
Abstract
Rice (Oryza sativa L.) yield enhancement is one of the prime objectives of plant breeders. Elucidation of the inheritance of grain weight, a key yield component trait, is of paramount importance for raising the yield thresholds in rice. In the present investigation, we employed Next-Generation Sequencing based QTL-seq approach to identify major genomic regions associated with grain weight using mapping populations derived from a cross between BPT5204 and MTU3626. QTL-seq analysis identified three grain weight quantitative trait loci (QTL) viz., qGW1 (35–40 Mb), qGW7 (10–18 Mb), and qGW8 (2–5 Mb) on chromosomes 1, 7 and 8, respectively and all are found to be novel. Further, qGW8 was confirmed through conventional QTL mapping in F2, F3 and BC1F2 populations and found to explain the phenotypic variance of 17.88%, 16.70% and 15.00%, respectively, indicating a major QTL for grain weight. Based on previous reports, two candidate genes in the qGW8 QTL were predicted i.e., LOC_Os08g01490 (Cytochrome P450), and LOC_Os08g01680 (WD domain, G-beta repeat domain containing protein) and through in silico analysis they were found to be highly expressed in reproductive organs during different stages of grain development. Here, we have demonstrated that QTL-seq is one of the rapid approaches to uncover novel QTLs controlling complex traits. The candidate genes identified in the present study undoubtedly enhance our understanding of the mechanism and inheritance of the grain weight. These candidate genes can be exploited for yield enhancement after confirmation through complementary studies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rice (Oryza sativa L.) is an important cereal crop and staple food for over three billion people on the planet. The stagnated yield levels since the Green revolution pose challenge to feed anticipated 9 billion people in the coming three decades [1, 2]. The grain yield potential of rice primarily depends on panicle number per plant, grain number per panicle, grain size and grain weight [3,4,5]. However, the elucidation of genetics of these yield components is complicated due to their polygenic inheritance besides being highly influenced by the environment. Hence, understanding of the genetic basis of yield traits at molecular level is required to further enhance yield ceiling in the present-day elite cultivars. As of now 639 genes [6] and 2060 QTLs (https://www.gramene.org) pertaining to yield and its component traits have been identified. Among them, GS3 for grain size/weight, DEP1 and Gn1a for grain number, MINI SEED 2 (MIS2) for grain size and shape, DEGENERATED PANICLE AND PARTIAL STERILITY 1 (DPS1) for anther cuticle and panicle development and GW8 for grain weight are important major QTLs governing the yield component traits [3, 5, 7].
Grain weight is one of the key component traits that determine the final yield in rice. The grain size is determined by few important QTLs such as GS3, GL3.1, GW2, GW5, GW6a and OsLG3 through regulating cell proliferation in spikelet hulls while TGW6 QTL in endosperm [8,9,10]. Further, the grain size is influenced by GLW7 and qNPT1/WTG1 QTLsby regulating cell expansion, whereas GS2, GS5, GL7/GW7 and GW8 influence by combining the regulation of cell division and cell expansion [8, 11,12,13]. From bulked QTL-seq analysis, a candidate genomic region (qTGW5.3) strongly associated with grain length and weight was identified and narrowed down to an interval of 1.13 Mb by screening the recombinants and their progeny test [14]. Yet the complete elucidation of inheritance of the grain weight is lacking due to its extremely complex nature. The key reason can be attributed to the insufficient genotyping data of the mapping populations or development of polymorphic DNA markers as the phenotyping for yield component traits and development of mapping populations from contrasting parental genotypes demands relatively less effort.
Bulked segregant analysis (BSA) is an elegant and rapid method to identify DNA markers linked to a trait of interest [15]. However, in BSA analyses, the availability of DNA markers was the key factor limiting effectiveness of the method. Furthermore, genotyping of each marker for the two extreme DNA bulks is still time-consuming and expensive. Rapid development of next-generation sequencing (NGS) technologies has facilitated to use the combination of BSA and whole genome re-sequencing of DNA pools to identify genomic regions for target traits. Using this concept, several methods have been developed for rapid identification of the QTLs such as SHOREmap [16], Next generation mapping (NGM) [17], MutMap [18], Isogenic mapping by sequencing [19], SNP-ratio mapping (SRM) [20], MutMap+ [21] MutMap-Gap [23], QTL-seq [22], SLAF-seq [24] and Indel-seq [25]. Among these techniques, QTL-seq that combines BSA with next-generation sequencing has successfully identified many QTLs in rice [14, 22, 26,27,28], chickpea [29] and [30], cucumber [31] and tomato [32] and [33].
Mining of candidate genes underlying a QTL is a challenging task. However, with the availability of high-quality reference genome sequence and other advances in sequencing technologies resulted in several public databases for genomics, transcriptomics, proteomics and metabolomics. Rice Annotation Project-Database (https://rapdb.dna.affrc.go.jp), RiceXPro (ricexpro.dna.affrc.go.jp), RicevarmapV2.0 (https://ricevarmap.ncpgr.cn/v2/vars_in_gene/) gramene (www.gramene.org) and Rice Genome Annotation Project (https://rice.plantbiology.msu.edu/) are some of the important rice databases, which enables effective functional annotation of genomic regions. Exploiting these public databases, now it is possible to pinpoint the candidate genes within the QTL regions. Therefore, the present research work was conducted to identify the genomic regions and candidate genes determining grain weight in rice by employing QTL-seq approach.
