
Abstract
Anther and tapetum-specific genes are important for understanding male gametophyte development, as well as for their use in the development of barnase/barstar-gene based male sterility and restorer system for hybrid seed production. An essential component of the system is the availability of tapetum-specific promoters. In the present study, anther-specific genes were identified in cotton using microarray-based differential expression analysis, some of which show expression specific to the anthers at a stage where tapetum tissue was fully developed. Validation of the identified genes using RT-PCR and in situ hybridization identified one novel gene (AEG—Anther Expressing Gene) encoding a putative lipid binding protein as having a tapetum-specific expression. Further, three paralogs of the gene were identified in the cotton genome out of which the gene AEG1 (Anther Expressing Gene1) was found to express in the tapetum layer. Analysis of transgenic plants developed in cotton using 1.5 Kb promoter region of the of AEG1 gene fused upstream to the reporter gene β-glucuronidase revealed a broad window of expression of the AEG1 promoter in the tapetum tissue from the tetrad stage of anthers till the degeneration of the tapetum cells. Low levels of expression were also observed in the root tissues. Expression was not observed in the stem and leaves. The broad window of expression of AEG1 promoter in the tapetum tissue makes it a suitable candidate for the expression of the barstar gene for effective fertility restoration in the barnase/barstar system.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cotton (Gossypium hirsutum) is a major fiber crop of the world. Cottonseed also provide edible oil and meal. Amongst the countries growing cotton, India has the largest area under cotton cultivation, around 12.25 million hectares in the year 2014 under this crop (Chaudhary and Gaur 2015). India was the first country to commercially grow cotton hybrids (Patel 1971); currently, 95% of the area under cotton cultivation is occupied by hybrid varieties. Most of the hybrid seed production in cotton is carried out by hand emasculation and pollination as the available cytoplasmic male sterility (CMS) systems impose yield penalties on the hybrids (Santhy et al. 2008). However, the manual method is time-consuming and labor intensive and negatively impacts the scope of dense planting of the crop as this requires much larger quantities of seed.
An alternative is to use a transgenic system of pollination control based on the use of the barnase/barstar genes, hitherto called barnase/barstar system (Mariani et al. 1990). In this system, the barnase gene encoding ribonuclease protein, Barnase is expressed in the tapetum tissue of developing anthers for generating male sterile lines in one of the combiners. The barstar gene coding for the Barstar protein that tightly binds to the Barnase protein and negates the effect of the latter is expressed in the tapetum tissue of the other parent for fertility restoration in the hybrids. Tapetum is a specialized tissue which secretes a large number of products into the anther locules for proper development of the microspores. The success of this strategy lies in the proper expression of the two genes. The expression of the barnase gene needs to be restricted only to the tapetum tissue (Jagannath et al. 2001) and the level of expression of the Barstar protein in the tapetum tissue should be able to completely negate the Barnase activity in the F1 plants (Bisht et al. 2004).
The barnase and barstar gene-based system is an effective transgenic technology that has been successfully used for hybrid seed production in several crop plants like B. napus (Mariani et al. 1990), B. juncea (Jagannath et al. 2001; Ray et al. 2007), maize (Mariani et al. 1992), and wheat (De Block et al. 1997). One of the major requirements for an effective barnase/barstar system is the availability of appropriate tapetum-specific promoters to drive the expression of the two genes.
Tapetum-specific promoters have been identified in different plants species like tobacco (Koltunow et al. 1990; Rogers et al. 1992), Arabidopsis (Scott et al. 1991a; Paul et al. 1992; Turgut et al. 1994; Lee et al. 2003), rice (Yokoi et al. 1997; Luo et al. 2006) and tomato (Twell et al. 1990; Chen et al. 1994; Van den Heuvel et al. 2002; Filichkin et al. 2004). Tapetum-specific promoter TA29 from tobacco has been used for the expression of barnase and barstar genes in Brassica napus (Mariani et al. 1990) and B. juncea (Jagannath et al. 2001; Ray et al. 2007) to develop an effective hybrid seed production system. Further, use of two tapetum-specific promoters TA29 from tobacco and A9 from A. thaliana to drive two independent barstar genes in the same construct independently has shown better fertility restoration of different barnase gene containing lines in B. juncea (Bisht et al. 2004). Most of the tapetum-specific promoters have been identified by cloning the differentially expressed cDNA and subtractive hybridization between cDNA from flower buds/stamens and tissues from the different vegetative organs (Tsuchiya et al. 1994; Ding et al. 2002; Li et al. 2006, Hsu et al. 2014). In cases were genomes were already sequenced like in Arabidopsis (Arondel et al. 2000; Ariizumi et al. 2002; Amagai et al. 2003) and rice (Endo et al. 2004; Swabe et al. 2008), microarray-based expression profiling of genes were carried out to identify anther/tapetum-specific genes.
In cotton, only three genes have been identified with expression in the anthers. Gene G9 and C10 have been found to express predominantly in the pollen cells (Chen et al. 2009; John and Petersen 1994), whereas Gh-ACS gene has been shown to express predominantly in the whole anthers including sporogenous cells, pollen mother cells and microspores as well as tapetal cells (Wang and Li 2009). Apart from these, there are probably no other studies on anther/tapetum specific genes in cotton.
The present study was undertaken to identify genes in cotton that express in the tapetal tissues. GeneChip Cotton Genome Array (Affymetrix), developed from cotton EST sequences, has been used for expression profiling of 21,854 genes in anthers at different developmental stages. A number of genes were identified as anther expressing genes. Based on the initial screening and further validations, a novel gene promoter with high level of expression in the tapetum tissues was identified and its expression pattern analyzed by developing transgenics in cotton.
Materials and methods
Plant material
Gossypium hirsutum L. Coker 310 FR (Kumar et al. 1998) was used for all the experiments. Cotton plants were grown in greenhouse at D/N temperature of 32/22 °C, 16/8 h (light/dark) photoperiod and approximate 70% relative humidity.
