
Abstract
The grain number is one of the highly complex and crucial traits determining the grain yield in rice. To identify grain number QTLs in rice, we employed the QTL-seq approach in the 297 F2 population derived from the cross between BPT5204 and NLR33892. In this investigation, five regions viz., qGN1, qGN3, qGN7, qGN9, and qGN12 on chromosomes 1,3,7,9, and 12, respectively, were identified as QTLs governing grain number per panicle in rice. To verify the QTLs identified in the present study, traditional QTL mapping was carried out using InDel markers and previously reported SSRs in the QTL region. From qGN12 QTL, RM6953 showed a significant association with the number of grains per panicle with a phenotypic variance of 24.58, 22.10, and 17.20% in F2, F2:3, and BC1F2 populations, respectively, at < 0.0001 P value, indicating major QTL. From genome resequencing data, a missense variance was observed in the exonic region of LOC_Os12g39330 gene controlling AP2 domain-containing protein which plays an important role in seed development in rice. Hence, this QTL can be a potential target for map-based cloning and marker-assisted transfer to enhance the grain number in rice.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rice (Oryza sativa L) is one of the major cereal crops feeding more than three billion population and accounts for 20% of the world’s total calorie intake. The doubling of the rice productivity was achieved by adopting semi-dwarf varieties for lodging resistance in the 1960s and hybrid rice technology in the 1970s. However, the rise in the yield levels were led to the excess usage of fertilizers and pesticides causing adverse environmental effects in rice-producing areas besides shooting up cost of the cultivation. Moreover, the demand for rice is continuously increasing to meet the food requirement of the rapidly growing global population (Singh et al. 2018and Wang et al. 2020). Hence, there is an urgent need to substantially enhance the productivity of rice with the existing ever-shrinking cultivable area while addressing climate change.
Rice yield or productivity is a complex trait multiplicatively determined by three main component traits viz., grain number, panicle number, and grain weight (Sakamoto and Matsuoka 2008; Xing and Zhang 2010; Wang et al. 2020). Of these, grain number per panicle is the major yield attributing trait in rice (Wang et al. 2020). Grain number per panicle is highly variable and mainly correlated with the architecture of the panicle that includes the number of primary and secondary branches, panicle length, and percentage of filled grains. Multiple genes are involved in regulating the inheritance of the grain number trait that shows continuous variation in the segregating populations. Hence, identification of the genetic basis responsible for grain number would be of great value in the breeding of high-yielding rice varieties. During the last two decades, many Quantitative Trait Loci (QTLs)/genes viz., Gn1a (Ashikari et al., 2005), DEP1 (Huang et al. 2009), qGN4.1(Deshmukh et al. 2010)OsSPL14 (Jiao et al. 2010), NAL1 (SPIKE, GPS, LSCHL4, and SS1), GNP1 (Wu et al. 2016a, b), qGN1c (Xu et al. 2019) controlling the grain number trait have been mapped, cloned, and functionally characterized in rice. However, the molecular mechanism of grain number trait formation is still far from a clear understanding as DNA marker development and genotyping are the most time-consuming and costly procedures in conventional QTL mapping.
Next-Generation Sequencing (NGS) technologies have facilitated the development of several efficient techniques for gene or locus mapping. Among them, NGS-based QTL-seq approach has successfully identified and mapped many QTLs in rice (Takagi et al. 2013; Daware et al. 2016; Ogiso-Tanaka et al. 2017; Yaobin et al. 2018; Kadambari et al. 2018; Arikit et al. 2019; Lei et al. 2020 and Bommisetty et al. 2020), chickpea (Singh et al. 2017), cucumber (Lu et al. 2014), and tomato (Illa-Berenguer et al. 2015 and Ruangrak et al. 2019).
Identification of QTL region that consists of numerous genes of targeted trait is a challenging task. However, with the availability of high-quality reference genome and advances in DNA sequencing technologies, now it is becoming very easy to map and precisely pinpointing the underpinning genes in QTL regions. Hence, the present research work was formulated to identify genomic regions determining grain number in rice employing QTL-seq approach in F2 mapping population and confirmed the identified QTLs additionally in BC1F2 and F3 populations.
