
Abstract
Purpose of Review
The hyper IgE syndromes (HIES) comprise a group of rare primary immunodeficiency disorders (PIDDs), which are characterized by extremely high serum IgE levels, eczema, recurrent skin and pulmonary infections. Both autosomal dominant (AD) HIES due to STAT3 mutations and autosomal recessive (AR) HIES due to PGM3, SPINK5, DOCK8 and TKY2 mutations have been reported. Here, we aim to summarize and compare the major clinical manifestations of different subtypes of HIES. We will also discuss otitis media, which usually do not get enough attention in HIES. Update and familiarity with these clinical features will help to make a better diagnose, assessment and treatment of HIES.
Recent Findings
Although hyper serum IgE levels have been identified in PGM3 deficiency and Comel–Netherton syndrome, PGM3 and SPINK5 genes were not included in the list of genetic etiologies of AR-HIES by the Expert Committee of the International Union of Immunological Societies until 2015. The identification of these HIES-causing genes greatly promoted the pathogenic mechanism studies of HIES. Also, in recent years, more clinical manifestations, which were often not of concern in HIES patients, have been shown to be highly related to HIES. For example, a significantly high frequency of vascular and gastrointestinal abnormities has been reported in STAT3-deficient AD-HIES patients. These new findings might help to provide new clues to the functional study of these HIES-related genes.
Summary
This review summarizes and compares the major clinical manifestations of different subtypes of HIES, and we suggest that the incidence and severity of otitis media should not be underestimated in HIES patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The hyperimmunoglobulin E syndromes (HIES; OMIM no. 147060) comprise a group of rare primary immunodeficiency disorders (PIDDs), which are characterized by extremely high serum IgE levels, eczema, and recurrent skin and pulmonary infections [1]. In 1966, HIES was firstly reported by Davis et al. [2] as Job’s syndrome in two unrelated girls with recurrent “cold” staphylococcal abscesses and pulmonary disease. The term of “hyper-IgE syndrome” was introduced by Buckley et al. in 1972, in which two patients with Job’s syndrome were reported to have very high serum IgE levels [3].
Both autosomal dominant (AD) and autosomal recessive (AR) modes of inheritance have been reported in HIES patients, of which AD-HIES was the most common form of this disease [4]. In addition to the typical characteristic of HIES mentioned above, patients with AD-HIES have also been reported to have some unique clinical manifestations, such as distinctive facial features, retained primary teeth and skeletal/connective tissue abnormalities, which are rarely reported in patients with AR-HIES [1]. The HIES-causing gene was not identified until 2006 when a homozygous deletion in Tyrosine kinase (TYK)2 was first reported in a boy with elevated IgE, eczema and recurrent infections [5], and the TYK2 gene was identified as a HIES-causing gene. Then, in 2007, dominant-negative mutations in the signal transducer and activator of the transcription factor 3 (STAT3) gene were identified as leading to AD-HIES [6]. Subsequently, in 2009, both homozygous and compound heterozygous mutations in the DOCK8 gene were identified in patients with AR-HIES [7]. More recently, the genes encoding phosphoglucomutase (PGM)3 and serine protease inhibitor Karzal type (SPINK)5, whose mutations lead to PGM3 deficiency and Comel–Netherton syndrome, respectively, were also included in the list of genetic etiologies of AR-HIES [8••]. The identification of these disease-causing genes greatly promoted the pathogenic mechanism studies of HIES.
In fact, HIES patients caused by different genetic etiologies share some clinical manifestations on the one hand, while, on the other hand, different subtypes of HIES have some their characteristic clinical features. In this review, we will mainly focus on the clinical features, including otitis media, of HIES.
Cutaneous Manifestations
Most HIES patients were reported to have a history of a newborn rash. The newborn rash usually started within the first month after birth and presented as the first clinical manifestation of HIES [9]. Eosinophilic infiltrate may be found by biopsy. The newborn rash usually starts at the face or scalp as pink papules, then developed into pustules within a few days of onset. Typically, the rash may ooze and spread to other parts of the body, then progress to include other parts of the body. In many cases, the rash has a protracted course that develops into eczema, often worsened by Staphylococcus aureus infection, while in others, there can be an improvement or resolution following treatment with oral antibiotics or topical hydrocortisone cream. Control of eczema is typically most efficient with antistaphylococcal therapy, either with antimicrobials or antiseptics. Although the frequency and severity of the newborn rash varies in HIES due to different gene deficiency, the early onset, severe newborn rash and eczema are considered as shared features of both AD- and AR-HIES [10, 11].