Staging of buds
Freshly harvested cotton buds of different lengths were fixed in FAA solution (4% para-formaldehyde, 50% ethanol and 5% acetic acid). Wax blocks of the fixed buds were prepared as described previously (Kim et al. 2002). 10 µm thick sections were cut using a rotary microtome (RM2245, Leica), de-waxed, rehydrated and stained with safranine before analyzing under BX51 microscope (Olympus).
RNA isolation, microarray experiments and semi-quantitative RT-PCR
Total RNA from leaf and boll tissue samples was isolated using the Spectrum Plant Total RNA Kit (Sigma), following the manufacturer’s instructions. Contaminating DNA was removed from the RNA using the DNaseA kit (Ambion). Quantity and quality of the isolated RNA were checked by Nanodrop ND1000 (Thermo) and Agilent RNA 6000 Nano kit on a Bioanalyzer machine (Agilent), respectively.
For microarray experiments, RNA samples with > 7 RIN (RNA Integrity Number) value were selected. 3 µg of total RNA was used for the labeling experiments using one-cycle target labeling kit (Affymetrix). In brief, cDNA was generated from the total RNA using T7-oligo(dT) primer, which was subsequently converted to double-stranded cDNA. The cleaned up double-stranded cDNA was used as a template for in vitro transcription reaction at 37 °C for 16 h using the T7 polymerase to generate biotin-labeled complementary RNA (cRNA). The developed cRNA was cleaned and QC was performed. Only QC passed cRNA samples were fragmented and used for the hybridization reaction. Hybridization, washing and scanning reactions were carried out following the manufacturer’s instructions. The labeled probes were hybridized to GeneChip Cotton Genome Array (Affymetrix). Hybridization and scanning of the chip were carried out at the Centre for Genomic Applications (TCGA), New Delhi, India. Three biological replicates were used separately for each tissue type. Final data for each chip was generated using GeneChip® Operating Software 1.2.1 (GCOS).
For semi-quantitative RT-PCR analysis, first-strand cDNA was synthesized from 1 µg of total RNA using the cDNA Archive kit (Perkin Elmer), using poly dT primers following the manufacturer’s instructions. The cDNA was used for the amplification of different candidate genes using primers designed from the available EST sequences (S4 Table) for 30 and 40 cycles of amplification. Ubiquitin gene was used as an internal control. The amplified fragments were cloned in pGEM-T Easy vector and sequenced.
Identification of transcriptional initiation and stop sites by RLM-RACE
Transcriptional initiation sites (TIS) were determined by carrying out 5′ RACE using the FirstChoice RNA ligase-mediated amplification of cDNA ends (RLM-RACE) kit (Ambion). Two nested reverse primers specific to the coding region of the AEG genes (S4 Table) and 5′ adapter-specific forward primers were used for the 5′ RACE amplification. For the amplification of 3′ ends of the AEG genes, two gene-specific forward primers and two 3′ adapter-specific reverse primers (S4 Table) were used. The amplified fragments were cloned in pGEM-T Easy vector and sequenced from both ends. The transcription start sites of the AEG genes were identified by aligning the obtained sequences with full-length AEG gene sequences.
Isolation of DNA, PCR amplification and genome walking
DNA was isolated using a modified CTAB method (Rawat et al. 2011) and for most cases further purified using DNeasy Plant Maxi Kit (Qiagen). In case of genome walking experiments, DNA was purified by CsCl density gradient centrifugation.
PCR reactions were carried out with purified genomic DNA using standard protocols. The PCR amplifications were performed with 50 ng of genomic DNA in a reaction volume of 25 µl. The reaction mix comprised of 200 µM dNTPs, 12.5 pmol of specific primers, 2.5 U of Taq polymerase, and 1× Taq buffer. For long-range PCR reactions, Takara HS Taq polymerase was used.
For genome walking, seven libraries of genomic DNA from G. hirsutum were constructed using the Genome Walker Kit (Clontech) as described earlier (Paritosh et al. 2013). Seven different libraries were developed using restriction enzymes—DraI, EcoRV, HincII, SspI, ScaI, MscI, and XmnI and fragments were ligated to a universal adaptor. The 5′ upstream region of the AEG1 gene was amplified using nested gene-specific reverse primers and adapter specific forward primers (S4 Table). Amplifications were carried out using Takara HS polymerase (Takara). The amplified fragments were resolved on an agarose gel, eluted and cloned in pGEM-T Easy vector, and sequenced. Final sequences were derived from three independent PCR reactions.
In situ hybridization
For in situ hybridization experiments, AEG1 gene cDNA was cloned in pGEM-T easy vector. T7 promoter was used for in vivo transcription of sense strand, whereas transcription from SP6 promoter was carried out for antisense strand. Reaction was carried out at 37 °C and RNA was DIG labeled using digoxigenin (DIG) RNA labeling kit (Roche). The transcribed RNA was purified by ethanol precipitation and quantified by dot blot analysis with known RNA concentrations.
Cotton buds were harvested in FAA solution and wax molds were prepared as described in the previous section. 8 µm thick wax sections were cut and placed on poly-l-lysine coated slides. After rehydration, bud tissues were treated with Proteinase K (1 µg/ml) for 30 min. The tissues were treated with pre-hybridization solution at 56 °C for 2 h and then with the hybridization solution containing the DIG labeled probes at 56 °C for 48 h. Washing was done with warm 0.1× SSC solutions; subsequently treated with anti-DIG antibody for 2 h. Staining of the tissues was done by treatment of the hybridized samples with Western blue solution overnight. Slides were rinsed in water and observed under a microscope.
Genetic transformation of cotton
Cotyledonary explants of 7-day-old seedlings of Coker 310FR line were used for genetic transformation. Genetic transformation was carried out with Agrobacterium tumefaciens strain GV3101 following the protocol and media described earlier (Chaudhary et al. 2003). Transgenic plants were maintained under controlled environmental conditions in the growth chamber [32 ± 2 °C, 16/8 h (light/dark) photoperiod, 80% relative humidity]. Transformation was carried out using the construct pnos-nptII-ocspA::AEG1P-gus-35SpA in binary vector pPZP200.