Materials and methods
Plant material and development of mapping population
In this study, F2 mapping population was developed using BPT5204 (Samba Mahsuri) and MTU3626 (Prabhat) as parents. BPT5204 is a fine-grained high yielding variety with excellent cooking quality developed at Agricultural College, Acharya NG Ranga Agricultural University (ANGRAU), Bapatla whereas MTU3626 is a high yielding variety with high grain weight developed at Regional Agricultural Research Station, ANGRAU, Maruteru.
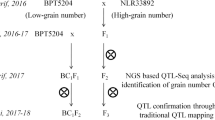
For development of mapping population, BPT5204 was crossed with male parent MTU3626 to develop F1 seeds during kharif (June to October), 2016 (Fig. 1). The F1 plants were confirmed for their heterozygosity using simple sequence repeat (SSR) markers that are polymorphic between parents (Supplementary Fig. 1). True F1s were selfed and simultaneously backcrossed with Samba Mahsuri to generate F2 and BC1F1 seeds, respectively. Later, F2 and BC1F1 plants were advanced to F3 and BC1F2 seeds, respectively by selfing. F2 mapping population along with parents were evaluated for yield and its component traits during kharif, 2017 while F3 and BC1F2 plants along with parents were raised during rabi (November to April), 2017–2018 at wetland farm, S. V. Agricultural College, Acharya NG Ranga Agricultural University, Tirupati (Supplementary Fig. 2). The 1000 grain weight was measured from the parents and the mapping populations and expressed in grams.

Development of mapping populations for mapping of grain weight QTLs in rice
Construction of extreme bulks for 1000 grain weight
From a total of 862 F2 plants of BPT5204 × MTU3626 cross, 15 plants each with the extreme high grain weight and low grain weight were selected to establish two bulks i.e. highest grain weight bulk (H-Bulk) and lowest grain weight bulk (L-Bulk), respectively. The genomic DNA from 100 mg fresh rice leaves of the parents and extreme F2 individuals for the grain weight trait were extracted using DNeasy Plant Mini Kit (QIAGEN Sciences) and quantification of DNA was performed using Quant-iTPicoGreen dsDNA reagent and kits (Invitrogen). The bulk DNA samples were prepared by mixing equimolar concentration of extracted DNA from 15 individuals of low-and high-mean grain weight traits and pooled together and two extreme bulks along with two parents were resequenced.
Whole genome resequencing and mapping of reads
A total of 4 libraries (2 bulks—H-Bulk and L-Bulk and 2 parents—BPT5204 and MTU3626) for Illumina sequencing were constructed from 10 µg of DNA sample and sequenced in Illumina Hi-Seq Ten X sequencer. The pair end (PE) short reads (150 × 2) obtained with the Phred quality score of ≥ 30 were used for aligning with the reference genome. Initially mapping of both parents was performed using Os-Nipponbare-Reference-IRGSP-1.0, and then post processing and filtering of the alignment files was executed. Further, called variants used to develop consensus assembly of the donor parent (MTU3626) by substituting the bases with confidence variants calls in the genome. All generated whole genome sequencing data of BPT5204 was used from our previous study [34] and MTU3626 with bulks, whole genome sequencing data is available in SRA database under the BioProject ID: PRJNA482942.
In total, 92.32, 107.41, 122.18 and 124.04 million paired-end (PE) reads for BPT5204, MTU3626, high bulk BPT5204 × MTU3626 and low bulk BPT5204 x MTU3626, respectively were generated (Table 1). GC content of H-bulk and L-bulk was 43.6% and 45.33%, respectively. The raw data was pre-processed using Adapter removal (version: 2.2.0). Cleaning of adapters and pre-processing of reads were performed and high-quality reads from parents and bulks used in mapping and QTL-seq analysis. After trimming and filtering 93230844, 70476388, 75200694 and 75200694 clean reads were procured in BPT5204, MTU3626, H-bulk and L-bulk, respectively. Alignment of clean reads of BPT5204 and MTU3626 with Os-Nipponbare-Reference-IRGSP-1.0 resulted in an average depth of 36.93X and 25.12X and genome coverage of 92.74% and 89.65% respectively, letting us to develop a reference-based assembly for MTU3626 male parent. The aligned samples and the reference genome sequence are used for variant calling with the help of samtools program (samtools version 0.1.18).
Calculation of SNP Index
Whole analysis was performed using QTLseq pipeline (https://genome-e.ibrc.or.jp/home/bioinformatics-team/mutmap) and associated tool i.e., BWA aligner [35], SAM tools [36] and Covel [37]. SNP-index (= count of alternate base/count of reads aligned) was calculated for all the SNP positions found in both bulks using reference genome positions. In total, 422,882 SNPs were identified across all 12 chromosomes (Table 2). However, low quality SNP positions with SNP-index of < 0.3 and read depth < 5 from the two sequences were excluded. If SNPs with SNP-index ≥ 0.3 were present in only one of the sequences obtained from the two DNA High bulk, we considered them as real SNPs and assumed their presence in the other Low bulk too. Further, ΔSNP-index was calculated by subtracting SNP-index of High bulk from SNP-index of Low bulk. SNP-index plots were generated using sliding window analysis with 2 Mb window size and increment of 10 Kb.The possible effects of the identified SNPs were annotated using In-house pipeline VAriMAT. SNPs with SNP index < 0.3 in both bulks were removed as they could be spurious SNPs caused by sequencing errors or alignment errors. The SNP index of remaining SNPs calculated from each bulk was physically plotted on all 12 rice chromosomes. The Δ(SNP index) calculated by subtracting the SNP index values in HGW-bulk by those in LGW-bulk together with the sliding windows of average SNP indices of SNPs located within a 2-Mb region and 1-Kb stepwise were also plotted.
