
Abstract
Duckweeds belong to the smallest aquatic flowering plant family, Lemnaceae, and have a rapid doubling time, making this group an excellent system to study reduced morphology and wide environmental adaptability at the molecular level. Despite the availability of genomic and transcriptomic data for duckweed member, Lemna minor, lack of an efficient genetic transformation system has limited its use in plant molecular biology research. The present study reports an efficient and rapid Rhizobium rhizogenes-mediated root transformation system for L. minor. Two different factorial experiments were designed to test the effect of explant type, age, culture media and inoculation methods on transformation efficiency. Leaf and root tip cut explants were inoculated with R. rhizogenes strain MSU 440 harboring pBIN-YFP vector using yellow fluorescent protein (YFP) as a reporter gene for identification of transgenic roots. In addition, two different culture media, full MS and 0.25X Hoagland, and four different infection methods, solid culture, centrifugation, liquid culture and sonication, were compared. After 8 weeks, about 17% of the root tip-cut explants infected via the solid culture method and maintained in 0.25X Hoagland medium had YFP-expressing roots. These transgenic L. minor roots were morphologically similar to normal roots and PCR analysis demonstrated that the YFP-expressing roots were positive for the integration-expected rol genes. The described optimized root transformation procedure is a valuable tool for pursuing high-throughput gene characterization studies in L. minor.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lemnaceae, commonly known as duckweeds, are the smallest and fastest growing angiosperms. Other than duckweed’s potential as a biomass source (Muradov et al. 2010), it has gained wide attention as a model system to study important biological processes, such as uranium clocks and rhythmic (Muranaka et al. 2015) phytotoxicity (Horemans et al. 2015), transfer of heavy metals to food chain (Lahive et al. 2014) and adaptation to environmental conditions (Appenroth and Adamec 2014). While such mechanisms have been studied at the morphological and biochemical level, studies on physiological mechanisms and genetic improvement awaited support from molecular work. Recent work on sequencing genomes of several duckweed species (Wang and Messing 2011; Wang et al. 2014 reviewed by Cao et al. 2015) and transcriptomics work under different growth and stress conditions (Wang and Messing 2014) have generated a considerable amount of sequencing data. However, lack of availability of a simple plant transformation system has limited the functional characterization of candidate genes and pathways identified through high-throughput sequencing studies. Chhabra and colleagues (Chhabra et al. 2011) have further improved the previously published (Yamamoto et al. 2001) Rhizobium tumefaciens (formally known as Agrobacterium tumefaciens)-mediated transformation protocol for L. minor. While transgenic fronds were obtained within 3 weeks, the procedure requires a preparatory period of about 10 weeks. Further, the calli generated from frond explants are used for R. tumefaciens inoculation, which is not ideal, as it is known that indirect organogenesis through hormonal treatments of calli may affect genetic stability and lead to somaclonal variations. The described work to develop a relatively simple and quick method for duckweed transformation will greatly facilitate high-throughput genetic studies.
Similar to R. tumefaciens, R. rhizogenes is a soil-borne bacterium that infects dicotyledonous plants. The bacterium responds to the chemical signals, particularly acetosyringone, released by wounded plant tissues (Winans 1992) and infects plant tissue through wounds. R. rhizogenes transfers and integrates a portion of its root-inducing (Ri) plasmid, consisting of root locus (rol) genes, into the host plant genome. Like the R. tumefaciens, Ti plasmid, the Ri plasmid gene products encode growth regulators. Rather producing than stimulating the development of a gall-like morphological structure; however, infection of host plant roots by R. rhizongenes leads to the proliferation of multi-branched roots called “hairy roots” at the site of infection (Chilton et al. 1982). R. rhizogenes-transformed roots are widely used in plant biology, including plant regeneration (Tepfer 1984), secondary metabolites production (Tian 2015), expression of foreign genes (Lonoce et al. 2016), studying biology of plant roots (Estrada-Navarrete et al. 2006; Plasencia et al. 2016), gene silencing (Limpens et al. 2004; Ron et al. 2014; Bandaranayake and Yoder 2013), assaying genes specific for rhizosphere interactions including nematodes (Li et al. 2011), nitrogen-fixing rhizobia (Kasai and Kanazawa 2011) mycorrhiza (Kuster et al. 2007) and root parasitic plants (Fernadez-Aparicio et al. 2011; Ishida et al. 2011; Bandaranayake et al. 2010).