Histochemical analysis of GUS expression
Histochemical assays for GUS activity in transgenic cotton were carried out according to an earlier described protocol (Jefferson et al. 1987) with some modifications. Buds of cotton of different sizes were collected in 90% acetone and stained overnight with 5-bromo-4-chloro-3-indolylglucuronide (X-gluc) solution containing 0.1% (w/v) X-gluc in phosphate buffer (pH 7.0) with 0.5 mM potassium ferrocyanide, 0.5 mM potassium ferricyanide and 10% tween 20. Wax blocks were prepared from the stained tissues and 10 µm thick sections were cut for analysis. Other tissues were excised and incubated with X-gluc overnight at 37 °C and the development of blue color was observed under a microscope (BX51 microscope, Olympus).
Results and discussion
Experimental design
To identify genes expressing specifically in the anthers, transcriptome profiles of anthers were compared with those in tissues of buds from which anthers were removed (hitherto called Rest of Bud; ROB), leaves and seedlings. As a first step, the correlation between different stages of anther development (with respect to the status of tapetum) and the size of bud was carried out. To identify genes that were up-regulated in anther tissue, microarray experiments were carried out using mRNA isolated from anthers at different stages of development, their corresponding ROB, leaf and seedling tissues. This was followed by comparative analysis of expression profiles. Further, genes that were specifically expressed in anthers, were identified by RT-PCR, based on the presence or absence of respective amplicons in the RNA isolated from the anther and other tissues. Expression of the identified candidate genes were then analyzed in anthers at different stages of tapetum development. Based on the above analysis, one candidate gene was identified and further characterized. The expression of the gene was also studied by in situ hybridization in sections of anthers of cotton. The upstream promoter region of the candidate gene was isolated and its expression was studied in transgenic cotton plants using β-glucuronidase (gus) as a reporter gene.
Correlation of cotton bud size with the development of tapetum layer
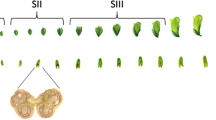
Transverse sections of buds of different sizes (5–8 mm) were observed to identify the stages of tapetum development (Fig. 1). A minimum of 30 buds for each size were analyzed. No discernible tapetum layer was observed in anthers of buds up to 5 mm in size (Fig. 1b, c). In buds of 6 mm and 7 mm, well-differentiated tapetum cell layer was observed (Fig. 1d, e). The tapetum cell layer was found to be degenerated in buds of 8 mm size and above (Fig. 1f). Therefore, microarray experiments were carried out on buds of three different stages—5 mm (Tapetum Not Developed, TND); 7 mm (Tapetum Fully Developed, TFD) and 8 mm (Tapetum Degenerated, TD).

Correlation of size of cotton buds with the development of tapetum layer. a Different sizes of cotton buds, from 4 to 8 mm. b, c The transverse section of 4 mm and 5 mm cotton buds, where tapetum layer (Tap) was not differentiated (Tapetum not developed, TND). d, e Fully developed tapetum layer (TFD) in 6 mm and 7 mm buds and f the cotton buds (8 mm) where tapetum is degenerated. Pollen mother cells, tetrad and pollen are depicted as ‘PMC’, ‘Tet’ and ‘P’ respectively
Differentially expressed genes of different stages of anther development
For microarray experiments, mRNA from eight different tissues were isolated viz. anthers at different stages of development (TND, TFD and TD), ROB of the respective stages (ROB–TND, ROB–TFD and ROB–TD), leaf and seedlings. Hybridization of the labeled cRNA was carried out on GeneChip cotton genome array to determine the expression values of 21,854 genes represented on the chip in the different tissues mentioned above. For each tissue, three biological replicates were analyzed.
The hybridization signal intensities for all the experiments from CEL files were scaled and normalized using GC-RMA algorithm (Wu et al. 2004). Microarray data have been deposited with the Gene Expression Omnibus (GEO) database (Accession number—GSE51779). Principal Component Analysis (PCA) of the samples was carried out and samples having < 92% correlation with the other biological replicates were removed from the analysis (S1 figure). Based on the above, one sample each from TND-ROB and TFD-ROB were not included in further analysis.
Of the 21,854 genes represented on the GeneChip cotton genome array, ~ 61% (~ 13,000) genes were found to express differentially in the tissues that were analyzed. Genes up-regulated in anthers of different stages were identified as those that had ≥ twofold expression in anthers as compared to other analyzed tissues. The result was validated by unpaired t test (p = 0.05) and ‘Benjamini and Hochberg’ false discovery rate analysis. A total of 1476 genes were observed to be up-regulated in anther tissues, of which 547 genes were up-regulated in anthers with tapetum fully developed (TFD, Fig. 2a). Information on the genes up-regulated at anthers in different stages of development has been provided in S2 Table.

Venn diagram representing the expression pattern in different tissues of cotton. Differentially up-regulated genes in anthers (a) and specifically expressing genes in anthers (b) as compared to rest of buds (ROB), leaf and seedling tissues. The data were obtained after analyzing the microarray-based expression profiling of 21,854 genes
Genes that specifically express at different stages of cotton anthers
Intensity values of each of the differentially up-regulated genes were analyzed with MAS5.0 algorithm. Anther-specific genes were defined as genes with positive intensity values in the anther tissues and with none or negative intensity values in all of the other analyzed tissue types. Genes showing no expression were identified based on the normalized expression values of the negative control probes (S2 Table). Highly variable (or less reliable) expression data were removed manually from the dataset of identified genes. In most of the cases, a gene was selected for further analysis only if expression levels in all the replicates showed a similar trend. Averaging ratios across experiments was considered a less stringent strategy because it could lead to the detection of false positive genes also. Therefore, available data were further sorted for those genes which had positive expression values in all the replicates of anther tissues and negative/marginal expression values in all the other tissue samples.