Materials and methods
Development and phenotyping of mapping populations for grain number per panicle
In the present study, BPT5204 (Samba Mahsuri) was crossed with NLR33892 (Pardhiva) to develop an F2 mapping population for mapping of grain number QTLs using the QTL-seq approach. BPT5204 is a fine-grained medium-duration high-yielding variety with moderate grain number developed at Agricultural College, Acharya NG Ranga Agricultural University (ANGRAU), Bapatla, Andhra Pradesh, India, whereas NLR33892 is a long duration, photosensitive variety, with high grain number, developed at Agricultural Research Station, ANGRAU, Nellore, Andhra Pradesh, India.
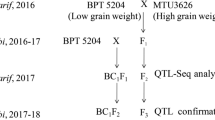
During Kharif (June to October), 2016, BPT5204 was crossed with high grain number male-parent NLR33892 to develop F1 seeds (Fig. 1). The F1 plants were screened with polymorphic simple sequence repeat (SSR) markers and true F1 plants were identified (Supplementary Fig. 1). During rabi (November to April), true F1s were selfed and simultaneously backcrossed with BPT5204 to generate F2 and BC1F1 populations, respectively. The F2 mapping population along with parents was evaluated for yield and its component traits during Kharif, 2017 while F2:3 and BC1F2 plants along with parents were raised during rabi (November to April), 2017–18 at wetland farm, S. V. Agricultural College, Acharya NG Ranga Agricultural University, Tirupati, Andhra Pradesh, India. A total number of grains including chaffy and filled grains per panicle was counted from each panicle in each plant.

Schematic representation of development of mapping population for identification of grain number QTLs in rice employing QTL-seq analysis
Preparation of bulks and DNA isolation for whole-genome resequencing
In this experiment, 297 F2 plants of the BPT5204 x NLR33892 cross were evaluated. Based on grain number per panicle, 15 plants with extremely low grain number and 15 plants with extremely high grain number were selected as Low grain number bulk (L-bulk) and High grain number bulk (H-bulk), respectively. The genomic DNA from 100 mg fresh rice leaves of the parents and the selected extreme F2 individuals for a number of grains per panicle were extracted using DNeasy Plant Mini Kit (QIAGEN Sciences), and quantification of DNA was performed using Quant-iTPicoGreen dsDNA reagent and kits (Invitrogen). The bulk DNA samples were prepared by mixing equimolar concentrations of extracted DNA from 15 individuals of extreme phenotypic traits and pooled together for whole-genome resequencing.
QTL-seq and QTL validation
QTL-seq and QTL validation was followed as per Bommisetty et al. (2020). All generated whole-genome sequencing data of BPT5204 were used from our previous study (Lachagari et al. 2019) and NLR33892 with bulks, whole-genome sequencing data are available in the SRA database under the BioProject ID: PRJNA484092.
Statistical analysis
Single-marker analysis is the simplest method to identify the marker–trait association. Linear regression is the most commonly used single-marker analysis method as the coefficient of determination (R2) from the marker explains the phenotypic variation arising from the QTL linked to the marker (Collard et al. 2005).
Results
Phenotypic evaluation of parents
Parents (BPT5204 and NLR33892) were analyzed for test of significance using paired t test(Fig. 2A, Fig. 2B, Fig. 3a and Table1) for plant height, number of tillers per plant, number of panicles per plant, panicle length, number of grains per panicle, number of filled grains per panicle, number of chaffy grains per panicle, spikelet fertility, biological yield per plant, grain yield per plant, harvest index, grain length, grain width, the ratio of grain length to width, and 1000 grain weight. It revealed highly significant differences between BPT5204 and NLR33892 for all the characters except for harvest index and grain size, indicating the presence of variability between the parental genotypes.