Here we introduce an R. rhizogenes-mediated transformation method that results in chimeric plants with transgenic roots and non-transgenic fronds ready for functional assays within 8 weeks. Two different types of explants, two different types of media and three different infection methods were tested.
Material and methods
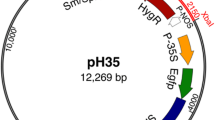
L. minor, known as common duckweed, was used as the plant material. The R. rhizogenes strain, MSU 440 harboring pBIN-YFP plasmid kindly provided by the Yoder lab at UC-Davis was used for all the experiments (Bevan 1984; Tomilov et al. 2006). The plasmid pBIN-YFP (11,775 bp) is a binary vector consists of kanamycin resistance gene for bacterial origin of replication and T-DNA region. Within the T-DNA borders there are kanamycin and YFP reporter genes for plant selection. When YFP is driven by CaMV 35S promoter (Tomilov et al. 2006) and kanamycin is driven by nos promoter (Bevan 1984). The bacteria were grown on MGL media (Walkerpeach and Velten 1994) at 28 °C.
First, the sterilization method, growth conditions, temperature, light intensity, solid media composition were optimized for L. minor growth. Each experiment consisted of three biological replicates with 15–30 individual plants in each replicate.
Optimization of surface sterilization
Four different sterilization methods previously used for Lemna spp were tested with some modifications (Table 1). After sterilization, individual plants were cultured in test tubes containing 0.25X Hoagland medium with 10 g/l sugar and maintained at 25 °C room at 16 h light and 8 h dark intervals. After 2 weeks, plants that survived without contamination were counted.
Optimization of L. minor growth
To determine the optimum temperature range (23–25) oC, (29–32) oC and (35–38) oC were tested at 85 µmolm−2 s−1 light intensity. The growth of L. minor was recorded for 5 consecutive days. Three different light intensities 10, 46, 85 µmolm−2 s−1were tested at (29–32) oC temperature.
Murashige and Skoog (MS) (Murashige and skoog 1962) medium has been used in previous transformation experiments. The growth performances of L. minor on solid Hoagland and MS medium were tested. Each experiment was replicated three times and maintained at 16 h light and 8 h dark conditions. The root growth was measured for 6 days in one-day intervals.
R. rhizogenes transformation
Four different R. rhizogenes infection methods previously used for other species (streak, liquid culture, sonication and centrifugation) and two different culture media (full MS with 3% sugar, 0.25 × Hoagland 1% sugar) were tested with root tip-cut fronds as explants.
Streak method
For the streak infection method (Walkerpeach and Velten 1994), an overnight liquid culture obtained from a single colony of MSU 440 with the plasmid was spread on a MGL plate with 50 µg/l kanamycin and allowed to grow at 29 °C for 12–24 h, until a creamy lawn of bacteria was observed (Fig. 1a). Then, from (Fig. 1b) Lemna explants, roots were wounded by cutting about 0.5 cm from the root tip with a sharp pair of scissors according to Fig. 1c and the cut surface was dipped carefully into the creamy culture (Fig. 1d). Plantlets were placed on either MS or Hoagland media consisted of 200 µm acetosyringone (Fig. 1e). The plates were maintained at 16 °C with 85 µmolm−2 s−1 light intensity for 2 days of co-cultivation.

R. rhizogenes inoculation: streak method. aR. rhizogenes culture on solid MGL media grown for 24 h. b Healthy sterile plants used as explants. c Method of explants root cut. d Surface of the wound, streak on the bacterial lawn. e Introduce transformed plants into MS or Hoagland media. Size bar − 1 cm
Liquid method
For the liquid culture infection method (Yamamoto et al. 2001), 200 ml of liquid MGL culture with the plasmid was grown at 160 rpm until an OD600 value of 0.5 was reached. Then, 200 µm of acetosyringone was added to the culture. Sharply cut explants roots (Fig. 1c) were introduced to the liquid bacterial culture and shaken for 20 min. Explants were then transferred to either liquid MS or Hoagland media and kept on the shaker for 3 days at 16 °C for co-cultivation.
