply towards glycogen synthesis [63]. This also resulted in decreased risk of progressive liver disease in individuals at high risk of NASH. All in all, a general concept emerging from these studies is that the risk of progressive NAFLD is strongly related and proportional to the impact of these genetic risk factors on hepatic fat accumulation, suggesting this is a major driver of liver damage progression [33].
However, other genetic variants may modify the effect of fat accumulation on inflammation and fibrosis. The best studied are represented by variants regulating oxidative stress and innate immunity in the liver. Indeed, variants in the mitochondrial Mn-superoxidase dismutase 2 (SOD2), and uncoupling protein 2 (UCP2), regulating fatty acid oxidation and redox status in the mitochondria [64, 65]. Concerning inflammation, variants in the interleukin 28 (IL28) locus encoding for the alternative interferon lambda-3 and lambda-4 proteins, and those in Mer T kinase (MERTK), regulating the activation of phagocytes and hepatic stellate cells [66]. Very recently, a common variant in 17-beta hydroxysteroid dehydrogenase 13 (HSD17B13) encoding form of this enzyme that is expressed at the surface of lipid droplets in hepatocytes has been identified. This polymorphism protects against progressive liver disease associated with fat accumulation particularly in carriers of the I148M PNPLA3 variant [67, 68].
7.7 The Role of Rare Inherited Mutations
Noteworthy, rare genetic mutations with a strong impact on the function of proteins involved in NAFLD pathogenesis may also contribute to determine the predisposition to develop advanced NAFLD and disease clustering in specific families. For example, mutations in Apolipoprotein B (APOB) favour disease progression again by causing lipid compartmentalization in hepatocytes [69]. This is caused by the inability to secrete VLDL from hepatocytes. However, as APOB is also involved in the secretion of chylomicrons, damage to the intestinal barrier due to accumulation of lipids in enterocytes, and malabsorption of liposoluble vitamins (and especially A, D, E may be relevant for the risk of NAFLD) may also occur. Another mechanism leading to progressive NAFLD is related to telomere shortening and cell senescence [70], usually mediating the effect of ageing on the risk of liver disease, and mutations in telomerase reverse transcriptase (TERT) have been associated with progressive NAFLD [71, 72]. Finally, it should not be forgotten that in children NAFLD may represent the manifestation of severe genetic disorders, such as lysosomal acid lipase deficiency caused by mutation of LIPA gene, which determine accumulation of cholesteryl esters and triglycerides in hepatocytes [73].
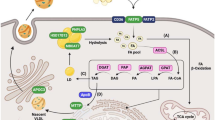
An overall picture of genetic loci associated with NAFLD development and progression, classified according to the role on encoded proteins in the accumulation of lipids (lipogenesis, lipid oxidation, lipid droplets formation and remodelling, lipid secretion within VLDL) and development of liver damage (lipotoxicity, inflammation and activation of fibrogenesis) is shown in Fig. 7.2.

Genetic loci involved in the development and progression of NAFLD, classified according to the mechanism by which the encoded proteins intervene in the pathogenesis of the disease. Red arrows indicate pathological processes/lipid fluxes, green arrows beneficial pathways. PPP1R3B Protein phosphatase 1 regulatory subunit 3B, GCKR glucokinase regulator, UCP2 uncoupling protein 2, SOD2 mitochondrial superoxide dismutase, LIPA lysosomal acid lipase, PNPLA3 patatin-like phospholipase domain-containing 3, MBOAT7 membrane bound O-acyl transferase 7, HSD17B13 17-beta hydroxysteroid dehydrogenase 13, TM6SF2 transmembrane 6 superfamily member 2, APOB apolipoprotein B, VLDL very-low-density lipoproteins, IFNL4 interferon lambda 4, MERTK Mer T kinase, TERT human telomerase reverse transcriptase
7.8 The Role of Epigenetic Changes
The term “epigenetic changes” refer to relatively stable alterations of nuclear DNA and the mechanisms of transcriptional regulation that can be transmitted through cell division. These are involved in mediating the effect of environmental factors on phenotype, and may possibly explain part of the missing heritability and variability of disease progression of common diseases such as NAFLD. Methylation of cytosine nucleotides at CpG-rich regulatory or promoter regions represents the first level of regulation of gene expression. Several post-translational modifications of histones also contribute to modulating the access of transcription and regulatory factors to the DNA. An important role of epigenetic factors in modulating the susceptibility to NAFLD is demonstrated by the effect of intrauterine exposure to high-fat diet in experimental models, leading to more severe hepatic fat accumulation and the development of NASH [74]. These experiments recapitulate the effect of an adverse foetal environment on the risk of NAFLD. Indeed, both intrauterine growth retardation and accelerated foetal growth are associated with an increased risk of NAFLD and NASH [75,76,77,78]. In keeping, hepatic DNA tends to be demethylated in patients with NAFLD [79]. Genes involved in the methylation process, lipid metabolism (including PNPLA3), inflammation and fibrogenesis showed stage-dependent regulation, suggesting that epigenetic changes are involved in the progression of liver disease [79, 80]. These alterations confer an especially high risk of liver disease in patients born with a strong genetic predisposition.
