
Abstract
We have parametrized an AMBER force field in a TEAMFF database to accurately represent intra- and intermolecular interactions of cellulose. Parameters are obtained by fitting quantum mechanics (QM) energetic data on a training set of 12 simple and substituted heterocycles, alcohols, ethers, and saccharides. The temperature-dependent Lennard–Jones parameters were optimized by fitting experimental data of 23 molecular liquids at different temperatures using molecular dynamics (MD) simulations. Validation on cellobiose, the monomer of cellulose, exhibits excellent agreement between the data obtained by molecular mechanics calculations using the present force field and QM calculations in terms of conformational energies, structures, and vibrational frequencies. The MD simulation results of cellulose Iβ crystal agree well with the experimental data, showing a 0.1% deviation in density and less than 1% deviations in all the cell parameters.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cellulose is one of the most naturally abundant biopolymers, with approximately 700 billion tons produced every year.1 It is the main component of algae and plant cell walls, vegetable tissues such as wood and cotton, and, after processing, paper.2 In material science and engineering, cellulose has a particular interest for its uses as a biofuel,3,4,5,6 its mechanical,7,8,9,10 and superhydrophobic11,12,13,14,15,16 properties, and its amphiphilic character and behaviors with ionic liquids.1,17,18,19,20,21
Experimental investigation of cellulose and cellulose fibers at a molecular level began as early as 1912 by von Laue.22 The first complete crystallographic unit was proposed in 1928 by Mayer and Mark,23 refined in 1937 by Meyer and Misch,24 and confirmed in 1938 by Gross and Clark.22 A more detailed description of the cellulose crystal structure and its internal hydrogen bond network was established for the α and β allomorphs by Nishiyama, Langan, and Chanzy by x-ray and neutron diffraction.25 For the cellulose Iβ allomorph, they reported a monoclinic unit cell that contains two parallel cellobiose chains that form a layered structure, held together by covalent bonds in c directions, hydrophobic interactions in a, and hydrogen bonds in b.
Using a force field approach to study cellulose, like other carbohydrates, remains challenging. Several all-atom force fields (AAFF)26,27,28,29,30,31,32 have been published, and their parameters have been validated mostly using the conformational energies of model compounds of mono- or disaccharides. However, the predicted cellulose Iβ crystal lattice does not match the experimental values. More seriously, as pointed by Matthews et al.,33 long (up to 0.8 μs) molecular dynamics (MD) simulations of cellulose Iβ using these AAFFs converge slowly, and the results show considerable deviations in conformations and lattice parameters. Other available force fields of cellulose are the united atom34 and coarse grain models7,35 that rely on constraining the cellobiose monomer to the configuration found experimentally.
The difficulty of developing an accurate AAFF for cellulose originates from numerous hydroxyl substituents, forming intra- and intermolecular hydrogen bond networks. In addition, the glucosidic (-O-) bonds between two glucose rings are relatively flexible. As a result, the potential energy surface (PES) is complex with multiple minima and conformers often in equilibrium with each other.36 It has been reported that the conformational changes in cellobiose (disaccharide) take place on a microsecond time-scale,26 and result in significant fluctuations of the internal degrees of freedom and changes in the intra-ring hydrogen bonds.37 In the solid, the intermolecular hydrogen bonds and van de Walls interactions, coupled with the intramolecular interactions, control the form of packing. Therefore, a good force field must accurately represent both the intra- and intermolecular interactions in cellulose, and accurately describe conformational changes.
To solve this problem, in this work, we have conducted a full parameterization for cellulose. Using saccharide-related molecules as model compounds, we applied quantum mechanics (QM) density functional theory (DFT) to explore the PES for ring deformations and inter-ring rotations. From those QM calculations, an accurate force field representation of the intramolecular PES was obtained. The intermolecular interactions were initially optimized using liquid phase simulation data, and later refined with cellulose Iβ crystal data. The parameterization procedure was iterated multiple times so that the couplings between the intra- and intermolecular interactions are considered.