Sonication
For the sonication infection method (Finer and Trick 1997) the MSU 440 culture was again grown to an OD600 of 0.5, followed by the addition of 200 µm acetosyringone. Then, 1.5 ml sterile tubes were filled with the prepared culture and a single wounded plant was introduced to each tube. After three minutes of sonication, the plants were transferred to either MS or Hoagland plates with sugar and kept in 16 °C for 3 days.
Centrifugation
For the centrifugation infection method (Thu et al. 2015), a transformation culture was prepared as described for the sonication method. Again, 1.5 ml sterile tubes were filled with the prepared culture and a single wounded plant was introduced to each tube. However, instead of sonication the tubes were then centrifuged at 12,000 rpm for 10 min and kept at room temperature for another 20 min before transferring to co-cultivation plates and maintaining for 3 days at 16 °C.
After the co-cultivation period, explants were transferred to plates containing the same medium (either MS or Hoagland) with 300 mg/l timentin, but without sugar or acetosyringone, and kept horizontally at 25 °C temperature with 85 µmolm−2 s−1 light intensity.
Further optimization of streak method
Since the streak method resulted in the highest transformation efficiency in initial experiments and with easy preparatory and handling steps, it was selected as the preferred method for continued optimization for media types, explant age, etc. Two different explant ages (two fronds and four fronds), two different media types (full MS with 3% sugar and 0.25X Hoagland with 1% sugar) and two different explant types (root tip cut and leaf cut) were tested in another factorial experiment.
Across all of the experiments, a single Petridish with 10–15 plants was considered a technical replicate. The number of times each experiment was repeated were counted as biological replicates. Number of YFP-expression roots per Petridish and number of YFP-expressing roots per plant were counted after 8 weeks from the date of transformation using an Olympus (SZX10 Japan) stereo zoom research florescence microscope equipped with a YFP filter set with excitation HQ490-500, DM505, and emission HQ515-610. Images were captured with a C-mount CCD camera.
Integration of rol genes into plant genome
PCR primers were designed based on previous literature (Table 2) to confirm integration of rolB, rolC, virD2 genes. Absence of Rhizobium surrounding the roots (potential source of false positive PCR results) was confirmed by transferring roots to sugar-containing media before performing PCR. Selected L. minor root tips were isolated from individual plants and put into 1.5 ml tubes containing 2 µl of TE buffer, one root tip per tube, then crushed with a pipette tip. The root tip-TE buffer mixture was then stored in the refrigerator for 1 h. 1 µl of the solution was added to the PCR mix containing, 5 µl of 5 × PCR buffer, 1.5 µl of 25 mM MgCl2, 0.4 µl of 10 mM dNTP, 3 µl of 10 mM spermidine, 0.5 µl of 10 mM forward primer; 0.5 µl of 10 mM reverse primer, 0.1 µl of Taq polymerase (Go-TaqPomega) and 13 µl of nuclease free water. The PCR program was set with initial denaturation at 94 °C for 5 min followed by 35 cycles at of 94 °C for 30 s, 62 °C for 30 s, 72 °C for 1 min and a final extension of 72 °C for 10 min. A well-grown R. rhizogenes MSU 440 colony was suspended in water and used as a positive control. The PCR products were subjected to agarose gel electrophoresis (1%) and analyzed with a size marker.
Results and discussion
Optimization of L. minor growth
It is essential to have an optimized sterilization protocol for any in vitro study including Rhizobium transformation. To achieve this objective, four different sterilization methods previously used for Lemna species were tested with some modifications (Table 1). However, the optimization was significant only for method two (Fig. 2a) where the time of both Clorox treatment and the ethanol treatment doubled. With this, success of optimized method increased above 90% compared to 14% achieved with method described by Thomson and Dennis (2013).

