
Abstract
Plastid transformation is conducted by homologous recombination. Plastid transformation using a vector-containing promoter and/or terminator sequences homologous to the plastid DNA for transgene expression often results in the generation of unintended extrachromosomal DNA molecules derived from the initial plastid DNA molecule via intramolecular recombination. All the extrachromosomal DNA molecules, including those lacking the ori sequence, found in the T0 plastid-transformed tobacco plants were still detected in the T3 plants. These results suggest that the extrachromosomal DNA molecules are newly generated from the initially transformed plastid DNA that is transmitted to the progeny during each generation. In addition, inward, outward, and overlap extension PCR of the extrachromosomal DNA molecules confirmed that these molecules were circular.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Plastid transformation in higher plants occurs through homologous recombination. Transformation vectors contain a pair of sequences homologous to the plastid DNA between which the transgene is inserted (Staub and Maliga 1992). Previously, these vectors were often constructed using promoter and/or terminator sequences homologous to the host plastid DNA for transgene expression. This technique results in the generation of extrachromosomal DNAs via unintended intramolecular recombination between the homologous promoter and/or terminator sequences derived from the vector and the plastid DNA (Gray et al. 2009).
The presence of extrachromosomal DNA was first described by Staub and Maliga (1994), who reported that plastid DNA sequences from the trnI gene, designated NICE1, were transmitted to the progeny. However, Corneille et al. (2003) observed that no extrachromosomal DNA species were generated after multiple unintended recombination events in transgenic plastids following CRE-lox recombination in plants grown from seed. Likewise, Gray et al. (2009) reported that the abundance of unintended DNA species was lower in T1 than T0 progeny.
Intramolecular recombination is successfully prevented by replacing homologous promoter and/or terminator sequences with heterologous sequences, which results in no extrachromosomal DNA species (Gray et al. 2009). However, it remains unclear whether extrachromosomal DNAs generated via intramolecular recombination are retained in progeny and whether intramolecular recombination only occurs during the plastid transformation process. In this study, we conducted plastid transformation of tobacco plants using a plastid transformation vector-containing promoter and terminator sequences homologous to the host plastid DNA and observed the fate of extrachromosomal DNAs in the progeny. We found that the extrachromosomal DNAs were retained even in the T3 generation and that the extrachromosomal DNAs could be newly generated in the progeny.
Materials and methods
Construction of a conventional plastid transformation vector
The pCtVG plastid transformation vector was employed for the insertion of the aadA and gfp transgenes connected to the psbA terminator under the control of the rrn promoter between trnI and trnA in the plastome. The pCtVG vector was constructed via insertion of the ribosome binding sequence linked to the mGFP4 gene into the XbaI site between the aadA gene and psbA terminator of the pSBLCtV2 vector (Guda et al. 2000). This vector has four homologous sequences to the tobacco plastome, which includes the trnI and trnA genes as border sequences flanking the transgene unit, and the rrn promoter and psbA terminator sequences for transgene expression, derived from the corresponding genes in the plastome (Fig. 1a).

Introduction of aadA and gfp in a dicistronic manner into tobacco plastome using the pCtVG vector. a The transgenes (aadA and gfp) are controlled by the rrn promoter and psbA terminator and bordered by trnI and trnA on the right and left sides, respectively. The transgenes are integrated into the plastid genome via homologous recombination between the same trnI and trnA sequences of the vector and the plastome. b–d PCR analysis of transplastomic plants. trnI-trnA, PCR amplification using the FI and RA primers; trnI-GFP, PCR amplification using the primers FI and GR1; aadA-trnA, PCR amplification using the primers A5 and RA. e Southern blot analysis of one transplastomic plant. 1–10, ten independent transplastomic plants (T0 generation); WT wild-type plant; T transplastomic plant (T0 generation)
Production of transplastomic plants
The vector DNA was introduced into leaf plastids using a particle delivery system (PDS-1000/He, BioRad). After bombardment, the leaf explants were cultured on Murashige and Skoog (1962) medium supplemented with 1 mg/L 6-benzyladenine, 0.1 mg/L α-naphthaleneacetic acid, and 500 mg/L spectinomycin dihydrochloride. To generate homoplasmic transplastomic plants, the leaf explants were excised from regenerated shoots and cultured on the same medium to induce shoots. This procedure was repeated once more, and finally the regenerated shoots were transferred to MS medium with 500 mg/L of spectinomycin dihydrochloride without growth regulators for rooting. The putative transplastomic plantlets were acclimated, transplanted to potting soil, and then grown to maturity in a growth chamber (27 °C, approximately 50 μmolm−2 s−1 from cool-white fluorescent lamps with a 16-h photoperiod).