Another layer of regulation is provided by non-coding RNAs. Indeed, NAFLD is associated with deregulation of many hepatic micro-RNAs (miRNA) [81, 82]. The most robustly validated alteration is represented by downregulation of miR-122, [81,82,83,84,85], which promotes lipogenesis [81], and in experimental model is associated with spontaneous development of NASH and HCC [83]. However, several miRNAs are altered during NASH, and their variability seem be involved in mediating the susceptibility to the disease [37, 86].
7.9 Interaction Between Genetic and Environmental Factors
The phenotypic expression of the disease is triggered by the interaction between the genetic background and environmental triggers. The most common one is represented by increased adiposity, leading to insulin resistance and hyperinsulinemia [87]. For example, at the general population level most of the carriers of PNPLA3, TM6SF2 and GCKR common risk variants are not affected by NAFLD, and most importantly do not develop progressive liver disease. However, the impact of the variants on hepatic fat content, the risk of NAFLD and that of cirrhosis increases exponentially with increasing BMI, indicating the presence of a synergism between these components of the disease [88]. Similarly, there seems to be an interaction between consumption of industrial fructose in soft drinks and the PNPLA3 I148M variant in determining the susceptibility to NAFLD [49]. On the other hand, omega-3 fatty acids would be less effective in reducing lipogenesis and liver fat in carriers of this variant [89, 90]. Importantly, in individuals at high genetic risk a healthy dietary patter modelled on the Mediterranean diet may reduce the risk of NAFLD [91], as well as regular physical activity may prevent disease development [49].
An overview of common inherited and acquired factors involved in the development and progression of NAFLD is presented in Fig. 7.3 [78].

Complementary role of inherited and acquired risk factors for NAFLD according to life stages. BMI body mass index, ALT alanine aminotransferases, T2D type 2 diabetes, PCOS polycystic ovary syndrome, PNPLA3 patatin-like phospholipase domain-containing 3, MBOAT7 membrane bound O-acyl transferase 7, GCKR glucokinase regulator, TM6SF2 transmembrane 6 superfamily member 2
7.10 Possible Future Clinical Applications of Genetics
Variants in PNPLA3 and TM6SF2 are strong risk factors for NAFLD, especially in individuals with strong predisposition, such as obese adolescents with severe insulin resistance developed after intrauterine growth retardation. Genotypization of these common variants is able to significantly improve the prediction of the risk of severe progressive NAFLD, hopefully allowing to tailor preventive lifestyle approaches in the future [78]. Furthermore, the number of common genetic risk variants for hepatic fat accumulation in PNPLA3, TM6SF2, and MBOAT7 nicely stratify the risk of NAFLD in the general population, interacting with adiposity [50, 62]. Notably, the same simple genetic instrument is able to predict the risk of HCC in patients with NAFLD independently of classic risk factors, possibly improving risk stratification for this condition, even in patients without severe fibrosis [92]. An emerging concept is that genetic risk variants for progressive liver disease related to NAFLD may protect at the same time from dyslipidemia and cardiovascular disease. This is particularly true for those that have inhibition of lipid secretion within VLDL as the main mechanism, such as those in TM6SF2 and APOB, and also the PNPLA3 I148M mutation. Therefore, they may be useful to dissociate the risk of hepatic vs. cardiovascular complications of insulin resistance, and help guiding surveillance of complications. This concept is exemplified in Fig. 7.4.