Optimization of sterilization and growth. a Sterilization methods. b Temperature. c Light intensity. d Growth of L. Minor at 85 µmol m−2 s−1 light intensity. e Solid media. Values presented are mean ± SD, n = 30 for a, n = 90 for b, n = 90 for c, n = 18 for e (n = total number of explants in all the replicates)
Previous studies with L. minor have suggested different temperature ranges for its optimum growth. For example, Landolt and Kandeler (1987) reported 26 °C as the optimum temperature, while Leng (1999) suggested that the growth and proliferation of L. minor increased with increasing temperature up to 33 °C and decreases thereafter. Further, studies done by Heide et al. (2006) concluded that 38 °C was lethal for L. minor. Therefore, we tested three different temperature ranges (23–25) oC, (29–32) oC and (35–38) oC and, of them, the number of fronds counted after 5 days (Fig. 3a–f) were significantly higher in the culture maintained at the temperature (29–32) oC (Fig. 2b).

Growth of L. minor under optimum conditions identified. a Day 0. b Day 3. c Day 5. d Day 6. e Day 7. f Day 8. Size bar − 1 cm
Further, three different light intensities were tested. L. minor did not prefer low light intensity, while there was no significant difference between 46 µmolm−2 s−1 and 85 µmolm−2 s−1 (Fig. 2c). Previous works have also suggested higher light intensities for L. minor growth (Tabou et al. 2013).
It has been suggested that the doubling time of L. minor is around 48 h at optimum conditions (Leng 1999). When L. minor plants were grown in 0.25X Hoagland medium with 1% sugar and maintained at (29–32) oC and 85 µmolm−2 s−1 light intensity, the population doubled every 48 h (Fig. 2d). Therefore, we considered those conditions as optimal for the growth of L. minor in our experimental setup.
MS medium has been used in previous transformation experiments of L. minor (Yamamoto et al. 2001), while both Hoagland and MS media have frequently been used in R. rhizogenes transformation and subsequent experiments with the roots of many species (Ron et al. 2014; Bandaranayake and Yoder 2018; ono et al. 2012). Therefore, root growth of L. minor was studied on 0.25X Hoagland with 1% sugar and full MS with 3% sugar in these experiments. The results showed that L. minor root growth was significantly higher on Hoagland medium than on MS medium (Fig. 2e). Interestingly, the tested 0.25X Hoagland medium contains a lower amount of N (52.5 mg/l) than that of the tested full MS (776.69 mg/l). Nitrogen in the tested Hoagland medium derived from CaNO3 and KNO3, while it was derived from NH4NO3 and KNO3 in the tested MS medium. Previous work of Porath and Pollock (1982) and Fang et al. (2007) showed that duckweed prefers uptake of NH4+ than NO3−; however, works by Oron et al. (1985) and Caicedo et al. (2000) showed that higher NH4+ concentrations inhibit the growth of duckweed. Oscarson et al. (1988) found out that root growth of L. minor decreases with the higher availability of nitrogen, a result that seems to be supported by the research finding reported here.
Suitable transformation method for L. minor
There are various methods developed for successful infection of R. rhizogenes. The preferred method varies mainly with the plant species. However, most scientists prefer methods optimized for speed and ease of use, especially for large scale studies. Hence, we tested four available R. rhizogenes infection methods with the objective of achieving the highest efficiency within the shortest time duration. Additionally, two different co-cultivation media were compared for each method.
In all the transformation methods describe here, resulting plants are chimeric, where leaves or top part of the plants are non-transgenic and roots generate from the infected cut surface are transgenic (Fig. 4a–e). The number of plants per plate with at least one YFP-expressing root was counted and the percentage was taken as the transformation efficiency (Fig. 5a). Further, when present, number of YFP-expressing roots per plant were counted and expressed as a fraction of total number of roots present and introduced as YFP expression efficiency (Fig. 5b). None of the plants survived more than 5 days after being subjected to sonication for 3 minutes and, therefore, the sonication treatment could not be continued.