The force field is one of the specific force fields in the TEAM force field database (TEAMFF), which consists of multiple force field tables which are independently developed. These force fields are grouped by force field types (e.g., AMBER,38 CHARMM,31,39 CFF,40 and TEAM41). Each group has a base force field that provides generic coverage, allowing force fields developed for specific compounds to have s high accuracy. On deployment, the force fields of a group are compatible and can be used together by combining the atom types and parameters.
In the following sections, we first explain the parameterization and simulation procedures, then present and discuss the parameterization and validation results in the gas and condensed phases, and finally summarize the main contributions of this work.
Methods
Functional Form and Atom Types
In this work, we choose the AMBER functional form, with a temperature-dependent dispersion term.42 The total energy is expressed as:
where \(b, \theta , \varphi ,\chi ,\mathrm{ and }\, r\) represent the bonds, angles, dihedral angles, improper-dihedral angles, and nonbonded atom–atom distances. The first four terms are called valence terms because they are described by the connectivity of the valence bonds. The last two terms, coulombic and Lennard–Jones (LJ) 12-6 functions, are nonbond terms representing all intra- and intermolecular nonbonded interactions, including hydrogen bonds. The well depth and diameter parameters are scaled with a scaling factor \({(f}_{\mathrm{disp}})\)42 and expressed as functions of temperature, and given by:
The charge parameters, \(q,\) are expressed in terms of partial atomic charges and bond-charge increments:
The pairwise LJ12-6 potential uses the Lorentz–Berthelot combination rule:
The atom types are defined following the TEAMFF Hierarchical Atom Definition scheme that takes into account the immediate environment of each atom and the essential features of the atom, such as hybridization, coordination number, ring size, and aromaticity.41 A list of the atom types is given in the support information, Table S1.
Parameterization
The valence terms are parametrized from the QM-DFT data. Molecules that form the training set were selected using a fragment-based approach.43 The QM-DFT calculations were carried out with Gaussian0944 software at the B3LYP/6-31G(d) level. The bond-charge increment parameters were determined from QM ESP charges, while the initial LJ parameters were taken from the default TEAMFF. With the charge and the LJ parameters fixed, the valence parameters were optimized using a Levenberg–Marquardt procedure to fit the QM-DFT data, including the energies and the first and second energy derivatives for all the training set molecules.
With the valence and charge terms fixed, the LJ parameters were optimized using a MD simulation to fit the liquid state experimental data. The training set for this part of the parametrization includes cyclic ethers, cyclic alcohols, and linear molecules with similar atom sequences. For each compound of the training set, the experimental liquid density (ρ) and heat of vaporization (HoV) at different temperatures were obtained from the NIST standard reference database.45 To avoid complications due to couplings with intramolecular interactions and strong polarization, large polyolic chains and small alcohols have been excluded.29
Generally, for each molecule, several temperature points have been selected to adequately cover its thermodynamic space (above the melting point, at the boiling point, and near the critical point).42 A series of MD simulations were carried out at each temperature point to calculate the liquid densities and HoV values to optimize the van der Waals parameters. While liquid densities can be directly determined from MD simulations, the HoV values are calculated by extracting the intermolecular energy \(({E}_{\mathrm{inter}})\) according to the ideal gas approximation:
The resulting MD data were used to fit the available experimental data using a least-squares procedure. At this stage, bulk liquid MD simulations, where the liquid was represented by a simulation box with a 3-D periodic condition, were carried out with the GROMACS simulation software46 in boxes constructed with Packmol47 that contained approximately 300 molecules. The simulation boxes underwent annealing, where the temperature of the system was raised at 800 K to relax the internal strain and to randomize the initial configuration, followed by a conjugate gradient energy minimization. Then, they were equilibrated at the target temperature and pressure conditions under an isothermal-isobaric (NPT) ensemble with Langevin temperature control and Berendsen pressure control for 0.2 ns in 1 fs time step. Once the system reached equilibrium, the density and HoV were sampled during 1 ns in 2 fs time step with using a Parrinello–Rhaman barostat. During the simulations, long-range electrostatics were modulated using a particle-mesh Ewald (PME) summation and long-range van der Waals using the dispersion correction with a cutoff of 1.2 nm. Bonds with hydrogen atoms were constrained with the LINCS algorithm.