Atopic dermatitis (AD) is clinically diagnosed based on chronic pruritic eczema with a distinct appearance and distribution. Because of impaired innate immunity and the barrier function of the skin, patients with AD are at high risk for disseminated viral infections and bacterial colonization, resulting in staphylococcal skin infections [12]. The rash observed in HIES has been reported to be very similar to AD clinically and histopathologically [13]. Erlewyn-Lajeunesse et al. mentioned that the distribution of the rash in HIES was atypical for atopic dermatitis [14, 15•]. Eberting et al. reported that a majority of (28/43, 65%) patients with a clinical diagnosis of HIES fulfilled the criteria for AD [10]. Therefore, differentiating HIES from AD is important for giving optimal treatment at an early age before serious complications occur [12]. The following are some differences between HIES and AD. Firstly, the distribution and timing of skin symptoms is different between HIES and AD. The skin involvement is usually located in the face and extensor surfaces in HIES patients and has an early onset before 1 month of age, while it is usually located in flexural surfaces of the body in AD and has a relatively late onset after 2–4 months of age. Secondly, AD is often associated with a history of other allergic disorders such as food allergy, asthma, and allergic rhinitis, which is rarely reported in AD-HIES. Thirdly, staphylococcal infections in AD are usually superficial, whereas deep-seated abscesses are prone to be developed in HIES patients. Fourthly, mucocutaneous candidiasis is also occurs frequently in HIES, but not in AD. In fact, although skin manifestations in HIES are not confined to AD-like skin lesions, the dermatitis in HIES invariably becomes infected or colonized with Staphylococcus aureus, thus providing the necessary conditions for an “autosensitization reaction” and a generalization of more severe dermatitis. Although these acute episodes of infection can be resolved, recurrences are often reported if patients fail to take prophylactic antibiotics [11].
Recurrent skin infections caused by bacterial, virus and fungi are also commonly observed in HIES patients. As described in the original report of Job’s syndrome, “cold’ abscesses caused mainly by S. aureus, which are characterized by a lack of pain and insufficient inflammatory response than expected of typical staphylococcal abscesses, is a typical feature of AD-HIES [16]. The abscess may be large and some patients need surgical incision and drainage [4]. A considerable number of AD-HIES patients are reported to have “cold’ abscesses, for example, up to 73% patients in a large French cohort study and 52.94% patients in our previous study in China [4, 17]. Unlike AD-HIES, AR-HIES has an increased susceptibility to typical staphylococcal skin abscesses [16]. In addition, most AR-HIES patients with DOCK8 deficiency have been reported to have recalcitrant, widespread and difficult-to-control cutaneous viral infections, which are absent in AD-HIES patients with STAT3 mutations. The most common viruses involved in DOCK8 deficiency are herpes simplex virus (HSV), human papillomavirus (HPV), molluscum contagiosum virus (MCV), and varicella-zoster virus [18]. In addition, HIES patients are susceptible to chronic mucocutaneous candidiasis, including thrush, onychomycosis and genitalia infection, which are usually effectively managed with a combination of oral and topical antifungal agents [13].
Pulmonary Manifestations
Infectious disease is the hallmark of primary immunodeficiency diseases, in which upper and/or lower respiratory tract infections have been most commonly reported in HIES patients [19]. Pneumonia often occurs in early childhood, and the most common infected organisms are Staphylococcus aureus, Streptococcus pneumoniae and Haemophilus influenzae. However, pneumonia has rarely been reported in patients with SPINK5 mutations [20]. It has been reported that AD-HIES patients had exaggerated parenchymal damage, thus leading to high incidence of pulmonary cysts (pneumatoceles) following pneumonia [21]. The incidence of pneumatoceles in AD-HIES patients was as high as 41.18% in our previous study and was 52% in a French national study [4, 17]. Although pneumatocele formation was previously presumed to be pathognomonic for STAT3-associated HIES, a considerable number of HIES patients with PGM3 mutations have been reported to suffer from pneumatocele [22]. In addition, pneumatocele has also been reported in a few patients with DOCK8 mutations. Secondary bronchiectasis has also been commonly reported in HIES patients following pneumonia. For patients with DOCK8 deficiency, although rare pneumatocele formation has been observed, a significantly high incidence of bronchiectasis has been reported [23]. High incidence of bronchiectasis caused by recurrent pulmonary infections has also been frequently observed in patients with PGM3 deficiency [22]. Prophylaxis combining oral antibiotics and IgG injections seems to be particularly beneficial in patients with recurrent pulmonary infections, and antifungal prophylaxis should be offered to patients with pneumatocele or bronchiectasis [17].