A total of 428 genes were observed to be anther-specific, which could be further grouped as genes expressed specifically in the anthers of TND (78 genes), TFD (88 genes), TD (19 genes), TFD + TD (48 genes), TND + TFD (26 genes), TND + TD (14 genes) and TND + TFD + TD (155) buds, respectively (Fig. 2b, S3 Table).
Validation of some of the anther-specific genes identified by microarray data analysis by RT-PCR
A total of 65 genes with specific anther expression were selected for experimental validation by RT-PCR analysis. Gene-specific primers were used to amplify the transcripts from TFD-anthers and TFD-ROB leaf and seedlings. Primers specific to various EST (gene) sequences were designed based on information available about the ESTs from which sequences have been taken for the Affymetrix chip used in this study. The sequences of the primers used are presented in S4 Table.
For each sample, three independent PCR reactions were carried out. Presence and absence of amplicons was checked after 30 and 40 cycles of reaction. The data is presented as a heat map in Fig. 3. Initially, amplifications were carried out only on cDNA from TFD-anthers and TFD-ROB tissues and genes observed to be expressing in the TFD-anther tissues and not expressing in TFD-ROB tissues were checked for their expression in the leaf and seedling tissues. A group of genes (Group A, Fig. 3) was found to be expressing in TFD-anthers and TFD-ROB both after 30 and 40 cycles of amplification. Further, it was observed that in several cases, while no amplification was observed in ROB after 30 cycles, amplification was observed after 40 cycles (Group B, Fig. 3). Thus, 40 cycles of PCR were found to be more stringent to identify genes that expressed in anther tissues only. Expression of genes, which did not show expression in ROB and expression in the anther tissues were then checked in leaf and seedling tissues, following 40 cycles of PCR. Out of the eight shortlisted genes, two genes were found to be expressing in the leaf and seedling tissues and were rejected. This analysis led to the identification of six genes (Group C, Fig. 3) that expressed only in TFD-anthers and not in the other analyzed tissues. The six genes are denoted herein by their Affymetrix probe id number (Table 1). A group of shortlisted up-regulated genes (group D, Fig. 3) was found to have no expression in the anther and ROB tissues. These genes may have been false positives in the microarray experiments due to variable expression patterns in different biological samples. The genes under Group C were carried forward for further analysis.

Heatmap representing the expression pattern of the selected candidate gene in TFD-anthers, TFD–ROB, leaf and seedling tissues. At first, PCR was carried out for 30 cycles and 40 cycles in TFD-anther and TFD–ROB, subsequently genes identified as anther-specific in the first step were checked for their expression in leaf and seedling tissues. Based on the expression pattern, four groups were identified. a Genes expressed in anthers and ROB both in 30 and 40 cycles of amplification. b Genes which were expressing in anthers but had no expression in ROB tissues in 30 cycles but expression was observed in the ROB tissues after 40 cycles of amplification. c Genes identified as anther-specific after 40 cycles of amplification. d Genes which had no expression in any of the analyzed tissue
Expressions of the short-listed six genes (Table 1) was analyzed by RT-PCR in anthers and ROB from buds at different stage of tapetum development (Fig. 4). It was observed that four genes—Ghi.1464, Ghi.9693, Gra.2743 and Ghi.7345 expressed in the anther tissue at all the three stages (TND, TFD and TD) and did not express in any of the other analyzed tissues. Gene with probe id Ghi.4591 showed a low level of expression in TFD-anther and initial stages of TD-anther tissues. Gene with probe id GhiAffx.42243 was found to express in the TFD-anthers only. Based on this analysis, expression of the gene represented by probe id GhiAffx.42243 was found to be of interest as its expression was not only restricted to anthers tissues, but was also present at the time when the tapetum layer is well developed. This gene was named as Anther Expressing Gene (AEG).

Analysis of transcripts of the six identified gene in the anthers and ROB tissues of cotton buds of different sizes and leaf and seedling tissues. Lane 1 and 20 show the 100 bp DNA marker, lane 2 shows amplification from the genomic DNA, Lane 3 represents PCR setup with isolated total RNA (negative control), Lane 4, 6, 8, 10, 12, 14 and 16 represent amplification profile from the rest of bud tissues of different sizes of buds, Lane 5, 7, 9, 11, 13, 15 and 17 represent amplification profile from the anther tissues of different sizes of buds. Lane 18 and 19 represent the amplification profile from the leaf and seedling, respectively GhiAffx.42243 in the figure represents the expression profile of the AEG1 gene. Arrows indicate the transcript-specific amplicons
Paralogs of AEG and their expression
Based on the available sequence information of the genes present on cotton microarray chip, primers were designed to amplify ~ 400 bp of coding region of the gene. Partial coding region of the gene AEG (~ 400 bp) was amplified from cotton genome and cloned in pGEM-T easy vector. After sequencing of more than 40 recombinant clones from two independent PCR reactions, three different types of sequences were obtained. The first sequence was found to be less than 96% identical as compared to the other two which showed > 98% sequence identity. Based on the identity of the sequences, they were named as Anther Expressing Gene1 (AEG1), Anther Expression Gene 2a (AEG2a) and Anther Expressing Gene 2b (AEG2b). PCR based analysis for the presence of these genes in the parental genomes of G. hirsutum (G. herbaceum var africanum and G. raimondii) showed that there is one ortholog of the gene in G. herbaceum and two orthologs of the gene are present in the G. raimondii genome. While the three paralogs of AEG genes, AEG1, AEG2a and AEG2b were identified in the present study by genome walking, this was also confirmed once the genome sequence of G. hirsutum was published (Zhang et al. 2015; Li et al. 2015). Further, no other similar gene was found to be present in the published genomic sequence.