QTLs responsible for changes observed in low and high bulks of grain weight were identified by comparing the ‘SNP-index’ between them. SNP-index represents the frequencies of parental alleles in the bulked individuals. In the BPT5204 × MTU3626 bulks, “SNP-index = 1” indicates that reads in the population are derived only from the MTU3626 genome, whereas “SNP-index = 0” indicates the reads are derived only from BPT5204. A SNP-index of 0.5 indicates an equal genome contribution from both parents and a deviation from 0.5 SNP-index indicates that contribution of that SNP to the phenotypic difference observed in the bulks. Differences in SNP-indices between high and low bulks were plotted through ΔSNP-index with significance of P < 0.05.
Confirmation of identified genomic regions
The QTLs detected in the F2 population using QTL-seq approach were confirmed in F2, F2 and BC1F2 populations of the cross between BPT5204 and MTU3626 using polymorphic markers developed from the InDel regions of QTL-seq analysis. Primers were designed for identified InDel markers using Primer3 and previously reported genomic SSR markers (physically/genetically mapped on selected QTL regions) of chromosomes (Supplementary Table 1).
Parents were screened with these markers to identify polymorphic markers. The PCR was carried out using a programmable thermal cycler (Eppendorf). 2 µl of 50 ng template DNA was pipetted into each of the wells in the PCR plate. Then, the master mix was prepared by taking each 0.5 µl of 10 pmol primer (both forward and reverse primer), 0.5 µl of 10 mM deoxy ribonucleotides (dNTPs), 1 µl of 10X PCR buffer with Mg2+(ABM, Canada) and 0.1 µl of 5 U/µl Taq DNA polymerase (ABM, Canada) and 4.4 µl of sterile distilled water was added to make up the volume to 8 µl. The master mix was centrifuged for short period of about 10 s for thorough mixing of the components. Subsequently, 8 µl of master mix was added to each of wells in the PCR plate well having 2 µl of template DNA to make the final volume to 10 µl. The PCR products were separated on 3.5% agarose gel electrophoresis.
Statistical analysis
Single-marker analysis is the simplest method for detecting markers associated with QTLs. The statistical methods used for single-marker analysis includes, t-tests, analysis of variance (ANOVA) and linear regression. Linear regression is most commonly used method for the coefficient of determination (R2) from the marker that explains the phenotypic variation arising from the marker linked to the QTL [38]. Linear regression analysis was used to study the marker-trait correlation in the study.
Results
Phenotyping of F2 mapping populations
The parents used for mapping, BPT5204 (Samba Mahsuri) and MTU3626 (Prabhat) were analysed for test of significance using paired t-test and observed highly significant differences for plant height, number of tillers per plant, number of panicles per plant, number of grains per panicle, number of filled grains per panicle, spikelet fertility, grain yield per plant, grain length, grain width, grain size and 1000 grain weight suggesting that there is substantial variability between BPT5204 and MTU3626 genotypes (Fig. 2 and Table 3). During kharif 2017, 862 F2 plants of the cross between BPT5204 and MTU3626, along with the parents were evaluated for yield and its component traits. The frequency distribution (Fig. 3b) of 1000 grain weight in F2 mapping population is symmetrical with bell shaped curve. The mean (20.79 g), median (21 g) and mode (20 g) were almost equal, which clearly showed the normal distribution of grain weight trait in F2 mapping population, indicating that this trait was controlled by many genes with small effect in the expression of the trait.

Morphological characteristics of the parents and F1 generations for a Plant height (BPT5204: 95.17 cm, MTU3626: 88.17 cm and F1: 93.28 cm) and b Panicle Length (BPT5204: 24.52 cm, MTU3626: 26.53 cm and F1: 26.33 cm). c Grain length traits (BPT5204: 6.50 mm, MTU3626: 8.40 mm). and d Grain width (BPT5204: 2.00 mm, MTU3626: 3.60 mm)

QTL-seq approach adopted for mapping genomic regions responsible for 1000 grain weight. a Crossing of BPT5204 (Fine-grained parent) and MTU3626(High grain weight parent) to obtain F1 (BPT5204 × MTU3626) progeny. b Frequency distribution based on thousand grain weight of 862 F2 plants. The F2 plants that were selected to build the highest and lowest thousand grain weight bulks are highlighted in the rectangles. c Three Δ(SNP-index) graphs for the chromosomes 1, 7 and 8 plotted using a sliding window of 4 Mb with a step of 10 kb. The significant genomic regions are highlighted in shaded colour (35–40 Mb on chromosome 1, 10–17 Mb on chromosome 7 and 2–5 Mb on chromosome 8)
QTL-seq analysis
A total of 5714 and 6494 SNPs were identified for H-bulk and L-bulk, respectively in selected target regions. Homozygous SNPs identified for H-bulk and L-bulk are 1040 and 1251, respectively (Supplementary Table 1). In total, 1896 SNPs were present in genic region at Read Depth ≥ 5 and among them only 962 SNPs were in the exonic regions of the gene. SNPs with less than 0.3 SNP index in both bulks were removed as they could be spurious SNPs. The SNP index of remaining SNPs calculated from each bulk was physically plotted onto all 12 rice chromosomes. The Δ(SNP index) calculated by subtracting the SNP index values in H-bulk by those in L-bulk together with the sliding windows of average SNP indices of SNPs located within a 2-Mb region and 1 Kb stepwise were also plotted.QTL-seq analysis revealed three genomic regions i.e., qGW1 (35–40 Mb), qGW7 (10–18 Mb), qGW8 (2–5 Mb) on chromosomes 1, 7 and 8 (Fig. 3c and Table 4) for grain weight at P < 0.01.