The crystal structure of the cellulose Iβ form of cellobiose was used to test and fine-tune the LJ parameters. The force field functions are commonly used and supported by different simulation software packages. While the simulations of liquids were carried out with GROMACS,46 mainly for its efficiency of simulations of amorphous systems, the simulations of crystals were performed using LAMMPS,48 for its flexibility of handling lattice structures. The initial unit cell and atom positions were taken from the x-ray and neutron diffraction data of Nishiyama et al.,25 available on the Cambridge Crystal Structure Database.49 The simulations were carried out on a \(4\times 4\times 4\) supercell, which had the size of \(31.14\times 32.60\times 41.52\) Å3, and 5,376 atoms. The periodic boundary conditions were applied in the X, Y, and Z directions. The long-range electrostatics and van der Waals cutoffs were set to be 1.2 nm. The experimental structure was relaxed by simulated annealing and conjugate gradient energy minimization. The minimization was then used to test and fine-tune the nonbond parameters to improve the fit of the lattice parameters.51 For true comparisons with the experimental data, MD simulations at the experimental temperature (298 K) and pressure (1 atm) were carried out using the same super-cell model. The equilibration was done initially by an isochoric-isothermal (NVT) simulation and then an NPT simulation, while the data collection was carried out by NPT simulation. In the NPT equilibration, the a, b, and c edges of the lattice were controlled independently with anisotropic pressure control. The data collection period was extended to 100 ns, with a time step of 2 fs; snapshots of this simulation are shown in Fig. 1 of the supporting information. As evident from the block-averaged cell parameters (Table III of the supporting information), the simulation converged under the simulation condition.

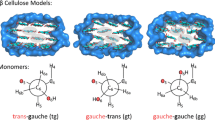
Model disaccharides. For better visualization, only hydrogen atoms involved in hydrogen bonds are shown.
After the LJ parameters were optimized by condensed phase simulations, the QM data fit was repeated to count in the small perturbations to the intramolecular interactions due to the revised LJ parameters. The entire process was repeated a couple of times to obtain the final force field parameters that yielded consistent results.
Results and Discussion
Fit QM Data
The training set for the QM calculations and the parameterization of the valence terms is shown in Table I. It includes small linear alcohols, substituted and unsubstituted heterocycles, and model disaccharides (Fig. 1) that describe the hydroxyl/oxygen and hydroxyl/hydroxyl interactions.
Linear alcohols were scanned along the O-C-C-C and the H-O-C-C dihedrals at intervals of 10° for 180°. Intra-ring hydrogen bonding between vicinal hydroxyls has been explored from single-ring compounds such as cyclohexanediol and the heterocycle tetrahydro-2H-pyran-2,3-diol. Sampling starts from two configurations, one with the hydroxyl groups located at the same side forming a 60° angle between them and internal hydrogen bonds, and the other with the hydroxyls at opposite sides at 150°, without any internal hydrogen bonding. Inter-ring hydroxyl interactions were sampled along the hydroxyl substituent and the methylhydroxyl of the model disaccharide 3 (Fig. 1).
More complex cycles, such as cyclohexanol and oxanol, have been used to sample the position of the rotations of the hydroxyl groups and its interactions with the heterocycle oxygen in a fixed chair configuration. Cyclohexanol and oxanol were scanned along the C-C-C-C/O-C-C-C and O-C-C-C/H-O-C-O directions, respectively. After fitting, carbon ring deformations and some possible hydroxyl positions were accurately captured.
The glucosidic bond was scanned from model disaccharides 1–3 along the O-C-O-C direction for 360°28,32 and refined with cellobiose. This underwent additional sampling along its conformational dihedrals and hydroxyl groups. The overall fit results of the QM training set are shown in Fig. 2 for the conformational energies and vibrational (normal mode) frequencies, and in Fig. 3 for structures in terms of bond-length, bond-angle and dihedral angles.

Comparison of energies (left) and normal mode frequencies (right) between QM calculations and MM fitting. Energies are measured in kcal/mol and frequencies in wavenumbers.

Comparison of structural parameters in bond-length (a), bond-angle (b), and torsion dihedral angle (c), obtained between QM and MM calculations from the training set. Length is measured in Å and angles in degrees.