Possible role of genetics for stratification of patients with NAFLD in those at higher risk of hepatic vs. cardiovascular complications, and disease management. PNPLA3 patatin-like phospholipase domain-containing 3, MBOAT7 membrane-bound O-acyl transferase 7, TM6SF2 transmembrane 6 superfamily member 2, APOB apolipoprotein B
Carriage of specific genetic risk factors may influence the response and in particular the side effect of drugs. For example, the PNPLA3 I148M variant has been reported to reduce the protective effect of statins on the risk of progressive NAFLD [93], to reduce the beneficial impact of dapagliflozin, an SGLT2 inhibitor, on hepatic fat accumulation, and to predict hepatotoxicity of glucagon receptor agonists and insulin peglispro, which is related to induction of hepatic fat accumulation [94,95,96]. Finally, drugs directly targeting protein mutated in NAFLD may prove beneficial to prevent progressive liver disease caused by fat accumulation. For example, silencing of the mutated PNPLA3 protein may potentially revert liver damage in carriers of the mutation by restoring dismissal of lipids from intracellular droplets, and possibly retinol metabolism [97]. This concept is presented in Fig. 7.5. Therefore, it could be envisioned that evaluation of genetic risk variants may help guiding pharmacological therapy for the disease. The clinical utility of these approached remains to be demonstrated in future studies.

Possible role of drugs silencing the mutated PNPLA3 in the prevention and treatment of liver disease in carriers of the I148M variant. Beneficial fluxes of lipids are indicated by green arrows, detrimental pathways as red arrows. The wild-type protein is involved in the remodelling and dismissal of lipids from lipid droplets under insulin resistance conditions. The mutated I148M variant is not enzymatically active and accumulate at the surface of lipid droplets because it is not ubiquitylated. At this level, it acquires the ability to impede lipid remodelling causing their retention and likely reduced turnover and activation of lipotoxicity. Pharmacological approaches that downmodulate the PNPLA3 I148M protein may contrast this pathophysiological mechanism. PNPLA3 patatin like phospholipase domain-containing 3, TAG triglycerides, Ub ubiquitin
References
Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64(1):73–84.
Estes C, Anstee QM, Teresa Arias-Loste M, Bantel H, Bellentani S, Caballeria J, et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016-2030. J Hepatol. 2018;69(4):896–904.
Marchesini G, Brizi M, Morselli-Labate AM, Bianchi G, Bugianesi E, McCullough AJ, et al. Association of nonalcoholic fatty liver disease with insulin resistance. Am J Med. 1999;107(5):450–5.
Yamaguchi K, Yang L, McCall S, Huang J, Yu XX, Pandey SK, et al. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology. 2007;45(6):1366–74.
Mashek DG, Khan SA, Sathyanarayan A, Ploeger JM, Franklin MP. Hepatic lipid droplet biology: getting to the root of fatty liver. Hepatology. 2015;62(3):964–7.
Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41(6):1313–21.
Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 2010;52(5):1836–46.
Adams LA, Lymp JF, St Sauver J, Sanderson SO, Lindor KD, Feldstein A, et al. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology. 2005;129(1):113–21.
Ekstedt M, Franzen LE, Mathiesen UL, Thorelius L, Holmqvist M, Bodemar G, et al. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology. 2006;44(4):865–73.
Ekstedt M, Hagstrom H, Nasr P, Fredrikson M, Stal P, Kechagias S, et al. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology. 2015;61(5):1547–54.
Adams LA, Harmsen S, St Sauver JL, Charatcharoenwitthaya P, Enders FB, Therneau T, et al. Nonalcoholic fatty liver disease increases risk of death among patients with diabetes: a community-based cohort study. Am J Gastroenterol. 2010;105(7):1567–73.
Targher G, Day CP, Bonora E. Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N Engl J Med. 2010;363(14):1341–50.
Fracanzani AL, Tiraboschi S, Pisano G, Consonni D, Baragetti A, Bertelli C, et al. Progression of carotid vascular damage and cardiovascular events in non-alcoholic fatty liver disease patients compared to the general population during 10 years of follow-up. Atherosclerosis. 2016;246:208–13.
Ballestri S, Zona S, Targher G, Romagnoli D, Baldelli E, Nascimbeni F, et al. Nonalcoholic fatty liver disease is associated with an almost twofold increased risk of incident type 2 diabetes and metabolic syndrome. Evidence from a systematic review and meta-analysis. J Gastroenterol Hepatol. 2016;31(5):936–44.