Confirmation of the QTL-seq derived QTLs in F2, F3 and BC1F2 populations
The polymorphic SSR markers in candidate genomic regions were identified and used for screening the F2 and BC1F2 population (Supplementary Table 2). RM3572 was significantly associated with grain weight trait (qGW8) in F2 mapping population obtained from the cross BPT5204 × MTU3626 (Fig. 4). The per cent contribution of the significantly associated marker RM3572 to total phenotypic variance was 17.88% with a P-value of < 0.0001. RM3572 marker showed significant association with grain weight in F3 and BC1F2 population with a phenotypic variance of 16.70% and 15.00%, respectively at < 0.0001 P-value (Fig. 5) (Table 5).

a Representative agarose gel picture showing segregation pattern of F2 and BC1F2 populations of the cross between BPT5204 and MTU3626. Primer-RM3572, 1–42 are F2 plants, 1–22 are BC1F2 Plants, L-50 bp Ladder, B-BPT5204; M-MTU3626. b Frequency of marker RM3572 genotypes in the F2 and F3 and BC1F2 populations of the cross between BPT5204 and MTU3626 for 1000 grain weight. RM3572 marker genotype: BPT5204-BPT5204 marker allele, MTU3626-MTU marker allele and Heterozygote-heterozygote marker allele

Linear regression analysis for confirmation of grain weight QTL, qGW8 in F2, F3 and BC1F2 populations of a cross between BPT5204 and MTU3626
Discussion
The grain weight is one of the key yield contributing traits in rice. However, the complete mechanism of the inheritance of this complex trait is far from comprehensive understanding in spite of several QTLs and candidate genes were identified. In the present investigation, an attempt was made to uncover QTLs for grain weight employing NGS-based QTL-seq analysis using mapping population derived from BPT5204 and MTU3626. The bold grain variety, MTU3626 shows as much as 29 g of 1000 grain weight-a highest among all cultivated varieties. Therefore, MTU3626 variety was used as one of the parent.
From ∆SNP index plots, three regions on chromosomes 1, 7 and 8 were identified as putative QTLs for 1000 grain weight at P < 0.01. These three QTLs were found to be novel as they were not reported earlier in thesame chromosomal regions. However, in these QTL regions, five yield component traits governing QTLs other than grain weight viz., white berry kernals [39], spikelet fertility QTL, sf1.1 [40], shattering habit at maturity QTL, qSH1 [41], plant height QTL, ph1.1 [40] and panicle length QTL, pl1.1 [42] on chromosome 1, number of spikelets per panicle governing QTL, qSSP7 [43] on chromosome 7 and mean seed fertility controlling QTL, qCTB8 [44] on chromosome 8 were previously reported (Supplementary Table 3).
In the present study, the QTL, qGW8 was detected on chromosome 8 between 2 and 5 Mb region for grain weight. Through single marker analysis, Illa-Berenguer et al. [32] reported significant markers linked to fruit weight QTL (fw3.3) identified from QTL-seq analysis in F2 populations of tomato. The marker-QTL linkage may not be consistent in different genetic backgrounds or in different testing environments, especially for complex traits [45]. Markers validation on independent populations of different genetic background is essential in determining the effectiveness and reliability of markers to predict phenotype of trait [46,47,48,49]. Hence, in the present study, genotypic data of F2 was analysed with the phenotypic data of F3 population and also BC1F2 population was screened with polymorphic markers for QTL confirmation. These results inferred that phenotypic variation of 1000 grain weight in F2, F3 and BC1F2 population was completely in accordance with its genotypic data. These results suggest the reliability of RM3572 marker association with the qGW8 QTL. It was further verified with previously reports and was found that no QTL for grain weight was identified in the selected region. Hence, qGW8 was considered as novel QTL for grain weight in rice. In identified QTL region, alleles from MTU3626 contributed to higher trait value and lower trait value. From the mean values of marker classes, MTU3626 was identified as the source of favourable allele for grain weight. Furthermore, this QTL explained more than 15% phenotypic variance in all three populations suggesting that this is a major QTL. Hence, it can be targeted for candidate gene identification and subsequently this QTL can also be transferred to low yielding varieties via marker-assisted breeding.