The primer pair, which was used to amplify the 400 bp of the AEG genes, was used to amplify cDNA prepared from TFD-anthers. Amplification, cloning and sequencing of the amplicons obtained from two independent PCR reactions revealed that the transcript of gene AEG2b was absent, whereas genes AEG1 and AEG2a were found to be expressing in the anther tissues. No intron was found in the amplified region. To locate the transcription initiation and stop sites of the two genes, RACE was performed on the library of the TFD-anthers. A total of 44 clones were sequenced and analyzed. One transcription initiation site (TIS) was identified for both AEG1 and AEG2a. The transcripts had 5′ UTR of 67 and 64 bp for AEG1 and AEG2a, respectively. On the other hand, two transcription stop sites (TSS) were identified for the gene AEG1 and three TSS for gene AEG2a (Fig. 5). Considering the longest ORF as coding unit, the 3′ UTR for AEG1 and AEG2a transcripts was observed to be 239 bp and 232 bp, respectively. The longest transcript for the gene AEG1 was observed to be 647 bp long and for gene AEG2a it was 651 bp in size.

Diagram representing the differences between the transcripts of AEG1 and AEG2a. Positions of the differences are shown on the top of each SNP. The transcription initiation and stop sites are shown with downward and upward arrows, respectively. * and # denotes the translational start and stop sites. Regions of 5′ and 3′ UTRs are indicated with black bars
Although multiple ORFs were predicted for the transcripts of AEG1 and AEG2a, the longest ORFs in both the genes encoded for 115 amino acid long peptide, which were 94% similar. Only six amino acid differences were observed between the two genes. BLASTX search of each of the proteins against the non-redundant protein database identified a putative lipid-binding protein from castor (Ricinus Communis, NCBI accession no.—XP_00252644.1) as the best match. It was interesting to note that in several earlier reports, genes expressed in tapetum were shown to encode for lipid-binding proteins, e.g., A9 (Scott et al. 1991a), At5g62080 (Huang et al. 2013), Satap35 (Staiger et al. 1994) and TomA108 (Chen et al. 2006).
As a greater number of cDNA clones were observed for the AEG1 gene as compared to AEG2a transcripts, it was probable that the expression of the gene AEG1 was more than AEG2a. Thus, further analysis was carried out only for AEG1 gene. The spatial expression of the gene was checked in cotton anthers by in situ hybridization experiments. Positive hybridization signals were observed in the tapetum layer with antisense probe (Fig. 6c) and no signal was observed in any other region of the anther, indicating that the expression of the gene is restricted to the tapetum in anthers (Fig. 6).

In situ hybridization based expression analysis of AEG1 gene in cotton anthers. a The transverse section of anther lobe before hybridization experiment. b The hybridization of the cotton buds with labelled sense probes. No hybridization was observed in the anthers. b The hybridization of cotton anthers with anti-sense labeled probes. Strong signal was detected in the tapetum layer, representing expression of the AEG1 gene in the tapetum tissues. Tapetum and pollen are depicted as ‘Tap’ and ‘P’, respectively
Isolation of the upstream promoter region of the gene AEG1
Based on sequences specific for AEG1 gene, primers were designed to isolate upstream regions of the gene using seven different Genome Walking Libraries. Amplicon from multiple walk reactions were cloned and sequenced to identify upstream and downstream regions of the genes. The obtained amplicon from multiple walk reactions were cloned and sequenced. Gene sequences were assembled using results from at least three independent PCRs to discern any Taq polymerase-based mutation. Different paralogs of AEG genes were identified based on gene-specific SNPs. Based on the amplicon from multiple walk reactions; more than 2.0 kb of upstream regions of the gene AEG1 was isolated. This was also validated once the cotton genome sequence was published (Zhang et al. 2015; Li et al. 2015). BLAST search of AEG1 sequence against the available cotton genome sequence showed its presence on chromosome A13 (Zhang et al. 2015). On the other hand, AEG2b was found to be present on chromosome D13 in both the reported genome sequences.
An in silico analysis was carried out on the 1.5 Kb promoter region of gene AEG1 (Fig. 7). TATA box with the sequence TATATAA was found to be located between − 101 and − 103 bp to transcription initiation site. The promoter also had motifs like—GTGANTG10 (Rogers et al. 2001) and POLLEN1LELAT52 (Filichkin et al. 2004). These motifs were also reported in many of the pollen and anther-specific promoters like g10, Lat56 and A9, TA29 (Chen et al. 2009; Scott et al. 1991b). Apart from these boxes, it was also observed that the promoter region contained multiple copies of ROOTMOTIFTAPOX1 (Elmayan and Tepfer 1995) in the promoter region.

Predicted cis-elements in the 1.5 kb of the promoter region of AEG1 and AEG2a gene of G. hirsutum. Description of the boxes is given below the figure. Boxes like GTGANTG10 and POLLEEN1LELAT52 are reported to be abundant in the anther/pollen specific promoters. ROOTMOTIFTAPOX1 was clustered between − 556 and − 391 of the promoter region of AEG1, whereas it was discretely arranged in AEG2a promoter. GATA box and GT1 consensus motifs were also identified in the promoter regions
Determination of spatial expression of the promoter of the gene AEG1
The activity of the AEG1 promoter was analyzed using a promoter-reporter construct in transgenic cotton plants. A 1.5 Kb region of the AEG1 promoter was cloned upstream to the reporter gene β-glucuronidase in the vector pZP200N (Bhullar et al. 2003) creating the vector AEG1P:gus.
A total of 17 independent cotton transgenic lines were developed with AEGP::gus construct. Promoter activity was monitored by histochemical staining for gus activity in different tissues of cotton plants, i.e., anthers, stem, root, leaves and buds (Fig. 8). It was observed that the activity of AEG1 promoter started in the tapetum layer from the time of the visible differentiation and persisted till the tapetum layer degenerated (Fig. 8f, h, j, l). This was observed in all the 17 plants that were developed. Apart from expression in the tapetum tissue of anthers, low level of expression was also observed in the roots of 9 out of the 17 transgenic lines developed (Fig. 8n).