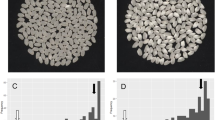
A. Morphology of BPT5204 and NLR33892 at maturity grown in paddy field B. Panicle length (BPT5204:24.52 cm and NLR33892:29.23 cm)

QTL-seq approach adopted for mapping genomic regions responsible for grain number per panicle (a) and (c) Image shows the morphological difference of BPT5204 (Low grain number parent) and NLR33892 (High grain number parent) (b) Frequency distribution of grain number per panicle of 297 F2 plants. (d) ∆SNP index for the chromosome number 12, plotted using a sliding window of 4 Mb with a step of 10 kb. The significant genomic regions are highlighted in shaded color (23–27 Mb). The statistical confidence interval under the null hypothesis of no QTLs is presented in the graphs (orange, P < 0.01 and green P < 0.05). (e) Grain size picture of the parents
Inheritance pattern of grain number per panicle in the F2 mapping population
The F2 mapping population comprising 297 plants along with BPT5204 and NLR33892 was evaluated for grain number per panicle trait during Kharif2017. The grain number per panicle varied from 102 to 582 in the F2 population, with a mean, median, and mode of 303.53, 316, and 326, respectively. Grain number per panicle recorded a skewness value of − 0.25 and a kurtosis value of 0.26. The continuous variation as well as the normal frequency distribution of grain number per panicle was observed in the mapping population (Fig. 3b). Further, grain number per panicle showed a bi-directional transgressive segregation beyond that of parental genotypes in the F2 mapping population. High variability and normal distribution of grain number per panicle in the F2 mapping population inferred the polygenic inheritance of the character.
Identification of QTLs for grain number per panicle by QTL-seq approach
The DNA sequencing outputs are summarized in Table 2. From whole-genome resequencing, 93,272,796, 10,6482,020, 124,559,926, and 91,651,766 raw reads were obtained for BPT5204, NLR33892, H-bulk and L-bulk, respectively. H-bulk and L-bulk had 43.179 and 44.39% of GC content, respectively. The mapped reads covered 92.37, 89.94, 90.02, and 89.53% of rice genome in BPT5204, NLR33892, H-bulk, and L-bulk, respectively. Later, the raw data were pre-processed using Adapter removal (version: 2.2.0). After processing, a total of 63,084,068 clean reads with 75.36% high-quality bases were obtained for H-bulk, while L-bulk recovered 63,084,068 clean bases with 78.01% high-quality base. The aligned samples and the reference genome sequence are used for variant calling and was performed using Samtools v0.1.18.
The H-bulk contained 8339 SNPs while L-bulk had 8337 SNPs. Homozygous SNPs present in H-bulk and L-bulk were 2171 and 2091, respectively. A total of 2945 SNPs were present in the gene region and of them, only 1541 SNPs fall in the exonic regions of the gene. The SNP index was calculated for each SNP (Abe et al. 2012). SNPs with SNP index < 0.3 in both bulks were removed as they could be spurious SNPs. The SNP index of remaining SNPs calculated from each bulk was physically plotted throughout 12 rice chromosomes (Fig. 4). The Δ(SNP index) was calculated by subtracting the SNP index values in HGW-bulk by those in LGW-bulk together with the sliding windows of average SNP indices of SNPs located within a 2-Mb region and 1 Kb stepwise were also plotted. Statistical confidence intervals of ∆(SNP index) for all the SNP positions with given read depths under the null hypothesis of no QTLs were calculated and plotted along with ∆(SNP index).

Plots of SNP index of two bulks (High grain number (HGN) bulk and Low grain number (LGN) bulk) and Δ(SNP index) compared between them. (A) Read coverage of HGN bulk. (B) Read coverage of LGN bulk. (C) SNP index of HGN bulk. (D) SNP index of LGN bulk. (E) Delta SNP index (F) SNP density with 50 Kb step with P < 0.05. (G) Candidate genomic regions containing QTLs for number of grains per panicle
Identification of genomic regions for grain number
In total, five genomic regions viz., qGN1, qGN3, qGN7, qGN9, and qGN12 on chromosomes 1, 3, 7, 9, and 12 were identified for grain number at P < 0.01 as shown in Fig. 4 and Table 3 (Supplementary Fig. 2).