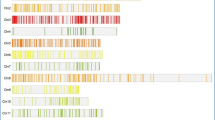
A total of 45 annotated genes are present in the genomic region (2–5 Mb) of qGW8 QTL as per the rice genome annotation project- database (RAP-DB). Among them, five genes viz., LOC_Os08g01490 (Cytochrome P450), LOC_Os08g01510 (Cytochrome P450), LOC_Os08g01520 (Cytochrome P450), LOC_Os08g01680 (WD domain, G-beta repeat domain containing protein) and LOC_Os08g01780 (OsIAA25-Auxin-responsive Aux/IAA gene family member) were considered as putative candidate genes controlling grain weight in rice as similar kind of genes in different genomic regions were reported to control grain weight in rice and other crops as per the previous literature (Table 6). In addition, the cytochrome P450 controlling genes, LOC_Os08g01520, LOC_Os08g01510 and LOC_Os08g01520 () found to be over expressed while LOC_Os08g01680 (WD domain, G-beta repeat domain containing protein) exhibited low expression in lemma and palea as per the RiceXPro (https://ricexpro.dna.affrc.go.jp/) (Fig. 6), hence, can be considered as probable candidate genes.

Gene expression analysis of candidate genes underlying grain weight QTL region. More intense red colour represents the high expression while more intense blue colour indicates low expression. X-axis—various tissues of rice inflorescence
Genes encoding cytochrome P450 are known to be involved in brassinosteroid biosynthesis [50]. Brassinosteroids (BRs) are a class of growth-promoting steroid plant hormone, which are crucial for normal growth and development, such as plant height, leaf angle, panicle architecture, and seed size [51]. Liu et al. [52] reported that GW5 could repress the kinase activity of GSK2 (Glycogen synthase kinase2) toward OsBZR1 (Oryza sativa BRASSINAZOLE RESISTANT1) and DLT (DWARF AND LOW-TILLERING), resulting in accumulation of their unphosphorylated forms and altered BR signalling. Therefore, GW5 might be a positive regulator of the BR signalling pathway in regulating grain width and weight in rice. Daware et al. [26] identified a cytochrome P450 and serine carboxy peptidase protein encoding genes in the 1000 grain weight QTL, OsqGW5.1 on chromosome 5 of rice using QTL-seq approach in the F4:5 population of the cross between IR64 and Sonasal. Rice encodes one each of Gα and Gβ, and five Gγ proteins [53] and Botella et al. [54]. Both Gα and Gβ proteins are positive regulators of cell proliferation and grain size in rice [55]. Loss of function of Gα (RGA1) or suppression of Gβ (RGB1) decreases grain size in rice [56], and [57], suggesting that growth of rice grain is also regulated by Gα and Gβ proteins. Utsunomiya et al. [57] reported that RICE G PROTEIN BETA SUBUNIT (RGB1) (Os03g0669100) belongs to G protein β subunit protein category, is a positive regulator of cell proliferation in rice. Ishimaru et al. [58] reported that TGW6 (THOUSAND GRAIN WEIGHT 6) encodes a novel protein with indole-3-acetic acid (IAA)-glucose hydrolase activity and positively regulates free IAA levels in rice grains and loss of function of TGW6 enhances rice grain weight. Through RNA-sequencing analysis, Hu et al. [59] reported that OsARF4 and OsSK41 at qTGW3 repress the expression of a common set of downstream genes, including some auxin-responsive genes, during rice grain development and loss of function of OsARF4 results in larger rice grains.
In conclusion, the present study demonstrates the rapid identification of QTLs employing QTL-seq approach and the prediction of candidate genes exploiting rice genome database. Hence, these genes also might have a positive role in controlling grain weight in rice. However, further confirmation of this novel QTL is warranted before exploiting to develop elite cultivars through MAS. The novel QTL identified in the present study undoubtedly enhance our understanding of the complex nature of the yield component traits in rice.
Abbreviations
- SM:
-
Samba Mahsuri
- HGW:
-
High grain weight
- QTLs:
-
Quantitative trait loci
- NGS:
-
Next generation sequencing
- NGM:
-
Next generation mapping
- BSA:
-
Bulked segregant analysis
- SNP:
-
Single nucleotide polymorphism
- QTLs:
-
Quantitative trait loci
- TGW:
-
Thousand grain weight
- ANOVA:
-
Analysis of variance
- RAP-DB:
-
Rice annotation project-database
References
Khush GS (2005) What it will take to feed 5.0 billion rice consumers in 2030. Plant Mol Biol 59(1):1–6
Ray DK, Mueller ND, West PC, Foley JA (2013) Yield trends are insufficient to double global crop production by 2050. PLoS ONE 8(6):e66428
Chun Y, Fang J, Zafar SA, Shang J, Zhao J, Yuan S, Li X (2020) MINI SEED 2 (MIS2) encodes a receptor-like kinase that controls grain size and shape in rice. Rice 13:7. https://doi.org/10.1186/s12284-020-0368-9