Cyclic molecules are complex, with several possible conformations, accompanied by significant bond-length, bond-angle, and dihedral fluctuations. Here, we have chosen simple molecules that display accessible deformations which are six-membered heterocycles, such as tetrahydropyran and dioxane, to sample the ring deformations. Due to their limited independent internal degrees of freedom, a complete scanning of their PES is possible using two dihedral angles.52 As shown in Fig. 4 and Table II, all distinct conformers were obtained for the unsubstituted heterocycles, tetrahydropyran and dioxane.

Comparison of PES of tetrahydropyran (top) and dioxane (bottom) between QM and FF. Dihedrals are measured in degrees, and the energy in kcal/mol, QM data are shown on the left and the MM fit on the right. The relative energy difference between two regions of different colors is at 2 kcal/mol with zero set in the blue area (chair conformer) (Color figure online).
Fit Liquid Data
The LJ parameters were optimized from the bulk liquid densities and HoV of 24 molecules, including alcohols, ethers, and cyclic compounds (Table III). The liquid phase fitting results of the densities and HoVs (Fig. 5) show a globally good agreement with the experimental values. Due to the limited availability of high-quality HoV experimental data, there are significantly more points for densities than for HoVs. More details about the fitting results of liquid properties can be found in the supporting information SI2.

Density (left) and HoV (right) fitting results. Experimental data is on the X-axis and simulation data on Y.
Condensed phase polarizability effects are apparent both in the density, and the HoV fits. In the density fit, the largest percentage deviation of the set (− 2.5%), appears at the 2,4,6-Trimethyl-1,3,5-trioxane, commonly known as Paraldehyde, near its melting point. In the HoV fit, the largest percentage deviation (− 8.05%) is located in the ethylene glycol near its boiling point.
Validation on Cellobiose
A first-level validation concerns the monomer of glucose, cellobiose, which is a disaccharide with significant internal hydrogen bonds and flexible linkage (-O*-) between the two rings; here, O* indicates the linkage oxygen. The internal hydrogen bonding stabilizes the cellobiose conformers.37 We obtained 7 conformers by scanning the C-C-O*-C and C-O*-C-O dihedral angles. The structures and relative energies of the conformers are shown in Fig. 6. The molecular mechanics (MM) calculation using the present force field accurately predicts the minimum energy structure (#0) and all conformers with various relative energies. Low energy conformers are due to more intramolecular hydrogen bonds. In high-energy conformers, the methyl hydroxyl group rotates and the intramolecular hydrogen bonds are broken. However, the conformers may be stabilized in the condensed phase due to intermolecular hydrogen bonds. Structure VII is a fixed structure that is cut from the crystal of glucose and the MM energy agrees well with the QM data, both located approximately 18–19 kcal/mol above the minimum. This energy cost is compensated in the crystal by intermolecular interactions.

Conformation energies and structures with hydrogen bonds (dashed lines) of cellobiose. The relative energies in kcal/mol are relevant to QM energy (orange) of structure 0. Conformer VII isa fixed structure cut from Iβ crystal of cellulose (Color figure online).
Figure 7 shows the normal mode frequencies of the QM and MM calculations. Over the entire frequency range, the MM data agree well with the QM data. It is interesting to see that small but systematical deviations are found in the middle frequency ranges. Close examination of the normal modes indicates that these modes are associated with couplings in bond stretching and angle distortions, for which more complex functional forms are required to fully describe.

Normal mode frequencies (in cm−1) of cellobiose. Quantum data are shown in blue and MM data in orange (Color figure online).
A statistical analysis of QM and MM structural parameters in terms of bond-lengths, bond-angles and dihedral angles of cellobiose is given in Table IV. For each type of internal coordinate, the number of data points and the ranges of data measured by the maximum and minimum values, as well as the standard deviation of the data distribution, are listed for QM and MM, respectively. The first point to make is that the molecule, although it is relatively small, presents a significant fluctuation in all the internal coordinates. The second point to make is that the MM calculation based on the present force field agrees well with QM calculation in terms of the predicted values and fluctuations.