Otitis Media
Otitis media is one of the most common infections in childhood. It is reported that up to 80% of children have suffered from at least one episode of otitis media within their first 3 years of life. Antibiotic treatment is usually effective and thus it is not considered to be a worrisome problem [24]. The different anatomy and the immature immune system of the eustachian tube contributes to the increased incidence of otitis media in children. Other factors, such as adenoidal hypertrophy, parental smoking, breast feeding and diet, are also reported to be correlated with the occurrence of acute otitis media in children [25]. However, in some cases, otitis media is an early manifestation of a severe underlying disease and should not be underestimated [26•]. It has been suggested that suspicions of immunodeficiency should increase when ear infections are frequent, suppurative, unresponsive to antibiotics, caused by unusual organisms, or seen in the context of other frequent infections, severe eczema, or failure to thrive [25]. In fact, recurrent otitis media has been considered as one of the most important warning signs of PIDDs, including selective IgA deficiency, common variable immunodeficiencies (CVID), severe combined immunodeficiencies, HIES and so on [26•].
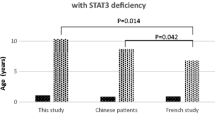
Most AD-HIES patients have been reported to have a history of otitis media. In our previous single-center studies of Chinese HIES patients, otitis media was one of the most common infectious diseases and about half the patients had suffered from recurrent otitis media [4]. The incidence was up to 73% in a French national AD-HIES study [17]. However, many reports in the literature related to AR-HIES did not pay enough attention to otitis media, and the incidence and the importance of it seems to be underestimated. Zhang et al. reported that 8 of 9 AR-HIES patients with PGM3 mutations suffered from otitis, including 3 of recurrent acute otitis media and 6 of chronic otitis externa, respectively [27]. Pedersen et al. reported that all three PGM3-HIES patients had recurrent otitis media [28]. Engelhardt et al. observed 6 of 27 (22.2%) DOCK8-deficient AR-HIES patients suffered from recurrent or chronic otitis media, and 7 of 27 patients (25.9%) were reported to have recurrent or suppurative otitis, while lacking further detailed information [7]. The first reported TYK2-deficient AR-HIES patient was also mentioned to have recurrent otitis media [5]. Furthermore, some HIES patients with otitis media were reported have progressive infections leading to complications such as mastoiditis and external otitis [29]. In fact, studies have shown that humoral immune deficiencies were most likely to present with recurrent otitis media as the chief complaint. In our single-center study, the incidence of recurrent otitis media was 41.5% in 174 patients with X-linked agammaglobulinemia [30]. In an analysis of CVID patients, 69% patients with relapsing or chronic otitis were reported [31]. It seems that the incidence of otitis media in HIES patients, especially AD-HIES patients, is not lower than these humoral immune-deficient disorders. Therefore, we suggest that the incidence and severity of otitis media should not be underestimated in HIES patients.
In addition, other ear, nose, and throat infections, including recurrent and chronic sinusitis, severe pharyngitis, laryngitis, and/or rhinitis tonsillitis, have also been reported in HIES patients [29] .
Gastrointestinal Manifestations
A majority of AD-HIES subjects develope gastrointestinal (GI) manifestations as part of their disease. A high overall rate of eosinophilic infiltration throughout the GI tract has been observed in AD-HIES, with the most common manifestations being gastroesophageal reflux disease, dysphagia, and abdominal pain. In addition, GI infections, including esophageal candidiasis, esophageal cryptococcus, and esophageal EBV, have been observed in some AD-HIES patients. These findings suggest that the STAT3 pathway might be involved in the pathogenesis of some GI disorders [32]. In addition, GI infections, which were usually accompanied by acute or chronic diarrhea, have also been reported in some AR-HIES patients with DOCK8, TYK2 or PGM3 mutations, but rarely in patients with SPINK5 mutations [7, 22, 33].