Huang R, Jiang L, Zheng J, Wang T, Wang H, Huang Y, Hong Z (2013) Genetic bases of rice grain shape: so many genes, so little known. Trends Plant Sci 18:218–226
Zafar SA, Patil SB, Uzair M, Fang J, Zhao J, Yuan S, Uzair M, Luo Q, Shi J, Schreiber L, Li X (2020) DEGENERATED PANICLE AND PARTIAL STERILITY 1 (DPS1) encodes a CBS domain containing protein required for anther cuticle and panicle development in rice. New Phytol 225:356–375
Wing RA, Purugganan MD, Zhang Q (2018) The rice genome revolution: from an ancient grain to Green Super Rice. Nat Rev Genet 19:505–517
Qian Q (2018) Chapter 11, gene network of grain size and number in rice. In: Sasaki T, Ashikari M (eds) Rice genomics, genetics and breeding. Springer Nature Singapore Pte Ltd., Singapore, pp 191–206
Li N, Li YH (2016) Signaling pathways of seed size control in plants. Curr Opin Plant Biol 33:23–32
Duan PG, Xu JS, Zeng DL, Zhang BL, Geng MF, Zhang GZ, Huang K, Huang LJ, Xu R, Ge S, Qian Q, Li YH (2017) Natural variation in the promoter of GSE5 contributes to grain size diversity in rice. Mol Plant 10(5):685–694
Yu JP, Xiong HY, Zhu XY, Zhang HL, Li HH, Miao JL, Wang WS, Tang ZS, Zhang ZY, Yao GX, Zhang Q, Pan YH, Wang X, Rashid MAR, Li JJ, Gao YM, Li ZK, Yang WC, Fu XD, Li ZC (2017) OsLG3 contributing to rice grain length and yield was mined by Ho-LAMap. BMC Biol 15:28
Yi CD, Wang DR, Jiang W, Li W, Cheng XJ, Wang Y, Zhou Y, Liang GH, Gu MH (2016) Development of functional markers and identification of haplotypes for rice grain width gene GS5. Chin J Rice Sci 30(5):487–492
Huang K, Wang DK, Duan PG, Zhang BL, Xu R, Li N, Li YH (2017) WIDE AND THICK GRAIN 1, which encodes an otubain-like protease with deubiquitination activity, influences grain size and shape in rice. Plant J 91(5):849–860
Wang S, Li S, Liu Q, Wu K, Zhang J, Wang S, Wang Y, Chen X, Zhang Y, Gao C, Wang F, Huang H, Fu X (2015) The OsSPL16-GW7 regulatory module determines grain shape and simultaneously improves rice yield and grain quality. Nat Genet 47(8):949–954
Yaobin Q, Cheng P, Cheng Y, Feng Y, Huang D, Huang T, Song X, Ying J (2018) QTL-seq identified a major QTL for grain length and weight in rice using near isogenic F2 population. Rice Sci 25:121–131
Michelmore RW, Paran I, Kesseli RV (1991) Identification of markers linked to disease resistance genes by bulked segregant analysis: a rapid method to detect markers in specific genomic regions by using segregating populations. . Proc Natl Acad Sci USA 88:9828–9832
Schneeberger K, Ossowski S, Lanz C, Juul T, Petersen AH, Nielsen KL, Jorgensen JE, Weigel D, Andersen SU (2009) SHOREmap: simultaneous mapping and mutation identification by deep sequencing. Nat Methods 6:550–551
Austin RS, Vidaurre D, Stamatiou G, Breit R, Provart NJ, Bonetta D, Zhang J, Fung P, Gong Y, Wang PW, McCourt P, Guttman DS (2011) Next-generation mapping of Arabidopsis genes. Plant J 67:715–725
Abe A, Kosugi S, Yoshida K, Natsume S, Takagi H, Kanzaki H, Matsumura H, Yoshida K, Mitsuoka C, Tamiru M, Innan H, Cano L, Kamoun S, Terauchi R (2012) Genome sequencing reveals agronomically important loci in rice using MutMap. Nat Biotechnol 30:174–178
Hartwig B, James GV, Konrad K, Schneeberger K, Turck F (2012) Fast isogenic mapping-by-sequencing of ethyl methane sulfonate-induced mutant bulks. Plant Physiol 160:591–600
Lindner H, Raissig MT, Sailer C, Shimosato-Asano H, Bruggmann R, Grossniklaus U (2012) SNP-ratio mapping (SRM): Identifying lethal alleles and mutations in complex genetic backgrounds by next-generation sequencing. Genetics 191:1381–1386
Fekih R, Takagi H, Tamiru M, Abe A, Natsume S, Yaegashi H, Sharma S, Sharma S, Kanzaki H, Matsumura H, Saitosh H, Mitsuoka C, Utsushi H, Uemura A, Kanzaki E, Kosugi S, Yoshida K, Cano L, Kamoun S, Terauchi R (2013) MutMap+: genetic mapping and mutant identification without crossing in rice. PLoS ONE 8:e68529
Takagi H, Abe A, Yoshida K, Kosugi S, Natsume S, Mitsuoka C, Uemura A, Utsushi H, Tamiru M, Takuno S, Innan H, Cano LM, Kamoun S, Terauchi R (2013) QTL-seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J 74:174–183
Takagi H, Uemura A, Yaegashi H, Tamiru M, Abe A, Mitsuoka C, Utsushi H, Natsume S, Kanzaki H, Matsumura H, Saitosh H, Yoshida K, Cano LM, Kamoun S, Terauchi R (2013) MutMap-Gap: wholegenome resequencing of mutant F2 progeny bulk combined with de novo assembly of gap regions identifies the rice blast resistance gene Pii. New Phytol 200:276–283