Dulai PS, Singh S, Patel J, Soni M, Prokop LJ, Younossi Z, et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: systematic review and meta-analysis. Hepatology. 2017;65(5):1557–65.
Golabi P, Stepanova M, Pham HT, Cable R, Rafiq N, Bush H, et al. Non-alcoholic steatofibrosis (NASF) can independently predict mortality in patients with non-alcoholic fatty liver disease (NAFLD). BMJ Open Gastroenterol. 2018;5(1):e000198.
Vilar-Gomez E, Calzadilla-Bertot L, Wai-Sun Wong V, Castellanos M, Aller-de la Fuente R, Metwally M, et al. Fibrosis severity as a determinant of cause-specific mortality in patients with advanced nonalcoholic fatty liver disease. Gastroenterology. 2018;155(2):443–457.e17.
Bhala N, Angulo P, van der Poorten D, Lee E, Hui JM, Saracco G, et al. The natural history of nonalcoholic fatty liver disease with advanced fibrosis or cirrhosis: an international collaborative study. Hepatology. 2011;54(4):1208–16.
Musso G, Gambino R, Cassader M, Pagano G. Meta-analysis: natural history of non-alcoholic fatty liver disease (NAFLD) and diagnostic accuracy of non-invasive tests for liver disease severity. Ann Med. 2012;43(8):617–49.
Angulo P, Kleiner DE, Dam-Larsen S, Adams LA, Bjornsson ES, Charatcharoenwitthaya P, et al. Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology. 2015;149(2):389–97.e10.
Hagstrom H, Nasr P, Ekstedt M, Hammar U, Stal P, Hultcrantz R, et al. Fibrosis stage but not NASH predicts mortality and time to development of severe liver disease in biopsy-proven NAFLD. J Hepatol. 2017;67:1265.
Romero-Gomez M, Zelber-Sagi S, Trenell M. Treatment of NAFLD with diet, physical activity and exercise. J Hepatol. 2017;67(4):829–46.
Singh S, Allen AM, Wang Z, Prokop LJ, Murad MH, Loomba R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: a systematic review and meta-analysis of paired-biopsy studies. Clin Gastroenterol Hepatol. 2015;13(4):643–54.e1-9; quiz e39–40.
McPherson S, Hardy T, Henderson E, Burt AD, Day CP, Anstee QM. Evidence of NAFLD progression from steatosis to fibrosing-steatohepatitis using paired biopsies: implications for prognosis and clinical management. J Hepatol. 2015;62(5):1148–55.
Pelusi S, Petta S, Rosso C, Borroni V, Fracanzani AL, Dongiovanni P, et al. Renin-angiotensin system inhibitors, type 2 diabetes and fibrosis progression: an observational study in patients with nonalcoholic fatty liver disease. PLoS One. 2016;11(9):e0163069.
Pais R, Charlotte F, Fedchuk L, Bedossa P, Lebray P, Poynard T, et al. A systematic review of follow-up biopsies reveals disease progression in patients with non-alcoholic fatty liver. J Hepatol. 2013;59(3):550–6.
Goldberg D, Ditah IC, Saeian K, Lalehzari M, Aronsohn A, Gorospe EC, et al. Changes in the prevalence of hepatitis C virus infection, nonalcoholic steatohepatitis, and alcoholic liver disease among patients with cirrhosis or liver failure on the waitlist for liver transplantation. Gastroenterology. 2017;152(5):1090–9.e1.
Baffy G, Brunt EM, Caldwell SH. Hepatocellular carcinoma in non-alcoholic fatty liver disease: an emerging menace. J Hepatol. 2012;56(6):1384–91.
Bugianesi E, Leone N, Vanni E, Marchesini G, Brunello F, Carucci P, et al. Expanding the natural history of nonalcoholic steatohepatitis: from cryptogenic cirrhosis to hepatocellular carcinoma. Gastroenterology. 2002;123(1):134–40.
Piscaglia F, Svegliati-Baroni G, Barchetti A, Pecorelli A, Marinelli S, Tiribelli C, et al. Clinical patterns of hepatocellular carcinoma in nonalcoholic fatty liver disease: a multicenter prospective study. Hepatology. 2016;63(3):827–38.