Somatic Non-immunological features
It is thought that AD-HIES can be distinguished from other type of HIES by its characteristic facial features, distinctive connective tissue, skeletal, and dental abnormalities [34]. Characteristic facial features have been observed in almost all AD-HIES patients with STAT3 mutations, including a high and prominent forehead, prognathism or retrognathism, enlargement of the interalar distance, thickening of the soft tissues of the ear or nose, high arched palate, and so on. Usually, the characteristic facial features clearly tend to increase with age. In addition, retention of primary teeth occurs in most AD-HIES patients, which may impair secondary dentition emergence [34]. Connective tissue and skeletal abnormalities in AD-HIES patients include osteoporosis, minimal trauma fractures, scoliosis, hyperextensibility of joints, degenerative spine disease, and craniosynostosis [34, 35]. In recent years, it has been reported that some patients with mutations in PGM3 also have characteristic dysmorphic face, musculoskeletal, and connective tissue manifestations. Characteristic face with wide nostrils and prominent lips, as well as scoliosis, have been observed in patients with PGM3 mutations [22]. However, these facial features, connective tissue, skeletal, and dental abnormalities are infrequent in other etiology of HIES [36].
Neurologic impairment from early life is a typical manifestation of HIES patients with PGM3 mutations. The most frequent clinical symptom is a developmental delay, ataxia, low intelligence quotient, followed by psychomotor retardation, hypotonia, dysarthria, sensorineural hearing loss, and myoclonus [22, 27, 28]. However, only a minority of patients with developmental delay and psychomotor retardation have been reported in other subtypes of HIES. In DOCK8-deficient patients, the central nervous system involvement includes vasculitis, brain infarction, meningitis, and so on [23].
Vascular anomalies and their associated vascular events have been reported to contribute to significant morbidity and mortality in HIES patients. Vascular abnormalities have been reported in all subtypes of HIES, including aneurysms (coronary, aortic, carotid and cerebral), pseudoaneurysms, congenital patent ductus venosus, superior vena cava syndrome, vasculitides, vascular ectasia, thrombosis and others [37]. Subsequently, a further study has reported that vascular abnormalities are highly prevalent in the STAT3-deficient AD-HIES adult patients by cardiac CT and MRI. Coronary artery tortuosity or dilation have been identified in 70% and aneurysms in 37% of STAT3-mutated HIES patients, which indicate that STAT3 is involved in vascular remodeling [38].
Malignancy
A history of hematologic malignancy has been reported in some AD-HIES patients with STAT3 mutations, including non-Hodgkin lymphoma (NHL), Burkitt lymphoma, disseminated anaplastic lymphoma kinase-negative anaplastic NHL and diffuse large B-cell lymphoma. Chemotherapy was reported to be effective in these patients. Other reported malignancies in AD-HIES include leukemia and cancers of the vulva, liver, and lung [34]. Malignancies are more commonly reported in DOCK8 deficiency than in AD-HIES. About 10–36% of patients with DOCK8 deficiency develop cancers, with squamous cell carcinomas, Burkitt lymphoma and diffuse large B-cell lymphoma occurring frequently [21]. The squamous cell carcinomas in DOCK8 deficiency are most likely related to HPV and difficult to cure [34]. In addition, patients with PGM3 mutations have also been reported to suffer from lymphoma [22].
Laboratory Findings
Individuals with HIES typically have quite elevated serum IgE levels. However, there is no clear correlation between disease severity and the serum level of IgE [34]. A value of 2000 U/ml is usually taken as a cut-off point, which has proved helpful in establishing a definitive diagnosis of AD-HIES [39]. Interestingly, serum IgE levels in some AD-HIES patients may decrease with age and fall within a normal range, while the severity of infectious complications in patients with AD-HIES do not correlate with serum IgE levels [39]. In addition to high serum IgE levels, eosinophilia is also commonly reported in AD-HIES patients due to STAT3 mutations and in AR-HIES patients with DOCK8 and PGM3 mutations. In addition, although total lymphocyte counts are usually normal in AD-HIES patients, a further subset analysis has shown that memory T and B cells are decreased in these patients. Moreover, patients with DOCK8 mutations are frequently reported to have lymphopenia, with T, B and NK cells all decreasing [34]. Decreases in serum IgM and specific antibodies are also frequently observed in patients with DOCK8 deficiency.