Confirmation of the QTL-seq-derived QTLs in F2, F3, and BC1F2 populations
To verify the QTLs identified in the present study, traditional QTL mapping was carried out using single-marker analysis for randomly selected 167 F2 plants of the total population. Twenty markers designed in the identified QTL regions and 20 previously reported genomic SSR markers (physically/genetically mapped on selected QTL regions of chromosomes 1, 3, 7, 9, and 12) were selected to screen parents (Supplementary Table 1). No polymorphism was observed between BPT5204 and NLR33892 for selected genomic regions on chromosomes 1, 3, 7, and 9 using InDel and SSR markers; this clearly explains that variation present in the segregation population was not under the control of genes present in these selected regions. One polymorphic marker, RM6953 located in the QTL region of chromosome 12, was used for genotyping of F2, F3, and BC1F2 population, and the resulting data were subjected to single-marker analysis (Fig. 5). Single-factor analysis of RM6953 revealed the significant association of the marker with grain number with a phenotypic variance of 24.58, 22.10, and 17.20% (Fig. 5 and Table 4) in F2, F2:3, and BC1F2 populations, respectively, at P value < 0.0001 and NLR33892 was the donor parent for high grain number trait (Table 4 and Fig. 5).

Representative gel picture showing segregation pattern of F2 (A) and BC1F2 (B) populations of the cross between BPT5204 and NLR33892. Linear regression analysis for RM6953 and number of grains per panicle in F2(C), F2:3(D), and BC1F2(E) populations of the cross between BPT5204 and NLR33892. Frequency of marker RM6953 genotype alleles in the F2, F2:3, and BC1F2 populations of the cross between BPT5204 and NLR33892 for number of grains per panicle (F)
Discussion
Rice is the major food crop feeding more than half of the global population. There is an urgent need to raise the productivity of rice to meet global food demand for the estimated nine billion people by 2050. Therefore, understanding the genetic basis, identification, and incorporation of major QTLs governing yield and its component traits into elite cultivars is essential to increase yield levels with existing agricultural land, water, and climatic conditions. Keeping this in view, the present investigation was conducted to identify QTLs governing grain number per panicle using QTL-seq analysis and their further confirmation by conventional linkage analysis.
The parents, BPT5204 and NLR33892 varieties, showed significant differences for grain number per panicle. In addition to continuous variation, grain number per panicle showed biparental transgressive segregation in the F2, F2:3, and BC1F2 population (Fig. 3b, Supplementary Fig. 3a, b). All these collectively inferred the quantitative genetic inheritance pattern of grain number per panicle in rice. Hence, this population was selected for mapping grain number QTLs with the help of NGS-based QTL-seq analysis.
In the present study, QTL-seq analysis identified five genomic regions viz., qGN1, qGN3, qGN7, qGN9, and qGN12 on chromosomes 1, 3, 7, 9, and 12, respectively, as candidate QTLs for a number of grains per panicle in rice based on ∆SNP index values at P value < 0.01 in an F2 population derived from BPT5204xNLR33892 cross. In the selected genomic region of chromosome 1, five yield traits QTLs viz., grains per panicle QTL gp1a (Hua et al. 2002), plant height QTL ph1.1 (Marri et al. 2005), panicles per plant QTL qPN1 (Tian et al. 2006), total number of spikelets per panicle QTL qTNSP-1–1 (Zhuang et al. 2002), and pollen sterility QTL Rf3 (Sattari et al. 2007) were reported previously (Supplementary Table 2). Chromosome 3 harbors two yield traits QTLs namely pollen sterility QTL S34(t) (Zhuang et al. 2005) and grain length QTL gl3a. Chromosomes 7, 9, and 12 had no previously reported yield QTLs in the selected genomic regions, suggesting they are novel and found in the present study for the first time for grain number in rice.
The grain number per panicle QTL, qGN12 (23 Mb to 27 Mb) was further confirmed through conventional QTL mapping in F2, F2:3, and BC1F2 populations of BPT5204 and NLR33892 cross. From the grain number trait mean values of the marker classes, the NLR33892 marker allele was identified as the source of the favorable allele. A total of 713 annotated genes were present in the qGN12 QTL region (23-27 Mb) as per the rice genome annotation project database (RGAP-DB). Among them, only seven genes viz., LOC_Os12g37690 (MYB family transcription factor), LOC_Os12g37970 (MYB family transcription factor), LOC_Os12g38400 (MYB family transcription factor), LOC_Os12g39330 (AP2 domain-containing protein), LOC_Os12g41060 (AP2 domain-containing protein), LOC_Os12g39640 (MYB family transcription factor) and LOC_Os12g40860 (Leucine-Rich Repeat family protein) were found to be potential candidate genes controlling grain number as similar kind of genes in different regions were found to control grain number in rice and other crops based on previous literature(Table 5).