Sun XW, Liu DY, Zhang XF, Li WB, Liu H, Hong W, Jiang C, Guan N, Ma C, Zeng H, Xu C, Song J, Huang L, Wang C, Shi J, Wang R, Zheng X, Lu C, Wang X, Zheng H (2013) SLAFseq: an efficient method of large-scale De Novo SNP discovery and genotyping using high-throughput sequencing. PLoS ONE 8:e58700
Singh VK, Khan AW, Saxena RK, Sinha P, Kale SM, Parupalli S, Kumar V, Chitikineni V, Suryanarayana V, Kumar CVS, Sharma M, Anuradha G, Yamini KN, Muniswamy S, Varshney RK (2017) Indel-seq: a fast forward genetics approach for identification of trait associated putative candidate genomic regions and its application in pigeonpea (Cajanus cajan). Plant Biotechnol J 15:906–914
Daware A, Das S, Srivastava R, Badoni S, Singh AK, Agarwal P, Parida SK, Tyagi AK (2016) An efficient strategy combining SSR markers- and advanced QTL-seq-driven QTL mapping unravels candidate genes regulating grain weight in rice. Front Plant Sci 7:1535–1545
Ogiso-Tanaka E, Tanaka T, Tanaka K, Nonoue Y, Sasaki T, Fushimi E, Koide Y, Okumoto Y, Yano M, Saito H (2017) Detection of novel QTLs qDTH4.5 and qDTH6.3, which confer late heading under short-day conditions, by SSR marker-based and QTL seq analysis. Breed Sci 67:101–109
Kadambari G, Vemireddy LR, Srividhya A, Nagireddy R, Jena SS, Gandikota M, Patil S, Veeraghattapu R, Deborah DAK, Reddy GE, Shake M, Dasari A, Rao PVR, Durgarani ChV, Neeraja CN, Siddiq EA, Sheshumadhav M (2018) QTL-Seq-based genetic analysis identifies a major genomic region governing dwarfness in rice (Oryza sativa L.). Plant Cell Rep 37:677–687
Das S, Upadhyaya HD, Bajaj D, Kujur A, Badoni S, Kumar V, Tripathi S, Gowda CL, Sharma S, Singh S, Tyagi AK, Parida SK (2015) Deploying QTL-seq for rapid delineation of a potential candidate gene underlying major trait-associated QTL in chickpea. DNA Res 22:193–203
Singh VK, Khan AW, Jaganathan D, Thudi M, Roorkiwal M, Takagi H, Garg V, Kumar V, Chiyikineni A, Gaur PM, Sutton T, Terauchi R, Varshney RK (2017) QTL-seq for rapid identification of candidate genes for 100-seed weight and root/total plant dry weight ratio under rainfed conditions in chickpea. Plant Biotechnol J 15:906–914
Lu HF, Lin T, Klein J, Wang SH, Qi JJ, Zhou Q, Sun JJ, Zhang ZH, Weng YQ, Huang SW (2014) QTL-seq identifies an early flowering QTL located near flowering locus T in cucumber. Theor Appl Genet 127:1491–1499
Illa-Berenguer E, Houten JV, Huang Z, Knaap EV (2015) Rapid and reliable identification of tomato fruit weight and locule number loci by QTL-seq. Theor Appl Genet 128:1329–1342
Ruangrak E, Du Y, Htwe NMPS, Pimorat P, Gao J (2019) Identification of early tomato fruit ripening loci by QTL-seq. J Agric Sci 11(2):51–70
Lachagari VBR, Gupta R, Lekkala SP, Mahadevan L, Kuriakose B, Chakravartty N, Mohan Katta AVSK, Santhosh S, Reddy AR, Thomas G (2019) Whole genome sequencing and comparative genomic analysis reveal allelic variations unique to a purple colored rice landrace (Oryza sativa ssp. indica cv. Purpleputtu). Front Plant Sci 10:513. https://doi.org/10.3389/fpls.2019.00513
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760
Li S, Qian Q, Fu Z, Zeng D, Meng X, Kyozuka J, Maekawa M, Zhu X, Zhang J, Li J, Wang Y (2009) Short panicle1 encodes a putative PTR family transporter and determines rice panicle size. Plant J 58:592–605
Kosugi S, Natsume S, Yoshida K, MacLean D, Cano L, Kamoun S, Terauchi R (2013) Coval: improving alignment quality and variant calling accuracy for next-generation sequencing data. PLoS ONE 8(10):e75402
Collard BCY, Jahufer MZZ, Brouwer JB, Pang ECK (2005) An introduction to markers, quantitative trait loci (QTL) mapping and marker-assisted selection for crop improvement: the basic concepts. Euphytica 142:169–196
Kajitani T, Yamamoto Y, Yano M, Politics S (2008) Detection of QTL related to the appearance quality of brown rice in rice using chromosomal fragment substitution line groups. Breed Res 10:91–99
Marri PR, Sarla N, Reddy LV, Siddiq EA (2005) Identification and mapping of yield and yield related QTLs from an Indian accession of Oryza rufipogon. BMC Genet 6:33
Onishi K, Horiuchi Y, Ishigoh-Oka N, Takagi K, Ichikawa N, Maruoka M, Sano Y (2007) A QTL cluster for plant architecture and its ecological significance in Asian wild rice. Breed Sci 57:7–16
Septiningsih EM, Prasetiyono J, Lubis E, Tai TH, Tjubaryat T, Moeljopawiro S, McCouch SR (2003) Identification of quantitative trait loci for yield and yield components in an advanced backcross population derived from the Oryza sativa variety IR64 and the wild relative O. rufipogon. Theor Appl Genet 107:1419–1432
Xing YZ, Tang WJ, Xue WY, Xu CG, Zhang Q (2008) Fine mapping of a major quantitative trait loci, qSSP7, controlling the number of spikelets per panicle as a single Mendelian factor in rice. Theor Appl Genet 116:789–796