Histochemical localization of GUS activity in transgenic AEG-Promoter:gus cotton (a–n). Left panel represents the observed GUS activity in untransformed control plants, whereas panel on the right represents the same in AEGP::gus transgenic plants. Transverse section of the anthers at stage TND (c, d), TFD (e–h) and TD (i–l) and whole tissues of leaf (a, b) and root (m, n) were analyzed for GUS activity by histochemical staining. Activity of AEG1 promoter was observed to be restricted in the tapetum layer in case of anthers. Low level of expression was also observed in the root tissues. Tapetum and pollen are depicted as ‘Tap’ and ‘P’, respectively
The expression in roots may be driven by ‘position effect’, which is dependent upon the site of integration of the transgene cassette. As this is observed in about 50% of the transgenic lines, it is possible that the promoter of the AEG1 carries putative cis-elements regulating expression in roots, which is activated when the promoter is located at certain ectopic genomic sites. This activity could probably be influenced by the state of chromatin. Similar phenomenon has been observed in the study involving the tapetum-specific promoter TA29 from Nicotiana tabacum (Sharma et al. 2018). As mentioned earlier, in silico analysis of the promoter had revealed the presence of a cis-element (ROOTMOTIFTAPOX1, Elmayan and Tepfer 1995) observed to be present in promoters of the genes expressing in the root predominantly (Zhang et al. 2008; Chen et al. 2014). Several copies of this element were observed in the AEG1, mainly located between − 500 and − 230 of the promoter region (Fig. 7). This root-specific motif is probably responsible for the expression in roots. The leaky expression in roots could possibly be abolished if these root-specific motifs were deleted or mutated.
The present study was initiated to identify a promoter, which can be used to express barnase gene in transgenic cotton. However, due to the leaky expression of the promoter in the roots of some of the transgenic plants, it cannot be used effectively for developing the barnase transgenic lines. Any leaky expression (other than that in the tapetum) of barnase gene, which encodes a RNase protein, would lead to the death of that tissue. To identify useful barnase lines, a large number of independent lines would have to be developed to identify those with no leaky expression in the roots. Attempts are currently underway to modify the promoter to abolish the leaky expression in the roots while maintain its expression in the tapetum tissues. Promoters, where the putative root-specific motifs are deleted or mutated have been developed and its activity being tested in transgenic lines. The availability of a modified tapetum-specific AEG1 promoter will allow the development of male sterile lines using the barnase gene.
Currently, the promoter can be used to drive the expression of barstar gene, the second component of the barnase–barstar system. Expression of the barstar protein in the roots is not likely to cause any negative effects. The long window expression of the promoter of AEG1 gene in the tapetum tissue will also be very useful for the expression of the barstar gene for effective fertility restoration.
NCBI database accession
The isolated sequences of the paralogs of AEG genes from G. hirsutum, G. herbaceum and G. raimondii were submitted in NCBI database with accession numbers—KF742407—KF742412.
References
Amagai M, Ariizumi T, Hatakeyama MEK, Shibata CKD, Toriyama K, Watanabe M (2003) Identification of anther-specific genes in a cruciferous model plant, Arabidopsis thaliana, by using a combination of Arabidopsis macroarray and mRNA derived from Brassica oleracea. Sex Plant Rep 15:213–220. https://doi.org/10.1007/s00497-002-0159-z
Ariizumi T, Amagai M, Shibata D, Hatakeyama K, Watanabe M, Toriyama K (2002) Comparative study of promoter activity of three anther-specific genes encoding lipid transfer protein, xyloglucan endotransglucosylase/hydrolase and polygalacturonase in transgenic Arabidopsis thaliana. Plant Cell Rep 21:90–96. https://doi.org/10.1007/s00299-002-0487-3
Arondel VV, Vergnolle C, Cantrel C, Kader J (2000) Lipid transfer proteins are encoded by a small multigene family in Arabidopsis thaliana. Plant Sci 157:1–12. https://doi.org/10.1016/S0168-9452(00)00232-6
Bhullar S, Chakravarthy S, Advani S, Datta S, Pental D, Burma PK (2003) Strategies for development of functionally equivalent promoters with minimum sequence homology for transgene expression in plants: cis-elements in a novel DNA context versus domain swapping. Plant Physiol 132(2):988–998. https://doi.org/10.1104/pp.103.020602
Bisht NC, Jagannath A, Gupta A, Burma PK, Pental D (2004) A two gene–two promoter system for enhanced expression of a restorer gene (barstar) and development of improved fertility restorer lines for hybrid seed production in crop plants. Mol Breed 14(2):129–144. https://doi.org/10.1023/B:MOLB.0000038002.45312.08
Chaudhary B, Gaur K (2015) Biotech cotton in India, 2002 to 2014. ISAAA series of biotech crop profiles. ISAAA, Ithaca
Chaudhary B, Kumar S, Prasad KVSK, Oinam GS, Burma PK, Pental D (2003) Slow desiccation leads to high-frequency shoot recovery from transformed somatic embryos of cotton (Gossypium hirsutum L. cv. Coker 310FR). Plant Cell Rep 21:955–960. https://doi.org/10.1007/s00299-003-0613-x
Chen R, Aguirre PJ, Smith AG (1994) Characterization of an anther- and tapetum-specific gene encoding a glycine-rich protein from tomato. J Plant Physiol 143:651–658. https://doi.org/10.1016/S0176-1617(11)81153-3