A Myb-1 factor (Blind gene), which regulates the development of secondary meristems and inflorescence in tomato, has been isolated by positional cloning. Deshmukh et al. (2010) reported downregulation of Myb-1 transcription factor in Pusa 1266 (compact panicle and high grain number) as compared to Pusa Basmati 1(low grain number) in the qGN4-1 region. The APETALA-2-Like transcription factor OsAP2-39 regulates plant growth and seed production by maintaining abscisic acid and gibberellin balance in rice (Yaish et al. 2010). The thick tassel dwarf1 (td1) encoding leucine-rich repeat receptor-like kinase (LRR-RLK) results in extra rows of kernels in maize ear and also increases spikelet density in the tassel (Bommert et al. 2005 and Liu et al. 2019).
Genes controlling MYB family transcription factor (LOC_Os12g37690, LOC_Os12g37970, LOC_Os12g38400, and LOC_Os12g39640), AP2 domain-containing protein (LOC_Os12g39330 and LOC_Os12g41060), and leucine-rich repeat family protein (LOC_Os12g40860) were found to be expressed in the panicle, flag leaf, and flower from RicevarmapV2.0 (Fig. 6 and Supplementary Table 3). LOC_Os12g38400, LOC_Os12g39640, and LOC_Os12g40860 genes were found to be highly expressed in the panicles. Sequence differences of the potential candidate genes between the parents revealed the presence of missense variance in the exonic region of LOC_Os12g39330 gene (Table 6). Among the seven candidate genes, the highest number of missense variants was observed in LOC_Os12g41060 through Ricevarmap2.0 (Supplementary Table3). Further it was found that both the genes were involved in the production of AP2 domain-containing protein which plays an important role in panicle development and seed development in rice (Yaish et al. 2010). Hence, these genes could be considered as candidate genes controlling high grain number in rice.

Gene Expression Atlas of the shortlisted candidate genes governing grain number per panicle. Intensity of the color represents the expression value of the genes in different tissues of rice genotypes
The grain number QTL identified in the present study exhibited as much as 20% phenotypic variance consistently in the F2, F2:3, and BC1F2 populations. Hence, this QTL can be considered as not only novel but also a major QTL, which can be targeted for map-based cloning to identify the underlying candidate gene. Moreover, this QTL can be transferred to low grain number varieties to enhance the yield through marker-assisted selection. Further, NLR33892 provided favorable allele, hence can be used as a potential donor for yield enhancement.
However, further validation of novel QTL is warranted before exploiting the genes predicted in the present study for deploying into elite cultivars. The novel QTLs identified in the present study undoubtedly enhance our understanding of the complex nature of the yield component traits.
Data availability
The data has been provided in the supplementary information.
Abbreviations
- QTL:
-
Quantitative trait loci
- SNP:
-
Single-nucleotide polymorphism
- InDel:
-
Insertion/deletion
- Kb:
-
Kilobase pair
- Mb:
-
Megabase pair
- BSA:
-
Bulk-segregant analysis
References
Abe A, Kosugi S, Yoshida K, Natsume S, Takagi H, Kanzaki H, Matsumura H, Yoshida K, Mitsuoka C, Tamiru M, Innan H, Cano L, Kamoun S, Terauchi R (2012) Genome sequencing reveals agronomically important loci in rice using MutMap. Nat Biotechnol 30:174–178
Arikit S, Wanchana S, Khanthong S, Saensuk C, Thianthavon T, Vanavichit A, Toojinda T (2019) QTL-seq identifies cooked grain elongation QTLs near soluble starch synthase and starch branching enzymes in rice (Oryza sativa L). Sci Report 9:8328