Transgenic L. minor roots. a, b Under white light. c, d, e Under florescent microscope. Size bar = 1 mm

Transformation efficiency and YFP expression. a The effect of inoculation method and co-cultivation medium. b Effect of inoculation method and culture media. c Effect of explant age and media. Values presented are mean ± SD of 3–5 technical replicates and three biological replicates n = 540 for a, n = 25 for b, n = 97 for b (n = total number of explants in all the replicates)
From the methods tested, the streak method resulted in the highest transformation efficiency of around 17%, followed by the liquid culture method with a transformation efficiency of approximately 15%, while the centrifugation method resulted in the lowest efficiency of about 2%. Of the three methods, we selected the streak method since it has relatively higher efficiency together with easy preparatory and handling steps. For the streak method, a lawn of bacteria covers the newly wounded plant surface as the wound is healed slowly at 16 °C. In the liquid culture and centrifugation methods, the number of bacteria cells in contact with the wound surface and the time duration are likely lower compared to the streak method. Of the two co-cultivation media tested, higher transformation efficiency was achieved when the inoculated plants were grown in 0.25X Hoagland medium with 1% sugar and subsequently transferred to the same medium without sugar, compared to the growth on full MS medium with 3% sugar as the co-cultivation medium and the same without sugar as the regeneration medium. However, there was no significant difference in percentage of YFP expressing roots between two media combinations (Fig. 5b).
In a separate two-factor experiment, the effect of growth stage, two fronds vs. four fronds, was compared with Hoagland and MS media combinations (Fig. 5c). When the two-frond stage was used, the transformation efficiency was 18% in Hoagland media and 14% in MS media. Transformation efficiency was significantly lower when older, four frond plants were selected as the explant material. These experiments confirm the observation that R. rhizogenes transformation efficiency is typically higher when younger explants are used (Piqueras et al. 2010), which may be due to the physiological activities of younger tissues that promote growth and development of transformed cells.
Of the two different explant types tested, root tip cut explants gave positive results, whereas leaf cut explants died within 5 days of bacterial infection. While leaf experiments are used in other species, such as pomegranate (Ono et al. 2012) tomato (Ron et al. 2014) and many other species, the relative fragility or the monocotyledonous nature of L. minor may have affected leaf cut explant survival after infection.
Confirmation of gene integration
Florescent markers are a reliable, effective and non-destructive way of identifying transgenic tissues. To confirm the integration of bacterial genes into the plant genome, presence of several R. rhizogenes Ri plasmid-specific genes were tested via PCR (Fig. 6). These genes and the primers have been successfully used in previous work (Medina-Bolivar et al. 2007; Triplett et al. 2008; Wang et al. 2010; Ono et al. 2012). Of them, rolB and rolC are located within the T-DNA boarders and virD2 is located outside. As such, the virD2 does not integrate into the plant genome and is amplified only from the positive control, R. rhizogenes MSU 440 bacterium. Therefore, the bacterial cells present outside the tissue are completely eliminated by growing the roots on no sugar media and by treating with antibiotics. The universal plant barcoding region rbcL was used as plant-specific sequence to confirm PCR results.

Confirmation of transgene integration via PCR. (B) R. rhizogenes bacteria culture. (W) Wild type L. minor plant. (S) (S’) and (L) transgenic L. minor YFP roots (S and S’ from streak method, L from liquid method)
Conclusions
This work has led to the identification of optimal methods for the growth and transformation of L. minor with R. rhizogenes. Of the methods tested, optimal transformation efficiency was achieved when two-frond stage L. minor root-cut explants were inoculated with 24 h old R. rhizogenes MSU 440 cultures growing on an agar plate. The explants recovered best when co-cultivated in 0.25X Hoagland with 1% sugar at 16 °C and 85 µmolm−2 s−1 light intensity with subsequent growth on 0.25X Hoagland medium without sugar at 23 °C for another 7 weeks. Using these optimized conditions, a transformation efficiency of at least 17% of L. minor explants expressing YFP in roots was achieved.