Comparing the Major Clinical Manifestation of Subtypes of HIES Due to STAT3, PGM3, SPINK5, DOCK8 or TYK2 Mutations
The hyper-immunoglobulin E syndromes are a group of rare multisystem primary immunodeficiency diseases caused by mutations in STAT3, PGM3, SPINK5, DOCK8 or TYK2. Although some clinical manifestations are similar in all subtypes of HIES, such as elevated serum IgE levels, eczema and recurrent skin and lung infections, different gene phenotypes are correlated with distinct disease manifestations characteristic for each subtype of HIES (Table 1).
AD-HIES patients characteristically have some nonimmunological features, such as distinctive facial features, retention of primary teeth, abnormal bone fractures, hyperextensibility and scoliosis. In addition, these patients also characteristically have ‘cold’ abscesses in skin and soft tissue, which are atypical staphylococcal abscesses lacking warmth and tenderness. In addition, most STAT3-HIES patients suffer from pneumatocele and bronchiectasis formation following recurrent pneumonia caused by pyogenic bacteria. Moreover, chronic mucocutaneous candidiasis due to reduced T17 responses, which occur in a large proportion of STAT3-HIES patients, is another hallmark of AD-HIES [40].
HIES caused by DOCK8 mutations account for the majority patients with AR-HIES. The shared manifestation of STAT3 and DOCK8 deficiency include high serum IgE levels, eosinophilia, eczema, recurrent staphylococcal skin abscesses, frequent pneumonia and candidiasis. However, DOCK8-HIES patients often suffer from severe and refractory cutaneous viral infections and various allergic diseases, including asthma, allergies to food and drugs and environmental allergies. In addition, DOCK8-HIES patients rarely develop pneumatocele as sequelae of their recurrent pneumonias [41]. AR-HIES patients due to PGM3 deficiency, can be discriminated from other subtypes of HIES by their neurological involvement, such as developmental delay, low intelligence quotient, failure to thrive, and psychomotor retardation. PGM3-HIES patients have also been reported to have high frequencies of eczema, atopic dermatitis, rhinitis, and multiple allergies [22]. AR-HIES patients with SPINK5 mutations characteristically have localized or generalized congenital ichthyosis, and hair shaft abnormalities, which are absent in other subtypes of HIES [42]. However, there is limited information about patients with TYK2 deficiency. Up to now, only two AR-HIES patients due to TYK2 mutations have been reported. Both patients had disseminated mycobacterial infection after BCG vaccination, recurrent sinopulmonary infections, and cutaneous viral infection with HPV, MCV or HSV, although with variable severity [21].
Conclusion
The hyper-IgE syndrome is a rare multisystemic disorder with a broad constellation of clinical manifestations. Clinical diagnosis criteria have been established in AD-HIES patients with STAT3 mutations, which is not suitable for AR-HIES patients. Therefore, genetic diagnosis is still considered to be the most important way to distinguish HIES due to different gene mutations. In addition, although otitis media is one of the most common infections in childhood, recurrent, suppurative and refractory otitis media is an important warning sign of congenital immunodeficiencies, including HIES.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Hashemi H, Mohebbi M, Mehravaran S, Mazloumi M, Jahanbaniardakani H, Abtahi SH. Hyperimmunoglobulin E syndrome: genetics, immunopathogenesis, clinical findings, and treatment modalities. J Res Med Sci. 2017;22(1):53.
Davis SD, Schaller SJ, Wedgwood RJ. Job’s Syndrome : recurrent, "cold", staphylococcal abscesses. Lancet. 1966;287(7445):1013–5.
Buckley R. Extreme hyperimmunoglobulinemia E and undue susceptibility to infection. Pediatrics. 1972;49(1):59–70.
Wu J, Chen J, Tian ZQ, Zhang H, Gong RL, Chen TX, et al. Clinical manifestations and genetic analysis of 17 patients with autosomal dominant Hyper-IgE Syndrome in Mainland China: new reports and a literature review. J Clin Immunol. 2017;37(2):166–79.
Minegishi Y, Saito M, Morio T, Watanabe K, Agematsu K, Tsuchiya S, et al. Human tyrosine kinase 2 deficiency reveals its requisite roles in multiple cytokine signals involved in innate and acquired immunity. Immunity. 2006;25(5):745–55.
Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. 2007;448(7157):1058.
Engelhardt KR, Mcghee S, Winkler S, Sassi A, Woellner C, Lopez-Herrera G, et al. Large deletions and point mutations involving the dedicator of cytokinesis 8 (DOCK8) in the autosomal-recessive form of hyper-IgE syndrome. J Allergy Clin Immunol. 2009;124(6):1289–302.e4.