Ashikari M, Wu J, Yano M, Sasaki T, Yoshimura A (1999) Rice gibberellin-insensitive dwarf mutant gene Dwarf 1 encodes the alpha-subunit of GTP-binding protein. Proc of National Academy Sci 96:10284–10289
Austin RS, Vidaurre D, Stamatiou G, Breit R, Provart NJ, Bonetta D, Zhang J, Fung P, Gong Y, Wang PW, McCourt P, Guttman DS (2011) Next-generation mapping of arabidopsis genes. Plant J 67:715–725
Bommert P, Lunde C, Nardmann J, Vollbrecht E, Running M, Jackson D, Hake S, Werr W (2005) Thick tassel dwarf 1 encodes a putative maize ortholog of the Arabidopsis CLAVATA1 leucinerich repeat receptor-like kinase. Development 132:1235–1245
Bommisetty R, Chakravartty N, Bodanapu R et al (2020) Discovery of genomic regions and candidate genes for grain weight employing next generation sequencing based QTL-seq approach in rice (Oryza sativa L). Mol Biol Report 47(11):8615–8627
Collard BCY, Jahufer MZZ, Brouwer JB, Pang ECK (2005) An introduction to markers, quantitative trait loci (QTL) mapping and marker-assisted selection for crop improvement: The basic concepts. Euphytica 142:169–196
Collins HMF, Panozzo SJ, Logue SP, Jefferies BAR (2003) Mapping and validation of chromosome regions associated with high malt extract in barley (Hordeum vulgare L). Austral J Agri Res. 54:1223–1240
Daware A, Das S, Srivastava R, Badoni S, Singh AK, Agarwal P, Parida SK, Tyagi AK (2016) An efficient strategy combining SSR markers- and advanced QTL-seq-driven QTL mapping unravels candidate genes regulating grain weight in rice. Front Plant Science 7:1535–1545
Deshmukh R, Singh A, Jain N, Anand S, Gacche R, Singh A, Gaikwad K, Sharma T, Mohapatra T, Singh N (2010) Identification of candidate genes for grain number in rice (Oryza sativa L). Funct Integr Genomics 10:339–347
GRiSP (Global Rice Science Partnership). (2013) Rice almanac, 4th edn. International Rice Research Institute, Los Baños
Haung X, Qian Q, Liu Z, Sun H, He S, Luo D, Xia G, Chu C, Li J, Fu X (2009) Natural variation at the DEP1 locus enhances grain yield in rice. Nat Genet 41(4):494–497
Hu Z, Lu SJ, Wang MJ, He H, Sun L, Wang H, Liu XH, Jiang L, Sun JL, Xin X, Kong W, Chu C, Xue HW, Yang J, Luo X, Liu JX (2018) A Novel QTL qTGW3 encodes the GSK3/SHAGGY-Like Kinase OsGSK5/OsSK41 that Interacts with OsARF4 to negatively regulate grain size and weight in rice. Mol Plant 11:736–749
Hua JP, Xing YZ, Xu CG, Sun XL, Yu SB, Zhang Q (2002) Genetic dissection of an elite rice hybrid revealed that heterozygotes are not always advantageous for performance. Genetics 162:1885–1895
Huang R, Jiang L, Zheng J, Wang T, Wang H, Huang Y, Hong Z (2013) Genetic bases of rice grain shape: so many genes, so little known. Trends Plant Sci 18:218–226
Illa-Berenguer E, Houten JV, Huang Z, Knaap EV (2015) Rapid and reliable identification of tomato fruit weight and locule number loci by QTL-seq. Theor Appl Genet 128:1329–1342
Jiao Y, Wang Y, Xue D, Wang J, Yan M, Liu G, Dong G, Zeng D, Lu Z, Zhu X, Qian Q, Li J (2010) Regulation of OsSPL14 by OsmiR156 defines ideal plant architecture in rice. Nat Genet 42:541–544
Kadambari G, Vemireddy LR, Srividhya A, Nagireddy R, Jena SS, Gandikota M, Patil S, Veeraghattapu R, Deborah DAK, Reddy GE, Shake M, Dasari A, Rao PVR, Durgarani ChV, Neeraja CN, Siddiq EA, Sheshumadhav M (2018) QTL-Seq-based genetic analysis identifies a major genomic region governing dwarfness in rice (Oryza sativa L.). Plant Cell Rep 37:677–687