References
Appenroth KJ (2015) International steering committee on duckweed research and applications, useful methods 2: sterilization of duckweed. 3:90–138
Appenroth K, Adamec L (2014) Specific turion yields of different clones of Spirodela polyrhiza depend on external phosphate thresholds. Plant Biol 17:125–129
Bandaranayake PCG, Yoder JI (2013) Trans-specific gene silencing of acetyl-CoA carboxylase in a root-parasitic plant. Mol Plant Microbe Interact 26:575–584
Bandaranayake PCG, Yoder JI (2018) Factors affecting the efficiency of Rhizobium rhizogenes root transformation of the root parasitic plant Triphysaria versicolor and its host Arabidopsis thaliana. Plant Methods 14(1):61
Bandaranayake PCG, Filappova T, Tomilov A, Tomilova NB, Jamison-McClung D, Ngo Q, Inoue K, Yoder JI (2010) A single-electron reducing quinoneoxidoreductase is necessary to induce haustorium development in the root parasitic plant Triphysaria. Plant Cell 22:1404–1419
Bevan M (1984) Binary Agrobacterium vectors for plant transformation. Nucl Acids Res 12(22):8711–8721. https://doi.org/10.1093/nar/12.22.8711
Bowker D, Duffield A, Denny P (1980) Methods for the isolation, sterilization and cultivation of Lemnaceae. Freshw Biol 10(4):385–388
Caicedo JR, van der Steennp NP, Arce O, Gijzen HJ (2000) Effect of total ammonia nitrogen concentration and pH on growth rates of duckweed (Spirodelapolyrrhiza). Water Res 34:3829–3835
Cao HX, Vu GTH, Wang W, Messing J, Schubert I (2015) Chromatin organisation in duckweed interphase nuclei in relation to the nuclear DNA content. Plant Biol 17:120–124
Chhabra G, Chaudhary D, Sainger M, Jaiwal PK (2011) Genetic transformation of Indian isolate of Lemna minor mediated by Agrobacterium tumefaciens and recovery of transgenic plants. Physiol Mol Biol Plants 17:129–136
Chilton MD, Tepfer DA, Petit A, David C, Delbart FC, Tempé J (1982) Agrobacterium rhizogenes inserts T-DNA into the genomes of the host plant root cells. Nature 295:432–434
Estrada-Navarrete G, Alvarado-Affantranger X, Olivares JE, Díaz-Camino E, Santana O, Murillo E, Guillén G, Sánchez-Guevara N, Acosta J, Quinto C, Li D, Gresshoff PM, Sánchez F (2006) Agrobacterium rhizogenes transformation of the Phaseolus spp. A tool for functional genomics. Mol Plant Microbe Interact 19:1385–1393
Fang YY, Babourina O, Rengel Z, Yang XE, Pu PM (2007) Ammonium and nitrate uptake by the floating plant Landoltia punctate. Ann Bot 99:365–370
Finer JJ, Trick HN (1997) Method for transforming plant tissue by sonication. U.S. Patent No. 5,693,512, 2 Dec 1997. U.S. Patent and Trademark Office, Washington, DC
Heide VDT, Roijackers RMM, Nes VEH, Peeters ETHM (2006) A simple equation for describing the temperature dependent growth of free-floating macrophytes. Aquat Bot 84:171–175
Horemans N, Van Hee M, Van Hoeck A, Saenen E, De Meutter T, Nauts R, Blust R, Vandenhove H (2015) Uranium and cadmium provoke different oxidative stress responses in Lemna minor. L Plant Biol 17:91–100
Ishida JK, Yoshida S, Ito M, Namba S, Shirasu K (2011) Agrobacterium rhizogenes-mediated transformation of the parasitic plant Phtheirospermumjaponicum. PLoS ONE 6:8
Jenner HA, Janssen-Mommen JPM (1993) Duckweed Lemna minor as a tool for testing toxicity of coal residues and polluted sediments. Arch Environ Contam Toxicol 25:3–11
Kasai M, Kanazawa A (2011) RNA silencing as a tool to uncover gene function and engineer novel traits in soybean. Breeding Sci 61:468–479
Kuster H, Vieweg MF, Manthey K, Baier MC, Hohnjec N, Perlick AM (2007) Identification and expression regulation of symbiotically activated legume genes. Phytochem 68:8–18