•• Picard C, Bobby GH, Alherz W, Bousfiha A, Casanova JL, Chatila T, et al. International Union of Immunological Societies: 2017 Primary Immunodeficiency Diseases Committee Report on Inborn Errors of Immunity. J Clin Immunol. 2018;38(1):96–128. COMMENT: This report details the categorization and listing of 354 inborn errors of immunity.
Gernez Y, Tsuang A, Smith TD, Shahjehan K, Hui Y, Maglione PJ, et al. Hemoptysis in a patient with elevated immunoglobulin E. J Allergy Clin Immunol Pract. 2016;4(6):1054–8. https://doi.org/10.1016/j.jaip.2016.08.003.
Eberting CL, Davis J, Puck JM, Holland SM, Turner ML. Dermatitis and the newborn rash of hyper-IgE syndrome.Digest of the World Core Medical Journals. Arch Dermatol. 2005;140(9):1119–25.
Minegishi Y, Saito M. Cutaneous manifestations of Hyper IgE syndrome. Allergol Int. 2012;61(2):191–6. https://doi.org/10.2332/allergolint.12-RAI-0423.
Schimke LF, Sawallebelohradsky J, Roesler J, Wollenberg A, Rack A, Borte M, et al. Diagnostic approach to the hyper-IgE syndromes: immunologic and clinical key findings to differentiate hyper-IgE syndromes from atopic dermatitis. J Allergy Clin Immunol. 2010;126(3):611–7.e1.
Minegishi Y, Saito M. Cutaneous manifestations of Hyper IgE Syndrome. Allergol Int. 2002;141(4):572–5.
Erlewyn-Lajeunesse MDS. Hyperimmunoglobulin-E syndrome with recurrent infection: a review of current opinion and treatment. Pediatr Allergy Immunol. 2010;11(3):133–41.
• Pichard DC, Freeman AF, Cowen EW. Primary immunodeficiency update: Part I. Syndromes associated with eczematous dermatitis. J Am Acad Dermatol. 2015;73(3):365–6. COMMENT: Discusses clinical manifestations of HIES caused by STAT3, DOCK8 and PGM3, especially eczematous dermatitis.
Lehman H. Skin manifestations of primary immune deficiency. Clin Rev Allergy Immunol. 2014;46(2):112–9. https://doi.org/10.1007/s12016-013-8377-8.
Chandesris MO, Melki I, Natividad A, Puel A, Fieschi C, Yun L, et al. Autosomal dominant STAT3 deficiency and Hyper-IgE Syndrome molecular, cellular, and clinical features from a French National Survey. Medicine. 2012;91(4):1–19.
Chu EY, Freeman AF, Jing H, Cowen EW, Davis J, Su HC, et al. Cutaneous manifestations of DOCK8 deficiency syndrome. Arch Dermatol. 2012;148(1):79–84.
Yong PF, Freeman AF, Engelhardt KR, Holland S, Puck JM, Grimbacher B. An update on the hyper-IgE syndromes. Arthritis Res Ther. 2012;14(6):228.
Sarri CA, Roussakischulze A, Vasilopoulos Y, Zafiriou E, Patsatsi A, Stamatis C, et al. Netherton syndrome: a genotype-phenotype review. Mol Diagn Ther. 2016;21(2):1–16.
Mogensen TH. Primary immunodeficiencies with elevated IgE. Int Rev Immunol. 2015;35(1):39–56.
Yang L, Fliegauf M, Grimbacher B. Hyper-IgE syndromes: reviewing PGM3 deficiency. Curr Opin Pediatr. 2014;26(6):697–703.
Engelhardt KR, Gertz ME, Keles S, Schäffer AA, Sigmund EC, Glocker C, et al. The extended clinical phenotype of 64 patients with dedicator of cytokinesis 8 deficiency. J Allergy Clin Immunol. 2015;136(2):402–12.
Urschel S. Otitis media in children with congenital immunodeficiencies. Curr Allergy Asthma Rep. 2010;10(6):425.
Wilson NW, Hogan MB. Otitis media as a presenting complaint in childhood immunodeficiency diseases. Curr Allergy Asthma Rep. 2008;8(6):519–24.