Lachagari VBR, Gupta R, Lekkala SP, Mahadevan L, Kuriakose B, Chakravartty N, Mohan Katta AVSK, Santhosh S, Reddy AR, Thomas G (2019) Whole genome sequencing and comparative genomic analysis reveal allelic variations unique to a purple colored rice landrace (Oryza sativa ssp indica cv Purpleputtu). Front Plant Sci 114:513
Lei L, Zheng H, Bi Y, Yang L, Liu H, Wang J, Sun J, Zhao H, Li J, Lai Y, Zou D (2020) Identification of a major qtl and candidate gene analysis of salt tolerance at the bud burst stage in rice (Oryza sativa L) using QTL-seq and RNA-seq. Rice 13:55
Li H, Durbin R (2009) Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25:1754–1760
Li S, Qian Q, Fu Z, Zeng D, Meng X, Kyozuka J, Maekawa M, Zhu X, Zhang J, Li J, Wang Y (2009) Short panicle1 encodes a putative PTR family transporter and determines rice panicle size. Plant J 58:592–605
Liu C, Zhou Y, Zhang X, Zhang J, Zhou Z, Weng J, Li X, Wang Z (2019) Natural variation in the THICK TASSEL DWARF1 (TD1) gene in the regulation of maize (Zea mays L) ear-related traits. Breed Sci. https://doi.org/10.1270/jsbbs.18170
Lu HF, Lin T, Klein J, Wang SH, Qi JJ, Zhou Q, Sun JJ, Zhang ZH, Weng YQ, Huang SW (2014) QTL-seq identifies an early flowering QTL located near flowering locus T in cucumber. Theor Appl Genet 127:1491–1499
Marri PR, Sarla N, Reddy LV, Siddiq EA (2005) Identification and mapping of yield and yield related QTLs from an Indian accession of Oryza rufipogon. BMC Genet 6:33
Ogiso-Tanaka E, Tanaka T, Tanaka K, Nonoue Y, Sasaki T, Fushimi E, Koide Y, Okumoto Y, Yano M, Saito H (2017) Detection of novel QTLs qDTH4.5 and qDTH6.3, which confer late heading under short-day conditions, by SSR marker-based and QTL seq analysis. Breed Sci 67:101–109
Ruangrak E, Du Y, Htwe NMPS, Pimorat P, Gao J (2019) Identification of early tomato fruit ripening loci by QTL-seq. J Agric Sci 11(2):51–70
Sakamoto T, Matsuoka M (2008) Identifying and exploiting grain yield genes in rice. Curr Opin Plant Biol 11:209–214
Sattari M, Kathiresan A, Gregorio G, Virmani S (2007) Comparative genetic analysis and molecular mapping of fertility restoration genes for WA, Dissi, and Gambiaca cytoplasmic male sterility systems in rice. Euphytica 160(3):305–315
Singh VK, Khan AW, Jaganathan D, Thudi M, Roorkiwal M, Takagi H, Garg V, Kumar V, Chiyikineni A, Gaur PM, Sutton T, Terauchi R, Varshney RK (2017) QTL-seq for rapid identification of candidate genes for 100-seed weight and root/total plant dry weight ratio under rainfed conditions in chickpea. Plant Biotech J 15:906–914
Singh VK, Ellur RK, Singh AK, Nagarajan M, Singh BD, Singh NK (2018) Effect QTL for grain number per panicle in genetic backgrounds of twelve different mega varieties of rice. Rice 1:1–8
Takagi H, Abe A, Yoshida K, Kosugi S, Natsume S, Mitsuoka C, Uemura A, Utsushi H, Tamiru M, Takuno S, Innan H, Cano LM, Kamoun S, Terauchi R (2013) QTL-seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J 74:174–183
Tian F, Li DJ, Fu Q, Zhu ZF, Fu YC, Wang XK, Sun CQ (2006) Construction of introgression lines carrying wild rice (Oryza rufipogon Griff.) segments in cultivated rice (Oryza sativa L.) background and characterization of introgressed segments associated with yield-related traits. Theor Appl Genet 112:570–580
Wang Y, Zhai L, Chen K, Shen C, Liang Y, Wang C, Zhao X, Wang S, Xu J (2020) Natural sequence variations and combinations of GNP1 and NAL1 determine the grain number per panicle in Rice. Rice 13:14
Wu Y, Fu Y, Zhao S, Gu P, Zhu Z, Sun C, Tan L (2016b) clustered primary branch 1, a new allele of controls panicle architecture and seed size in rice. Plant Biotechnol J 14:377–386