Validation on Cellulose Iβ Crystal
The crystal structure of cellulose Iβ has been resolved and published,25 and several research groups have published their computational results with different force fields. In Table V, we list a comparison of unit cell parameters on cellulose Iβ crystals obtained by using energy minimization (MM) and molecular dynamics simulation (MD) using the present force field, together with the experimental data and other simulation data for comparison. The MD simulation up to 100 ns appears well converged, as evident from the data given in supporting information Table SI3. The data show that the density obtained by energy minimization is about 1% higher, which is reasonable, while the MD simulation at ambient temperature and pressure yields excellent agreement with the experimental density with − 0.1% deviation. In terms of cell edge parameters, the a and c edges are slightly underestimated, whereas the b edge is slightly overestimated. Our MD predictions are overall closer to the experimental data than other predictions found in the literature.
As shown in Fig. 8, cellulose Iβ is monoclinic with the polymer backbone aligned with the c direction, and layers joined by hydrophobic interactions and hydrogen bonds in the a and b directions. Presumably, edge a is mostly affected by the hydrophobic interactions,35,54 edge b is mostly affected by the intralayer hydrogen bond network, and edge c is mostly influenced by the intramolecular structures. Since our force field is derived by using multiple datasets including liquid data of relevant molecules, the deviations indicate the limit of transferability of the force field parameters. We speculate that the problems might be lessened by including a polarizable function, since the polarization is significantly different between small molecular liquids and crystalline polymers.

Cellulose Iβ unit cell. Lattice is shown in dashed black lines, and the lattice directions are labelled with pink letters. Fragmented atoms are bonded to atoms located outside the unit cell. The C axis is placed in the Z direction, B in the Y and A in the XY plane. The unit cell and the atom coordinates are taken from the Nishiyama x-ray and neutron diffraction data25 deposited at the Cambridge Crystallographic Data Center (CCDC) (Color figure online).
Conclusion
We have presented a new AAFF for the simulation of cellulose. The force field is a part of the TEAMFF force field database in AMBER functional form, and can be used with popular simulation engines. The parameters are derived by fitting large amounts of QM energetic data generated from a training set of 12 molecules. The temperature-dependent LJ parameters have been optimized by simultaneously fitting the experimental density and the HoV of 23 relevant molecular liquids at various temperatures. Validation on glucoside, the monomer of cellulose, indicates that excellent agreements in conformational energies, molecular structures, and vibrational frequencies between MM calculations using the present force field and QM calculations are obtained. Finally, the force field is tested on the prediction of the crystal Iβ structure of cellulose. The density agrees perfectly with the experimental data, and all cell parameters have less than 1% deviations compared with the experimental data. This is a superior performance compared with previously published force fields.
Notes
The force field parameter set is available for download at https://github.com/sungroup-sjtu.
References
B. Medronho, A. Romano, M.G. Miguel, L. Stigsson, and B. Lindman, Cellulose 19, 581. (2012).
D. Ye, S. Rongpipi, S.N. Kiemle, W.J. Barnes, A.M. Chaves, C. Zhu, V.A. Norman, A. Liebman-Peláez, A. Hexemer, M.F. Toney, A.W. Roberts, C.T. Anderson, D.J. Cosgrove, E.W. Gomez, and E.D. Gomez, Nat. Commun. 11, 4720. (2020).
J.-Y. Kim, H.W. Lee, S.M. Lee, J. Jae, and Y.-K. Park, Bioresour. Technol. 279, 373. (2019).
J.V. Vermaas, L. Petridis, X. Qi, R. Schulz, B. Lindner, and J.C. Smith, Biotech. Biofuels 8, 217. (2015).
T.M. Shiga, W. Xiao, H. Yang, X. Zhang, A.T. Olek, B.S. Donohoe, J. Liu, L. Makowski, T. Hou, M.C. McCann, N.C. Carpita, and N.S. Mosier, Biotech. Biofuels 10, 310. (2017).