Younossi ZM, Otgonsuren M, Henry L, Venkatesan C, Mishra A, Erario M, et al. Association of nonalcoholic fatty liver disease (NAFLD) with hepatocellular carcinoma (HCC) in the United States from 2004 to 2009. Hepatology. 2015;62(6):1723–30.
Ajmera V, Park CC, Caussy C, Singh S, Hernandez C, Bettencourt R, et al. Magnetic resonance imaging proton density fat fraction associates with progression of fibrosis in patients with nonalcoholic fatty liver disease. Gastroenterology. 2018;155(2):307–310.e2.
Dongiovanni P, Stender S, Pietrelli A, Mancina RM, Cespiati A, Petta S, et al. Causal relationship of hepatic fat with liver damage and insulin resistance in nonalcoholic fatty liver. J Intern Med. 2018;283(4):356–70.
McPherson S, Pais R, Valenti L, Schattenberg J, Dufour JF, Tsochatzis E, et al. Further delineation of fibrosis progression in NAFLD: evidence from a large cohort of patients with sequential biopsies. J Hepatol. 2017;64:S593.
Jensen T, Abdelmalek MF, Sullivan S, Nadeau KJ, Green M, Roncal C, et al. Fructose and sugar: a major mediator of non-alcoholic fatty liver disease. J Hepatol. 2018;68(5):1063–75.
Mitchell T, Jeffrey GP, de Boer B, MacQuillan G, Garas G, Ching H, et al. Type and pattern of alcohol consumption is associated with liver fibrosis in patients with non-alcoholic fatty liver disease. Am J Gastroenterol. 2018;113:1484.
Eslam M, Valenti L, Romeo S. Genetics and epigenetics of NAFLD and NASH: clinical impact. J Hepatol. 2018;68(2):268–79.
Makkonen J, Pietilainen KH, Rissanen A, Kaprio J, Yki-Jarvinen H. Genetic factors contribute to variation in serum alanine aminotransferase activity independent of obesity and alcohol: a study in monozygotic and dizygotic twins. J Hepatol. 2009;50(5):1035–42.
Loomba R, Schork N, Chen CH, Bettencourt R, Bhatt A, Ang B, et al. Heritability of hepatic fibrosis and steatosis based on a prospective twin study. Gastroenterology. 2015;149(7):1784–93.
Guerrero R, Vega GL, Grundy SM, Browning JD. Ethnic differences in hepatic steatosis: an insulin resistance paradox? Hepatology. 2009;49(3):791–801.
Caussy C, Soni M, Cui J, Bettencourt R, Schork N, Chen CH, et al. Nonalcoholic fatty liver disease with cirrhosis increases familial risk for advanced fibrosis. J Clin Invest. 2017;127(7):2697–704.
Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40:1461–5.
Dongiovanni P, Donati B, Fares R, Lombardi R, Mancina RM, Romeo S, et al. PNPLA3 I148M polymorphism and progressive liver disease. World J Gastroenterol. 2013;19(41):6969–78.
Liu YL, Patman GL, Leathart JB, Piguet AC, Burt AD, Dufour JF, et al. Carriage of the PNPLA3 rs738409 C>G polymorphism confers an increased risk of non-alcoholic fatty liver disease associated hepatocellular carcinoma. J Hepatol. 2013;61(1):75–81.
Dongiovanni P, Romeo S, Valenti L. Hepatocellular carcinoma in nonalcoholic fatty liver: role of environmental and genetic factors. World J Gastroenterol. 2014;20(36):12945–55.
Mandorfer M, Scheiner B, Stattermayer AF, Schwabl P, Paternostro R, Bauer D, et al. Impact of patatin-like phospholipase domain containing 3 rs738409 G/G genotype on hepatic decompensation and mortality in patients with portal hypertension. Aliment Pharmacol Ther. 2018;48:451.
Valenti L, Alisi A, Galmozzi E, Bartuli A, Del Menico B, Alterio A, et al. I148M Patatin-like phospholipase domain-containing 3 gene variant and severity of pediatric nonalcoholic fatty liver disease. Hepatology. 2010;52(4):1274–80.
Sookoian S, Pirola CJ. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology. 2011;53(6):1883–94.