• Costa-Carvalho BT, Grumach AS, Franco JL, Espinosa-Rosales FJ, Leiva LE, King A, et al. Attending to warning signs of primary immunodeficiency diseases across the range of clinical practice. J Clin Immunol. 2014;34(1):10–22. COMMENT: Summarizes the warning signs of PIDDs, including otitis media.
Fleisher TA. Autosomal recessive phosphoglucomutase 3 (PGM3) mutations link glycosylation defects to atopy, immune deficiency, autoimmunity, and neurocognitive impairment. J Allergy Clin Immunol. 2014;133(5):1400–9.e5.
Stray-Pedersen A, Backe P, Sorte H, Mørkrid L, Chokshi N, Erichsen HC, et al. PGM3 mutations cause a congenital disorder of glycosylation with severe immunodeficiency and skeletal dysplasia. Am J Hum Genet. 2014;95(1):96–107.
Su HC. Dedicator of cytokinesis 8 (DOCK8) deficiency. Curr Opin Allergy Clin Immunol. 2010;10(6):515–20.
Chen XF, Wang WF, Zhang YD, Zhao W, Wu J, Chen TX. Clinical characteristics and genetic profiles of 174 patients with X-linked agammaglobulinemia: report from Shanghai, China (2000–2015). Medicine. 2016;95(32):e4544.
Urschel S, Kayikci L, Wintergerst U, Notheis G, Jansson A, Belohradsky BH. Common variable immunodeficiency disorders in children: delayed diagnosis despite typical clinical presentation. J Pediatr. 2009;154(6):888–94.
Arora M, Bagi P, Strongin A, Heimall J, Zhao X, Lawrence MG, et al. Gastrointestinal manifestations of STAT3-deficient hyper-IgE syndrome. J Clin Immunol. 2017;37(7):1–6.
Kilic SS, Hacimustafaoglu M, Boissondupuis S, Kreins AY, Grant AV, Abel L, et al. A patient with tyrosine kinase 2 deficiency without hyper-IgE syndrome. J Pediatr. 2012;160(6):1055–7.
Freeman AF, Holland SM. Clinical manifestations of hyper IgE syndromes. Dis Markers. 2010;29(3-4):123.
Sowerwine KJ, Holland SM, Freeman AF. Hyper-IgE syndrome update. Ann N Y Acad Sci. 2012;1250(1):25–32.
Sanal O, Jing H, Ozgur T, Ayvaz D, Straussalbee DM, Ersoyevans S, et al. Additional diverse findings expand the clinical presentation of DOCK8 deficiency. J Clin Immunol. 2012;32(4):698–708.
Yavuz H, Chee R. A review on the vascular features of the hyperimmunoglobulin E syndrome. Clin Exp Immunol. 2010;159(3):238–44.
Chandesris MO, Azarine A, Ong KT, Taleb S, Boutouyrie P, Mousseaux E, et al. Frequent and widespread vascular abnormalities in human STAT3 deficiency. Artery Res. 2011;5(4):163.
Szczawinskapoplonyk A, Kycler Z, Pietrucha B, Heropolitanskapliszka E, Breborowicz A, Gerreth K. The hyperimmunoglobulin E syndrome - clinical manifestation diversity in primary immune deficiency. Orphanet J Rare Dis. 2011;6(1):1–11.
Farmand S, Sundin M. Hyper-IgE syndromes: recent advances in pathogenesis, diagnostics and clinical care. Curr Opin Hematol. 2015;22(1):12–22.
Aydin SE, Kilic SS, Aytekin C, Kumar A, Porras O, Kainulainen L, et al. DOCK8 deficiency: clinical and immunological phenotype and treatment options - a review of 136 patients. J Clin Immunol. 2015;35(2):189.
Sun JD, Linden KG. Netherton syndrome: a case report and review of the literature. Digest of the World Core Medical Journals. Int J Dermatol. 2010;45(6):693–7.
Grant numbers
This research was supported by grants from National Natural Science Foundation of China (81571605 and 81701626).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare no conflicts of interest relevant to this manuscript.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Otitis
Rights and permissions
About this article

Cite this article
Wu, J., Hong, L. & Chen, TX. Clinical Manifestation of Hyper IgE Syndrome Including Otitis Media. Curr Allergy Asthma Rep 18, 51 (2018). https://doi.org/10.1007/s11882-018-0806-6
Published:
DOI: https://doi.org/10.1007/s11882-018-0806-6