R. Tiwari, L. Nain, N.E. Labrou, and P. Shukla, Crit. Rev. Microbiol. 44, 244. (2018).
X. Wu, R.J. Moon, and A. Martini, Cellulose 21, 2233. (2014).
G. Lo Re, S. Spinella, A. Boujemaoui, F. Vilaseca, P.T. Larsson, F. Adås, L.A. Berglund, and A.C.S. Sustain, Chem. Eng. 6, 6753. (2018).
G. Molnár, D. Rodney, F. Martoïa, P.J.J. Dumont, Y. Nishiyama, K. Mazeau, and L. Orgéas, Proc. Nat. Acad. Sci. USA 115, 7260. (2018).
G. Liu, W. Li, L. Chen, X. Zhang, D. Niu, Y. Chen, S. Yuan, Y. Bei, and Q. Zhu, Colloids Surf. A 594, 124663. (2020).
D.W. Wei, H. Wei, A.C. Gauthier, J. Song, Y. Jin, and H. Xiao, J. Bioresour. Bioprod. 5, 1. (2020).
S. Nie, H. Guo, Y. Lu, J. Zhuo, J. Mo, and Z.L. Wang, Adv. Mater. Tech. 5, 2000454. (2020).
C. Chen, M. Liu, Y. Hou, L. Zhang, D. Wang, H. Shen, C. Ding, C. Li, S. Fu, and A.C.S. Sustain, Chem. Eng. 8, 8505–8518. (2020).
S. Chen, Y. Song, F. Xu, and A.C.S. Sustain, Chem. Eng. 6, 5173. (2018).
N. Dizge, E. Shaulsky, and V. Karanikola, J. Membr. Sci. 590, 117271. (2019).
W. Huang, X. Tang, Z. Qiu, W. Zhu, Y. Wang, Y.-L. Zhu, Z. Xiao, H. Wang, D. Liang, J. Li, and Y. Xie, ACS Appl. Mater. Int. 12, 40968. (2020).
T. Ishida, J. Phys. Chem. B 124, 3090. (2020).
T. Endo, S. Yoshida, and Y. Kimura, Cryst. Growth Des. 20, 6267. (2020).
P.B. Sánchez, S. Tsubaki, A.A.H. Pádua, and Y. Wada, Phys. Chem. Chem. Phys. 22, 1003. (2020).
C.A. Pena, A. Soto, A.W.T. King, H. Rodríguez, and A.C.S. Sustain, Chem. Eng. 7, 9164. (2019).
A.S.M. Wittmar, D. Koch, O. Prymak, and M. Ulbricht, ACS Omega 5, 27314. (2020).
D.N.S. Hon, Cellulose 1, 1. (1994).
K.H. Meyer, and H. Mark, Ber. Dtsch. Chem. Ges. 61, 593. (1928).
K.H. Meyer, and L. Misch, Helv. Chim. Acta 20, 232. (1937).
Y. Nishiyama, P. Langan, and H. Chanzy, J. Am. Chem. Soc. 124, 9074. (2002).
W. Plazinski, A. Lonardi, and P.H. Hünenberger, J. Comput. Chem. 37, 354. (2016).
H.S. Hansen, and P.H. Hünenberger, J. Comput. Chem. 32, 998. (2011).
R.D. Lins, and P.H. Hunenberger, J. Comput. Chem. 26, 1400. (2005).
D. Kony, W. Damm, S. Stoll, and W.F. Van Gunsteren, J. Comput. Chem. 23, 1416. (2002).
K.N. Kirschner, A.B. Yongye, S.M. Tschampel, J. González-Outeiriño, C.R. Daniels, B.L. Foley, and R.J. Woods, J. Comput. Chem. 29, 622. (2008).
K. Vanommeslaeghe, E. Hatcher, C. Acharya, S. Kundu, S. Zhong, J. Shim, E. Darian, O. Guvench, P. Lopes, I. Vorobyov, and A.D. Mackerell, J. Comput. Chem. 31, 671. (2010).
O. Guvench, E. Hatcher, R.M. Venable, R.W. Pastor, and A.D. MacKerell, J. Chem. Theor. Comput. 5, 2353. (2009).
J.F. Matthews, G.T. Beckham, M. Bergenstråhle-Wohlert, J.W. Brady, M.E. Himmel, and M.F. Crowley, J. Chem. Theor. Comput. 8, 735. (2012).