Kuroki M, Saito K, Matsuba S, Yokogami N, Shimizu H, Ando I, Sato Y (2007) A quantitative trait locus for cold tolerance at the booting stage on rice chromosome 8. Theor Appl Genet 115(5):593–600
Boranayaka MB, Lokesha R, Diwan JR, Patil R (2018) Marker validation in F2 population of rice (Oryza sativa L.) for water and nitrogen use efficiency. Int J Curr Microbiol Appl Sci 7(1):1275–1278
Lin H, Yanagihara S, Zhuang J (1998) Identification of QTL for salt tolerance in rice via molecular markers. Chin J Rice Sci 12(2):72
Cakir M, Gupta S, Platz GJ, Ablett GA, Loughman R, Emebiri LC, Poulsen D, Li CD, Lance RCM, Galwey NW, Jones MGK, Appels R (2003) Mapping and validation of the genes for resistance to Pyrenophora teres f. teres in barley (Hordeum vulgare L.). Aust J Agric Res 54:1369–1377
Collins HM, Panozzo JF, Logue SJ, Jefferies SP, Barr AR (2003) Mapping and validation of chromosome regions associated with high malt extract in barley (Hordeum vulgare L.). Aust J Agric Res 54:1223–1240
Speilmeyer W, Sharp PJ, Lagudah ES (2003) Identification and validation of markers linked to broad-spectrum stem rust resistance gene Sr2 in wheat (Triticum aestivum L.). Crop Sci 43:333–336
Gupta PK, Rustgi S, Kumar N (2006) Genetic and molecular basis of grain size and grain number and its relevance to grain productivity in higher plants. Genome 49:565–571
Wu Y, Fu Y, Zhao S, Gu P, Zhu Z, Sun C, Tan L (2016) CLUSTERED PRIMARY BRANCH 1, a new allele of DWARF11, controls panicle architecture and seed size in rice. Plant Biotechnol J 14:377–386
Liu J, Chen J, Zheng X, Wu F, Lin Q, Heng Y, Tian P, Cheng Z, Yu X, Zhou K, Zhang X, Guo X, Wang J, Wang H, Wan J (2017) GW5 acts in the brassinosteroid signalling pathway to regulate grain width and weight in rice. Nature Plants 3:17043
Urano D, Chen J, Botella JR, Jones AM (2013) Heterotrimeric G protein signalling in the plant kingdom. Open Biol 3:120186
Botella JR (2012) Can heterotrimeric G proteins help to feed the world? Trends Plant Sci 17:563–568
Oki K, Fujisawa Y, Kato H, Iwasaki Y (2005) Study of the constitutively active form of the alpha subunit of rice heterotrimeric G proteins. Plant Cell Physiol 46:381–386
Ashikari M, Wu J, Yano M, Sasaki T, Yoshimura A (1999) Rice gibberellin-insensitive dwarf mutant gene Dwarf 1 encodes the alpha-subunit of GTP-binding protein. Proc Natl Acad Sci USA 96:10284–10289
Utsunomiya Y, Samejima C, Takayanagi Y, Izawa Y, Yoshida T, Sawada Y, Fujisawa Y, Kato H, Iwasaki Y (2011) Suppression of the rice heterotrimeric G protein beta-subunit gene, RGB1, causes dwarfism and browning of internodes and lamina joint regions. Plant J 67:907–916
Ishimaru K, Hirotsu N, Madoka Y, Murakami N, Hara N, Onodera H, Kashiwagi T, Ujiie K, Shimizu B, Onishi A, Miyagawa H, Katoh E (2013) Loss of function of the IAA-glucose hydrolase gene TGW6 enhances rice grain weight and increases yield. Nat Genet 45:707–711
Hu Z, Lu SJ, Wang MJ, He H, Sun L, Wang H, Liu XH, Jiang L, Sun JL, Xin X, Kong W, Chu C, Xue HW, Yang J, Luo X, Liu JX (2018) A novel QTL qTGW3 encodes the GSK3/SHAGGY-like kinase OsGSK5/OsSK41 that interacts with OsARF4 to negatively regulate grain size and weight in rice. Mol Plant 11:736–749
Acknowledgements
BRY is grateful to the Institute of Frontier Technology and S.V. Agricultural College, Tirupati, ANGRAU for providing the facilities to conduct the experiment.
Funding
LRV is grateful to the Department of Biotechnology, Government of India for providing financial assistance [DBT-NER/AGRI/29/2015(GROUP- 4) dated 19/10/2016].
Author information
Authors and Affiliations
Contributions
BRY, LRV and HKR planned the experiment. BRY and LRV contributed to the development of mapping populations. BRY performed the field experiments. JBN, BMM, EGR, GK, SBN and KI helped in phenotyping. SA, SC, RP and SP provided required materials and helped in maintaining the experiment. NC, RB, VBRL and SG sequenced the parents and bulks. NC, VBRL analysed the QTL-seq data. BRY, LRV SG, VBRL, and NC wrote the manuscript. VBRL, GT, SG, PSK and SPL reviewed the manuscript. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declared that there is no potential conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Bommisetty, R., Chakravartty, N., Bodanapu, R. et al. Discovery of genomic regions and candidate genes for grain weight employing next generation sequencing based QTL-seq approach in rice (Oryza sativa L.). Mol Biol Rep 47, 8615–8627 (2020). https://doi.org/10.1007/s11033-020-05904-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-020-05904-7