Wu Y, Fu Y, Zhao S, Gu P, Zhu Z, Sun C, Tan L (2016a) CLUSTERED PRIMARY BRANCH 1, a new allele of DWARF11, controls panicle architecture and seed size in rice. Plant Biotechnol J 14:377–386
Xing Y, Zhang Q (2010) Genetic and molecular bases of rice yield. Annual Rev Plant Biol 61:421–442
Xing YZ, Tang WJ, Xue WY, Xu CG, Zhang Q (2008) Fine mapping of a major quantitative trait loci, qSSP7, controlling the number of spikelets per panicle as a single Mendelian factor in rice. Theor Appl Genet 116:789–796
Xu Z, Miao Y, Chen Z, Gao H, Wang R, Zhao D, Zhang B, Zhou Y, Tang S (2019) Identification and fine mapping of qGN1c, a QTL for grain number per panicle, in rice (Oryza sativa). Mol Breeding 39:129
Yaish MW, El-kereamy A, Zhu T, Beatty PH, Good AG, Bi Y, Rothstein SJ (2010) The APETALA-2-Like transcription factor OsAP2-39 controls key interactions between abscisic acid and gibberellin in rice. PLoS Genet 6(9):e1001098
Yaobin Q, Cheng P, Cheng Y, Feng Y, Huang D, Huang T, Song X, Ying J (2018) QTL-seq identified a major qtl for grain length and weight in rice using near isogenic F2 population. Rice Sci 25:121–131
Zafar SA, Patil SB, Uzair M, Fang J, Zhao J, Yuan S, Uzair M, Luo Q, Shi J, Schreiber L, Li X (2020) DEGENERATED PANICLE AND PARTIAL STERILITY 1 (DPS1) encodes a CBS domain containing protein required for anther cuticle and panicle development in rice. New Phytol 225:356–375
Zhang X, Wang J, Huang J, Lan H, Wang C, Yin C, Wu Y, Tang H, Qian Q, Li J, Zhang H (2012) Rare allele of OsPPKL1 associated with grain length causes extra-large grain and a significant yield increase in rice. Proc Natl Acad Sci 109:21534–21539
Zhang Z, Xu P, Hu F, Zhou J, Li J, Deng X, Ren G, Li F, Tao D (2005) A new sterile gene from Oryza glaberrima on chromosome 3. Rice Genetics Newsletter. 11:22–26
Zhuang JY, Fan YY, Rao ZM, Wu JL, Xia YW, Zheng KL (2002) Analysis on additive effects and additive-by-additive epistatic effects of QTLs for yield traits in a recombinant inbred line population of rice. Theor Appl Genet 105:1137–1145
Acknowledgements
BRY is grateful to the Institute of Frontier Technology and S.V. Agricultural College, Tirupati, ANGRAU for providing the facilities to conduct the lab and field experiments, respectively.
Funding
VLR is grateful to the Department of Biotechnology, Government of India for providing financial assistance [DBT-NER / AGRI/29/2015 (GROUP- 4)dated 19/10/2016].
Author information
Authors and Affiliations
Contributions
BRY, VLR, and HKR planned the experiment. BRY and LRV contributed to the development of mapping populations. BRY performed the field experiments. JBN helped in the phenotyping of the mapping population. RP, SP, and BVBR provided the required materials and helped in maintaining the experiment. NC, RB, VBRL, and SG sequenced the parents and bulks. NC, VBRL analyzed the QTL-seq data. BRY wrote the manuscript. VLR, VBRL, and SPL reviewed the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declared that there is no potential conflict of interest.
Human participants and/or animals
Research is not involving human participants and/or animals.
Informed consent
Nil.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Bommisetty, R., Chakravartty, N., Hariprasad, K.R. et al. Identification of a novel QTL for grain number per panicle employing NGS-based QTL-seq approach in rice (Oryza sativa L.). Plant Biotechnol Rep 17, 191–201 (2023). https://doi.org/10.1007/s11816-023-00816-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11816-023-00816-x