Nobili V, Liccardo D, Bedogni G, Salvatori G, Gnani D, Bersani I, et al. Influence of dietary pattern, physical activity, and I148M PNPLA3 on steatosis severity in at-risk adolescents. Genes Nutr. 2014;9(3):392.
Stender S, Kozlitina J, Nordestgaard BG, Tybjaerg-Hansen A, Hobbs HH, Cohen JC. Adiposity amplifies the genetic risk of fatty liver disease conferred by multiple loci. Nat Genet. 2017;49(6):842–7.
BasuRay S, Smagris E, Cohen J, Hobbs HH. The PNPLA3 variant associated with fatty liver disease (I148M) accumulates on lipid droplets by evading ubiquitylation. Hepatology. 2017;66(4):1111–24.
Mitsche MA, Hobbs HH, Cohen JC. Phospholipase domain-containing protein 3 promotes transfers of essential fatty acids from triglycerides to phospholipids in hepatic lipid droplets. J Biol Chem. 2018;293(18):6958–68.
Donati B, Motta BM, Pingitore P, Meroni M, Pietrelli A, Alisi A, et al. The rs2294918 E434K variant modulates patatin-like phospholipase domain-containing 3 expression and liver damage. Hepatology. 2016;63(3):787–98.
Mondul A, Mancina RM, Merlo A, Dongiovanni P, Rametta R, Montalcini T, et al. PNPLA3 1148M variant influences circulating retinol in adults with nonalcoholic fatty liver disease or obesity. J Nutr. 2015;145(8):1687–91.
Pirazzi C, Valenti L, Motta BM, Pingitore P, Hedfalk K, Mancina RM, et al. PNPLA3 has retinyl-palmitate lipase activity in human hepatic stellate cells. Hum Mol Genet. 2014;23(15):4077–85.
Pingitore P, Dongiovanni P, Motta BM, Meroni M, Lepore SM, Mancina RM, et al. PNPLA3 overexpression results in reduction of proteins predisposing to fibrosis. Hum Mol Genet. 2016;25(23):5212–22.
Kozlitina J, Smagris E, Stender S, Nordestgaard BG, Zhou HH, Tybjaerg-Hansen A, et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2014;46(4):352–6.
Dongiovanni P, Petta S, Maglio C, Fracanzani AL, Pipitone R, Mozzi E, et al. Transmembrane 6 superfamily member 2 gene variant disentangles nonalcoholic steatohepatitis from cardiovascular disease. Hepatology. 2015;61(2):506–14.
Liu YL, Reeves HL, Burt AD, Tiniakos D, McPherson S, Leathart JB, et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat Commun. 2014;5:4309.
Speliotes EK, Yerges-Armstrong LM, Wu J, Hernaez R, Kim LJ, Palmer CD, et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011;7(3):e1001324.
Santoro N, Zhang CK, Zhao H, Pakstis AJ, Kim G, Kursawe R, et al. Variant in the glucokinase regulatory protein (GCKR) gene is associated with fatty liver in obese children and adolescents. Hepatology. 2011;55:781–9.
Mancina RM, Dongiovanni P, Petta S, Pingitore P, Meroni M, Rametta R, et al. The MBOAT7-TMC4 variant rs641738 increases risk of nonalcoholic fatty liver disease in individuals of European descent. Gastroenterology. 2016;150(5):1219–30.e6.
Dongiovanni P, Meroni M, Mancina RM, Baselli G, Rametta R, Pelusi S, et al. Protein phosphatase 1 regulatory subunit 3B gene variation protects against hepatic fat accumulation and fibrosis in individuals at high risk of nonalcoholic fatty liver disease. Hepatol Commun. 2018;2(6):666–75.
Al-Serri A, Anstee QM, Valenti L, Nobili V, Leathart JB, Dongiovanni P, et al. The SOD2 C47T polymorphism influences NAFLD fibrosis severity: evidence from case-control and intra-familial allele association studies. J Hepatol. 2012;56(2):448–54.
Fares R, Petta S, Lombardi R, Grimaudo S, Dongiovanni P, Pipitone R, et al. The UCP2 -866 G>A promoter region polymorphism is associated with nonalcoholic steatohepatitis. Liver Int. 2015;35(5):1574–80. https://doi.org/10.1111/liv.12707. Epub 2014 Nov 20.