Z. Gong, and H. Sun, J. Chem. Eng. Data 64, 3718. (2019).
Z. Wu, D.J. Beltran-Villegas, and A. Jayaraman, J. Chem. Theor. Comput. 16, 4599. (2020).
L. Perić-Hassler, H.S. Hansen, R. Baron, and P.H. Hünenberger, Carbohydr. Res. 345, 1781. (2010).
G.L. Strati, J.L. Willett, and F.A. Momany, Carbohydr. Res. 337, 1833. (2002).
J. Wang, R.M. Wolf, J.W. Caldwell, P.A. Kollman, and D.A. Case, J. Comput. Chem. 25, 1157. (2004).
B.R. Brooks, R.E. Bruccoleri, B.D. Olafson, D.J. States, S. Swaminathan, and M. Karplus, J. Comput. Chem. 4, 187. (1983).
M.J. Hwang, T.P. Stockfisch, and A.T. Hagler, J. Am. Chem. Soc. 116, 2515. (1994).
Z. Jin, C. Yang, F. Cao, F. Li, Z. Jing, L. Chen, Z. Shen, L. Xin, S. Tong, and H. Sun, J. Comput. Chem. 37, 653. (2016).
Z. Gong, H. Sun, and B.E. Eichinger, J. Chem. Theor. Comput. 14, 3595. (2018).
M.P. Oliveira, M. Andrey, S.R. Rieder, L. Kern, D.F. Hahn, S. Riniker, B.A.C. Horta, and P.H. Hünenberger, J. Chem. Theor. Comput. 16, 7525. (2020).
M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman, G. Scalmani, V. Barone, G.A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B.G. Janesko, R. Gomperts, B. Mennucci, H.P. Hratchian, J.V. Ortiz, A.F. Izmaylov, J.L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V.G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J.A. Montgomery, Jr., J.E. Peralta, F. Ogliaro, M. Bearpark, J.J. Heyd, E. Brothers, K.N. Kudin, V.N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J.C. Burant, S.S. Iyengar, J. Tomasi, M. Cossi, J.M. Millam, M. Klene, C. Adamo, R. Cammi, J.W. Ochterski, R.L. Martin, K. Morokuma, O. Farkas, J.B. Foresman, and D.J. Fox, Gaussian 09, Revision A.02. (Wallingford: Gaussian, 2018).
D. Siderius, NIST Standard Reference Simulation Website. (Gaithersberg, MD: NIST, 2017).
D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A.E. Mark, and H.J.C. Berendsen, J. Comput. Chem. 26, 1701. (2005).
L. Martínez, R. Andrade, E.G. Birgin, and J.M. Martínez, J. Comput. Chem. 30, 2157. (2009).
S. Plimpton, J. Comput. Phys. 117, 1. (1995).
C.R. Groom, I.J. Bruno, M.P. Lightfoot, and S.C. Ward, Acta Crystallogr. B 72, 171. (2016).
M. Diem, and C. Oostenbrink, J. Chem. Theor. Comput. 16, 5985. (2020).
H. Sun, J. Phys. Chem. B 102, 7338. (1998).
L. Paoloni, S. Rampino, and V. Barone, J. Chem. Theor. Comput. 15, 4280. (2019).
C.C. Sun, J. Pharm. Sci. 94, 2132. (2005).
S. Neyertz, A. Pizzi, A. Merlin, B. Maigret, D. Brown, and X. Deglise, J. Appl. Polym. Sci. 78, 1939. (2000).
Acknowledgements
We acknowledge the computational resources resource provided by the High-Performance Computing (HPC) Center of Shanghai Jiao Tong University.
Funding
This work was funded by the National Natural Science Foundation of China [Grant Nos. 21673138, 21603141, 21973060].
Author information
Authors and Affiliations
Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. All authors contributed equally.
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Charvati, E., Zhao, L., Wu, L. et al. A New Parameterization of an All-Atom Force Field for Cellulose. JOM 73, 2335–2346 (2021). https://doi.org/10.1007/s11837-021-04732-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11837-021-04732-9