Chen RD, Zimmermann E, Xu SX, Liu GS, Smith AG (2006) Characterization of an anther- and tapetum-specific gene and its highly specific promoter isolated from tomato. Plant Cell Rep 25:231–240. https://doi.org/10.1007/s00299-005-0056-7
Chen D, Ding Y, Guo W, Zhang T (2009) Molecular cloning and characterization of a flower-specific class III peroxidase gene in G. hirsutum. Mol Biol Rep 36:461–469. https://doi.org/10.1007/s11033-007-9202-3
Chen L, Jiang B, Wu C, Sun S, Hou W, Han T (2014) GmPRP2 promoter drives root-preferential expression in transgenic Arabidopsis and soybean hairy roots. BMC Plant Biol 14:245. https://doi.org/10.1186/s12870-014-0245-z
De Block M, Debrouwer D, Moens T (1997)) The development of a nuclear male sterility system in wheat. Expression of the barnase gene under the control of tapetum specific promoters. Theor Appl Genet 95:125–131. https://doi.org/10.1007/s001220050540
Ding ZJ, Wu XH, Wang T (2002) The rice tapetum-specific gene RA39 encodes a type Iribosome-inactivating protein. Sex Plant Reprod 15:205–212. https://doi.org/10.1007/s00497-002-0156-2
Elmayan T, Tepfer M (1995) Evaluation in tobacco of the organ specificity and strength of the rolD promoter, domain A of the 35S promoter and the 35S2 promoter. Transgenic Res 4(6):388–396. https://doi.org/10.1007/BF01973757
Endo M, Tsuchiya T, Saito H, Matsubara H, Hakozaki H, Masuko H, Kamada M, Higashitani A, Takahashi H, Fukuda H, Demura T, Watanabe M (2004) Identification and molecular characterization of novel anther-specific genes in Oryza sativa L. by using cDNA microarray. Genes Genet Syst 79:213–226. https://doi.org/10.1266/ggs.79.213
Filichkin SA, Leonard JM, Monteros A, Liu PP, Nonogaki H (2004) A novel endo-beta-mannanase gene in tomato LeMAN5 is associated with anther and pollen development. Plant Physiol 134:1080–1087. https://doi.org/10.1104/pp.103.035998
Hsu SW, Liu MC, Zen KC, Wang CS (2014) Identification of the tapetum/microspore-specific promoter of the pathogenesis-related 10 gene and its regulation in the anther of Lilium longiflorum. Plant Sci 215:124–133. https://doi.org/10.1016/j.plantsci.2013.11.006
Huang MD, Chen TL, Huang AH (2013) Abundant type III lipid transfer proteins in Arabidopsis tapetum are secreted to the locule and become a constituent of the pollen exine. Plant Physiol 163:1218–1229. https://doi.org/10.1104/pp.113.225706
Jagannath A, Bandyopadhyay P, Arumugam N, Gupta V, Burma PK, Pental D (2001) The use of a Spacer DNA fragment insulates the tissue-specific expression of a cytotoxic gene (barnase) and allows high-frequency generation of transgenic male sterile lines in Brassica juncea L. Mol Breed 8:11–23. https://doi.org/10.1023/A:1011916216191
Jefferson RA, Kavanagh TA, Bevan MW (1987) GUS fusions: beta-glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO J 6(13):3901–3907. https://doi.org/10.1002/j.1460-2075.1987.tb02730.x
John ME, Petersen MW (1994) Cotton (Gossypium hirsutum L.) pollen-specific polygalacturonase mRNA: tissue and temporal specificity of its promoter in transgenic tobacco. Plant Mol Biol 26:1989–1993. https://doi.org/10.1007/BF00019509
Kim MK, Choi JW, Jeon JH, Franceschi VR, Davin LB, Lewis NG (2002) Specimen block counter-staining for localization of GUS expression in transgenic arabidopsis and tobacco. Plant Cell Rep 21:35–39. https://doi.org/10.1007/s00299-002-0475-7
Koltunow AM, Truettner J, Cox KH, Wallroth M, Goldberg RB (1990) Different temporal and spatial gene expression patterns occur during anther development. Plant Cell 2:1201–1224. https://doi.org/10.1105/tpc.2.12.1201
Kumar S, Sharma P, Pental D (1998) A genetic approach to in vitro regeneration of non-regenerating cotton (Gossypium hirsutum L.) cultivars. Plant Cell Rep 18(1):59–63. https://doi.org/10.1007/s002990050532
Lee YH, Chung KH, Kim HU, Jin YM, Kim HI, Park BS (2003) Induction of male sterile cabbage using a tapetum-specific promoter from Brassica campestris L. ssp. pekinensis. Plant Cell Rep 22:268–273. https://doi.org/10.1007/s00299-003-0688-4
Li N, Zhang DS, Liu HS, Yin CS, Li XX, Liang WQ et al (2006) The rice tapetum degeneration retardation gene is required for tapetum degradation and anther development. Plant Cell 18:2999–3014. https://doi.org/10.1105/tpc.106.044107
Li F, Fan G, Lu C, Xiao G, Zou C, Kohel RJ et al (2015) Genome sequence of cultivated Upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat Biotechnol 33:524–530. https://doi.org/10.1038/nbt.3208
Luo H, Lee JY, Hu Q, Nelson-Vasilchik K, Eitas TK, Lickwar C, Kausch AP, Chandlee JM, Hodges TK (2006) RTS, a rice anther-specific gene is required for male fertility and its promoter sequence directs tissue-specific gene expression in different plant species. Plant Mol Biol 62:397–408. https://doi.org/10.1007/s11103-006-9031-0
Mariani C, De Beuckeleer M, Truettner J, Leemans J, Goldberg RB (1990)) Induction of male sterility in plants by a chimaeric ribonuclease gene. Nature 347:737–741
Mariani C, Gossele V, De Beuckeleer M, De Block M, Goldberg RB, De Greef W, Leemans J (1992)) A chimaeric ribonuclease-inhibitor gene restores fertility to male sterile plants. Nature 357:384–387
Paritosh K, Pental D, Burma PK (2013) Structural and transcriptional characterization of rbcS genes of cotton (Gossypium hirsutum). Plant Mol Biol Rep 31(5):1176–1183. https://doi.org/10.1007/s11105-013-0576-1
Patel CT (1971) Hybrid 4, a new hope towards self sufficiency in cotton in India. Cotton Dev 1(2):1–6
Paul W, Hodge R, Smartt S, Draper J, Scott R (1992) The isolation and characterization of the tapetum-specific Arabidopsis thaliana A9 gene. Plant Mol Biol 19:611–622. https://doi.org/10.1007/BF00026787