Petta S, Valenti L, Tuttolomondo A, Grimaudo S, Dongiovanni P, Pipitone RM, et al. IFNL4 rs368234815 δG>TT variant is associated with histological liver damage in patients with non-alcoholic fatty liver disease. Hepatology. 2017;66(6):1885–93.
Petta S, Valenti L, Marra F, Grimaudo S, Tripodo C, Bugianesi E, et al. MERTK rs4374383 polymorphism affects the severity of fibrosis in non-alcoholic fatty liver disease. J Hepatol. 2016;64(3):682–90.
Abul-Husn NS, Cheng X, Li AH, Xin Y, Schurmann C, Stevis P, et al. A protein-truncating HSD17B13 variant and protection from chronic liver disease. N Engl J Med. 2018;378(12):1096–106.
Di Filippo M, Moulin P, Roy P, Samson-Bouma ME, Collardeau-Frachon S, Chebel-Dumont S, et al. Homozygous MTTP and APOB mutations may lead to hepatic steatosis and fibrosis despite metabolic differences in congenital hypocholesterolemia. J Hepatol. 2014;61(4):891–902.
Donati B, Valenti L. Telomeres, NAFLD and chronic liver disease. Int J Mol Sci. 2016;17(3):383.
Donati B, Pietrelli A, Pingitore P, Dongiovanni P, Caddeo A, Walker L, et al. Telomerase reverse transcriptase germline mutations and hepatocellular carcinoma in patients with nonalcoholic fatty liver disease. Cancer Med. 2017;6(8):1930–40.
Calado RT, Regal JA, Kleiner DE, Schrump DS, Peterson NR, Pons V, et al. A spectrum of severe familial liver disorders associate with telomerase mutations. PLoS One. 2009;4(11):e7926.
Pericleous M, Kelly C, Wang T, Livingstone C, Ala A. Wolman’s disease and cholesteryl ester storage disorder: the phenotypic spectrum of lysosomal acid lipase deficiency. Lancet Gastroenterol Hepatol. 2017;2(9):670–9.
Bruce KD, Cagampang FR, Argenton M, Zhang J, Ethirajan PL, Burdge GC, et al. Maternal high-fat feeding primes steatohepatitis in adult mice offspring, involving mitochondrial dysfunction and altered lipogenesis gene expression. Hepatology. 2009;50(6):1796–808.
Breij LM, Kerkhof GF, Hokken-Koelega AC. Accelerated infant weight gain and risk for nonalcoholic fatty liver disease in early adulthood. J Clin Endocrinol Metab. 2014;99(4):1189–95.
Nobili V, Marcellini M, Marchesini G, Vanni E, Manco M, Villani A, et al. Intrauterine growth retardation, insulin resistance, and nonalcoholic fatty liver disease in children. Diabetes Care. 2007;30(10):2638–40.
Suomela E, Oikonen M, Pitkänen N, Ahola-Olli A, Virtanen J, Parkkola R, et al. Childhood predictors of adult fatty liver. The cardiovascular risk in young Finns study. J Hepatol. 2016;65:784–90.
Valenti L, Romeo S. Destined to develop NAFLD? The predictors of fatty liver from birth to adulthood. J Hepatol. 2016;65(4):668–70.
Murphy SK, Yang H, Moylan CA, Pang H, Dellinger A, Abdelmalek MF, et al. Relationship between methylome and transcriptome in patients with nonalcoholic fatty liver disease. Gastroenterology. 2013;145(5):1076–87.
Kitamoto T, Kitamoto A, Ogawa Y, Honda Y, Imajo K, Saito S, et al. Targeted-bisulfite sequence analysis of the methylation of CpG islands in genes encoding PNPLA3, SAMM50, and PARVB of patients with non-alcoholic fatty liver disease. J Hepatol. 2015;63(2):494–502.
Cheung O, Puri P, Eicken C, Contos MJ, Mirshahi F, Maher JW, et al. Nonalcoholic steatohepatitis is associated with altered hepatic microRNA expression. Hepatology. 2008;48(6):1810–20.
Gerhard GS, DiStefano JK. Micro RNAs in the development of non-alcoholic fatty liver disease. World J Hepatol. 2015;7(2):226–34.
Hsu SH, Wang B, Kota J, Yu J, Costinean S, Kutay H, et al. Essential metabolic, anti-inflammatory, and anti-tumorigenic functions of miR-122 in liver. J Clin Invest. 2012;122(8):2871–83.