Lahive E, O'Halloran J, Jansen MAK (2015) A marriage of convenience; a simple food chain comprised of Lemna minor (L.) and Gammarus pulex (L.) to study the dietary transfer of zinc. Plant Biol. 17(Suppl 1):75–81. https://doi.org/10.1111/plb.12179
Landolt E, Kandeler R (1987) Biosystematics investigation in the family of duckweeds (lemnacea). The family of the Lemnacea: a monographic study 2: Zurich: VeroffGeobotInst ETH
Leng RA (1999) Duckweed—a tiny aquatic plant with enormous potential for agriculture and environment. FAO, Rome
Li JR, Todd TC, Lee J, Trick HN (2011) Biotechnological application of functional genomics towards plant-parasitic nematode control. Plant Biotech J 9:936–944
Limpens E, Ramos J, Franken C, Raz V, Compaan B, Franssen H, Bisseling T, Geurts R (2004) RNA interference in Agrobacterium rhizogenes-transformed roots of Arabidopsis and Medicagotruncatula. J Exp Bot 55:983–992
Lonoce CR, Salem C, Marusic PV, Jutras A, Scaloni AM, Salzano S, Lucretti H, Steinkellner E, Benvenuto Donini M (2016) Production of a tumour-targeting antibody with a human-compatible glycosylation profile in N-benthamiana hairy root cultures. Biotechnol J 11:1209–1220
Medina-Bolivar F, Condori J, Rimando AM, Hubstenberger J, Shelton K, O’Keefe SF, Bennett S, Dolan MC (2007) Production and secretion of resveratrol in hairy root cultures of peanut. Phytochemistry 68(14):1992–2003
Muradov N, Fidalgo B, Gujar AC, T-Raissi A, (2010) Pyrolysis of fast-growing aquatic biomass—Lemna minor (duckweed): characterization of pyrolysis products. Biores Technol 21:8424–8428
Muranaka T, Okada M, Yomo J, Kubota S, Oyama T (2015) Characterisation of circadian rhythms of various duckweeds. Plant Biology 1:66–74
Murashige T, Skoog F (1962) A revised medium for rapid growth and bio assays with tobacco tissue cultures. Plant Physiol 15:473–496
Ono NN, Bandaranayake PCG, Tian L (2012) Establishment of pomegranate (Punica granatum) hairy root cultures for genetic interrogation of the hydrolyzable tannin biosynthetic pathway. Planta 236:931–941
Oron G, Wildschut LR, Porath D (1985) Waste water recycling by duckweed for protein production and Effluent renovation. Water Sci Technol 17(4–5):803–817. https://doi.org/10.2166/wst.1985.0181
Oscarson P, Ingemarsson B, Ugglas M, Larsson CM (1988) Characteristics of NO 3 − uptake in Lemna and Pisum. Plant Soil 111:203–205
Piqueras A, Albuquerque N, Folta KM (2010) Explants used for the generation of transgenic plants. In: Kole C, Michler CH, Abbott AG, Hall TC (eds) Transgenic crop plants. Springer Berlin Heidelberg 1(2):31–56
Plasencia A, Soler M, Dupas A, Ladouce N, Silva-Martins G, Martinez Y, Lapierre C, Franche C, Truchet I, Grima-Pettenati J (2016) Eucalyptus hairy roots, a fast, efficient and versatile tool to explore function and expression of genes involved in wood formation. Plant Biotech J 14:1381–1393
Porath D, Pollock J (1982) Ammonia stripping by duckweed and its feasibility in circulating aquaculture. Aquat Bot 13:125–131
Ron M, Kajala K, Pauluzzi G, Wang DX, Reynoso MA, Zumstein K, Garcha J, Winte S, Masson H, Inagaki S, Federici F, Sinha N, Deal RB, Bailey-Serres J, Brady SM (2014) Hairy root transformation using agrobacterium Rhizogenes as a tool for exploring cell type-specific gene expression and function using tomato as a model. Plant Physiol 166:455–U442
Tabou TT, Baya DT, Eyul’anki DM, Vasel JL (2014) Monitoring the influence of light intensity on the growth and mortality of duckweed (Lemna minor) through digital images processing. Biotechnol Agron Soc Environ 18:37–48