Rawat P, Singh AK, Ray K, Chaudhary B, Kumar S, Gautam T, Kanoria S, Kaur G, Kumar P, Pental D, Burma PK (2011) Detrimental effect of expression of Bt endotoxin Cry1Ac on in vitro regeneration, in vivo growth and development of tobacco and cotton transgenics. J Biosci 36:363–376. https://doi.org/10.1007/s12038-011-9074-5
Ray K, Bisht NC, Pental D, Burma PK (2007) Development of barnase/barstar transgenics for hybrid seed production in Indian oilseed mustard (Brassica juncea L. Czern & Coss) using a mutant acetolactate synthase gene conferring resistance to imidazolinone-based herbicide ‘Pursuit’. Curr Sci 93:1390–1396
Rogers HJ, Harvey A, Lonsdale DM (1992) Isolation and characterization of a tobacco gene with homology to pectate lyase which is specifically expressed during microsporogenesis. Plant Mol Biol 20:493–502. https://doi.org/10.1007/BF00040608
Rogers HJ, Bate N, Combe J, Sullivan J, Sweetman J, Swan C et al (2001) Functional analysis of cis-regulatory elements within the promoter of the tobacco late pollen gene g10. Plant Mol Biol 45(5):577–585. https://doi.org/10.1023/A:1010695226241
Santhy V, Khadi BM, Singh P, Kumari PRV, Deshmukh RK, Vishwanathan Q (2008) Hybrid seed production in cotton. 2008. CICR bulletin number 35
Scott R, Dagless E, Hodge R, Paul W, Soufleri I, Draper J (1991a) Patterns of gene expression in developing anthers of Brassica napus. Plant Mol Biol 17:195–207. https://doi.org/10.1007/BF00039494
Scott R, Hodge R, Paul W, Draper J (1991b) The molecular biology of anther differentiation. Plant Sci 80:167–191. https://doi.org/10.1016/0168-9452(91)90281-C
Sharma PA, Verma N, Burma PK (2018) Analysis of activity driven upstream regulatory modules (URM) of tapetum specific genes TA29 and A9 at ectopic locations in tobacco transgenics. J Plant Biochem Biotechnol. https://doi.org/10.1007/s13562-018-0453-y
Staiger D, Kappeler S, Muller M, Apel K (1994) The proteins encoded by two tapetum-specific transcripts, Sa tap35 and Sa tap44, from Sinapis alba L. are localized in the exine cell wall layer of developing microspores. Planta 192:221–231. https://doi.org/10.1007/BF01089038
Swabe K, Suzuki G, Takahashi H, Shiono K, Endo M, Yano K et al (2008) Separated transcriptomes of male gametophyte and tapetum in rice: validity of a laser microdissection (LM) microarray. Plant Cell Physiol 49:1407–1416. https://doi.org/10.1093/pcp/pcn124
Tsuchiya T, Toriyama K, Ejiri S, Hinata K (1994) Molecular characterization of rice genes specifically expressed in the anther tapetum. Plant Mol Biol 26:1737–1746. https://doi.org/10.1007/BF00019488
Turgut K, Barsby T, Craze M, Freeman J, Hodge R, Paul W, Scott R (1994) The highly expressed tapetum-specific A9 gene is not required for male fertility in Brassica napus. Plant Mol Biol 24:97–104. https://doi.org/10.1007/BF00040577
Twell D, Yamaguchi J, McCormick S (1990) Pollen-specific gene expression in transgenic plants: coordinate regulation of two different tomato gene promoters during microsporogenesis. Development 109:705–713
Van den Heuvel KJ, Van Lipzig RH, Barendse GW, Wullems GJ (2002) Regulation of expression of two novel flower-specific genes from tomato (Solanum lycopersicum) by gibberellin. J Exp Bot 53:51–59. https://doi.org/10.1093/jxb/53.366.51
Wang XL, Li XB (2009) The GhACS1 gene encodes an acyl-CoA synthetase which is essential for normal microsporogenesis in early anther development of cotton. Plant J 57:473–486. https://doi.org/10.1111/j.1365-313x.2008.03700.x
Wu Z, Irizarry R, Gentleman RC, Murillo FM, Spencer F (2004) A model based background adjustment for oligonucleotide expression arrays. Working paper 1 (Johns Hopkins University, Department of Biostatistics working papers. http://www.bepress.com/jhubiostat/paper)
Yokoi S, Tsuchiya T, Toriyama K, Hinata K (1997) Tapetum-specific expression of the Osg6B promoter-β-glucuronidase gene in transgenic rice. Plant Cell Rep 16:363–367. https://doi.org/10.1007/BF01146774
Zhang C, Barthelson RA, Lambert GM, Galbraith DW (2008) Global characterization of cell-specific gene expression through fluorescence-activated sorting of nuclei. Plant Physiol 147:30–40. https://doi.org/10.1104/pp.107.115246
Zhang T, Hu Y, Jiang W, Fang L, Guan X, Chen J et al (2015) Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat Biotechnol 33:531–537. https://doi.org/10.1038/nbt.3207
Acknowledgements
Research work was supported by grants from the Department of Biotechnology (DBT) and NMITLI program of Council for Scientific and Industrial Research (CSIR), Government of India. KP acknowledges the fellowship from the Council for Scientific and Industrial Research, India. DP acknowledges an award of J. C. Bose fellowship (Department of Science and Technology, Government of India). The authors acknowledge the help of Dr. Anjana N Dev for editing the manuscript.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: DP KP PKB. Performed the experiments: KP (Microarray experiments and analysis, identification of genes, sequence analysis) AKS (development of transgenic cotton) AKM (development of promoter:reporter construct and in situ experiments). Analyzed the data: KP AKS DP PKB. Wrote the paper: KP AKS DP PKB.
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
11816_2018_501_MOESM1_ESM.pdf
S1 Figure. Principal component analysis and correlation plot of the different samples used for the microarray experiments (PDF 1306 KB)
Rights and permissions
About this article

Cite this article
Paritosh, K., Singh, A.K., Mehrotra, A.K. et al. Identification and characterization of the promoter of a gene expressing mainly in the tapetum tissue of cotton (Gossypium hirsutum L.). Plant Biotechnol Rep 12, 377–388 (2018). https://doi.org/10.1007/s11816-018-0501-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11816-018-0501-z