Csak T, Bala S, Lippai D, Satishchandran A, Catalano D, Kodys K, et al. microRNA-122 regulates hypoxia-inducible factor-1 and vimentin in hepatocytes and correlates with fibrosis in diet-induced steatohepatitis. Liver Int. 2015;35(2):532–41.
Pirola CJ, Fernandez Gianotti T, Castano GO, Mallardi P, San Martino J, Mora Gonzalez Lopez Ledesma M, et al. Circulating microRNA signature in non-alcoholic fatty liver disease: from serum non-coding RNAs to liver histology and disease pathogenesis. Gut. 2015;64(5):800–12.
Zarrinpar A, Gupta S, Maurya MR, Subramaniam S, Loomba R. Serum microRNAs explain discordance of non-alcoholic fatty liver disease in monozygotic and dizygotic twins: a prospective study. Gut. 2016;65(9):1546–54.
Valenti L, Bugianesi E, Pajvani U, Targher G. Nonalcoholic fatty liver disease: cause or consequence of type 2 diabetes? Liver Int. 2016;36(11):1563–79.
Stender S, Smagris E, Lauridsen BK, Kofoed KF, Nordestgaard BG, Tybjaerg-Hansen A, et al. Relationship between genetic variation at PPP1R3B and liver glycogen and triglyceride levels. Hepatology. 2018;67(6):2182–95.
Nobili V, Bedogni G, Donati B, Alisi A, Valenti L. The I148M variant of PNPLA3 reduces the response to Docosahexaenoic acid in children with non-alcoholic fatty liver disease. J Med Food. 2013;16(10):957–60.
Scorletti E, West AL, Bhatia L, Hoile SP, McCormick KG, Burdge GC, et al. Treating liver fat and serum triglyceride levels in NAFLD, effects of PNPLA3 and TM6SF2 genotypes: results from the WELCOME trial. J Hepatol. 2015;63(6):1476–83.
Ma J, Hennein R, Liu C, Long MT, Hoffmann U, Jacques PF, et al. Improved diet quality associates with reduction in liver fat, particularly in individuals with high genetic risk scores for nonalcoholic fatty liver disease. Gastroenterology. 2018;155(1):107–17.
Donati B, Dongiovanni P, Romeo S, Meroni M, McCain M, Miele L, et al. MBOAT7 rs641738 variant and hepatocellular carcinoma in non-cirrhotic individuals. Sci Rep. 2017;7(1):4492.
Dongiovanni P, Petta S, Mannisto V, Mancina RM, Pipitone R, Karja V, et al. Statin use and non-alcoholic steatohepatitis in at risk individuals. J Hepatol. 2015;63(3):705–12.
Guzman CB, Duvvuru S, Akkari A, Bhatnagar P, Battioui C, Foster W, et al. Coding variants in PNPLA3 and TM6SF2 are risk factors for hepatic steatosis and elevated serum alanine aminotransferases caused by a glucagon receptor antagonist. Hepatol Commun. 2018;2(5):561–70.
Pillai S, Duvvuru S, Bhatnagar P, Foster W, Farmen M, Shankar S, et al. The PNPLA3 I148M variant is associated with transaminase elevations in type 2 diabetes patients treated with basal insulin peglispro. Pharmacogenomics J. 2018;18(3):487–93.
Eriksson JW, Lundkvist P, Jansson PA, Johansson L, Kvarnstrom M, Moris L, et al. Effects of dapagliflozin and n-3 carboxylic acids on non-alcoholic fatty liver disease in people with type 2 diabetes: a double-blind randomised placebo-controlled study. Diabetologia. 2018;61:1923.
Valenti L, Dongiovanni P. Mutant PNPLA3 I148M protein as pharmacological target for liver disease. Hepatology. 2017;66(4):1026–8.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Valenti, L., Pelusi, S. (2020). The Natural History of NAFLD: Environmental vs. Genetic Risk Factors. In: Bugianesi, E. (eds) Non-Alcoholic Fatty Liver Disease. Springer, Cham. https://doi.org/10.1007/978-3-319-95828-6_7
Download citation
DOI: https://doi.org/10.1007/978-3-319-95828-6_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-95827-9
Online ISBN: 978-3-319-95828-6
eBook Packages: MedicineMedicine (R0)