Tepfer D (1984) Genetic transformation of several species of higher plants by Agrobacterium rhizogenes: phenotypic consequences and sexual transmission of the transformed genotype and phenotype. Cell 37:959–967
Thomson EL, Dennis JJ (2013) Common Duckweed (Lemna minor) is a versatile high-throughput infection model for the Burkholderia cepacia complex and other pathogenic bacteria. PloS one 8(11):e80102
Thu P, Huong P, Tien V, Ham L, Khanh T (2015) Regeneration and transformation of gene encoding the Hemagglutinin antigen of the H5N1 virus in frond of duckweed (Spirodela polyrhiza L). J Agricult Stud 3(1):48
Tian L (2015) Using hairy roots for production of valuable plant secondary metabolites. Filaments Bioprocesses 149:275–324
Tomilov AA, Tomilova NB, Yoder JI (2006) Agrobacterium tumefaciens and Agrobacterium rhizogenes transformed roots of the parasitic plant Triphysaria versicolor retain parasitic competence. Planta 225:1059–1071
Triplett B, Moss S, Bland J, Dowd M (2008) Induction of hairy root cultures from Gossypium hirsutum and Gossypium barbadense to produce gossypol and related compounds. Vitro Cell Dev Biol Plant 44:508–517
Walkerpeach CR, Velten J (1994) Agrobacterium-mediated gene transfer to plant cells: Cointegrate and binary vector systems. Plant Molecular Biology Manual, S Gelvin, R Schilperoorteds (Dordrecht, The Netherlands: Kluwer) 1–19
Wang W, Messing J (2011) High-throughput sequencing of three Lemnoideae (Duckweeds) chloroplast genomes from total DNA. PLoS ONE 6(9):e24670
Wang W, Messing J (2015) Status of Duckweed genomics and transcriptomics. Plant Biol 17:10–15
Wang CT, Liu H, Gao XS, Zhang HX (2010) Overexpression of G10H and ORCA3 in the hairy roots of Catharanthus roseus improves catharanthine production. Plant Cell Rep 29:887–894
Wang W, Haberer G, Gundlach H, Gläßer C, Nussbaumer T, Luo MC, Lomsadze A, Borodovsky M, Kerstetter RA, Shanklin J, Byrant DW, Mockler TC, Appenroth KJ, Grimwood J, Jenkins J, Chow J, Choi C, Adam C, Cao XH, Fuchs J, Schubert I, Rokhsar D, Schmutz J, Michael TP, Mayer KF, Messing J (2014) The Spirodela polyrhiza genome reveals insights into its neotenous reduction fast growth and aquatic lifestyle. Nat Commun 5:3311
Winans SC (1992) 2-Way chemical signaling in Agrobacterium—plant interactions. Microbiol Rev 56:12–31
Yamamoto YT, Rajbhandari N, Lin X, Bergmann BENA, Nishhimura Y, Carolina N (2001) Genetic transformation of duckweed Lemnagibba andLemna minor. vitro Cell DevBiol Plant 37:349–353
Acknowledgements
We thank Dr. Denneal Jamison-McClung, Director of UC Davis Biotechnology Program for helpful comments provided for improving the manuscript. We would like to thank Dr. Bhagya Chandrasekara for her support on the molecular analysis and staff members of the Agricultural Biotechnology Center, University of Peradeniya for their support and encouragement throughout the research period.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Both authors, R.W.M.K. Kanchanamala and P.C.G. Bandaranayake declare that no conflicts of interest exist regarding the materials included in the manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kanchanamala, R.W.M.K., Bandaranayake, P.C.G. An efficient and rapid Rhizobium rhizogenes root transformation protocol for Lemna minor. Plant Biotechnol Rep 13, 625–633 (2019). https://doi.org/10.1007/s11816-019-00558-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11816-019-00558-9