Southern blot analyses
Total DNA was extracted from the leaves of one wild-type plant and independent transplastomic plants at almost the same developmental stage using Plant DNeasy® (Qiagen, Germany), followed by digestion with BamHI, BglII, or EcoRI, and undigestion. Approximately, 5 μg of digested or undigested DNA were electrophoresed on 0.8 % agarose gel and blotted onto Zeta-Probe® GT Blotting membranes (Bio-Rad). For hybridization, [α-32P] dCTP-labled probes were generated via random priming (RediprimeTM II labeling system; Amersham) in accordance with the manufacturer’s instructions. A 0.5 kb DNA fragment for aadA probe, 0.45 kb DNA fragment for psbA probe, 0.39 kb DNA fragment for trnI probe, and 0.15 kb DNA fragment for trnA probe were prepared via PCR amplification with the primer pairs A5/A3, PAF2/PAR1, trnIF3/trnIR4, and trnAF1/trnAR1, respectively. Prehybridization, hybridization, and washing of the membrane were conducted in accordance with the manufacturer’s instructions.
PCR analyses
The primers utilized in this study are listed in Table 1. Total DNA, primers, and the reaction buffers were incubated in a DNA thermal cycler (GeneAmp PCR system 9700; Applied Biosystems) under the following conditions: 94 °C for 5 min followed by 30 cycles of 94 °C for 1 min, 55 °C for 1 min, and 72 °C for 1–2 min, followed by a final 7 min extension at 72 °C before electrophoresis.
PCR amplification of the 2.8 and 21 kb subgenomic molecules
PCR primers were designed to amplify the 2.8 and 21 kb subgenomic circles (Table 1). For the 2.8 kb circle, PCR was conducted using three primer sets (trnIF3/rrn16R1, rrn16F5/rrn16R1, and trnIF4/trnIR7) which bind to the trnI and 16S rRNA genes (Fig. 5c). The resultant product was cloned into pGEM®-T Easy vector (Promega) for sequencing. The 21 kb subgenomic circle was amplified with three primer sets (LIP/RIP, LOP/ROP, LOP/OvRtP) facing in inward and outward orientations, and the OvRtP primer overlapped the LOP primer with a reverse orientation on the trnH gene from the genomic DNA of a transplastomic plant via long-range PCR with TaKaRa LA Taq polymerase. Cycle conditions for long-range PCR were as follows: 94 °C for 2 min, 5 cycles of 98 °C for 10 s, 65 °C for 20 s, and 68 °C for 18 min, then, 25 cycles of 98 °C for 10 s, and 68 °C for 18 min, with a final extension step of at 72 °C for 10 min, in a T-gradient (Biometra, T1 Thermocycler, USA). The amplified PCR products containing the 21 kb circle were electrophoresed on 0.8 % agarose gel.
DNA sequencing
The purified PCR products were directly subjected to sequencing or were cloned into pGEM®-T Easy Vector System prior to sequencing. For sequencing, the universal oligonucleotides T7 and SP6 primers, and/or the gene-specific oligonucleotides PAF2, PAR1, A5, and GR1 were used. Sequencing was conducted using an Applied Biosystems automatic DNA sequencer (ABI370) at the Genotech Co. Sequencing Center (Daejeon, Korea).
Northern blot analysis
Total RNA was extracted using Trizol from the leaves of wild-type and independent transplastomic plants in accordance with the manufacturer’s instructions. Five micrograms of total RNA were electrophoresed into 1.4 % agarose formaldehyde denaturing gel and transferred to Zeta-Probe® GT membranes (Bio-Rad). After UV cross-linking, the RNA blot was hybridized with the gfp probe. The 0.5 Kb gfp-specific DNA fragment was amplified with pCtVG vector as a template and the GFP-F (5′-GAA GGT GAT GCA ACA TAC GGA AAA-3′) and GFP-R (5′-GTT TGT CTG CCG TGA TGT ATA CGT T-3′) primers by PCR. Prehybridization and hybridization were conducted overnight at 65 °C, and the membranes were washed at 65 °C in accordance with the manufacturer’s instructions. RNA loading was monitored via methylene blue staining of the membranes.
Results
Generation of transplastomic plants and detection of aadA and gfp by PCR
Transplastomic plants were generated using the homologous recombination vector pCtVG to insert the aadA and gfp transgenes into the site between trnI and trnA in the plastome. Due to the presence of the gfp gene, transplastomic plants emitted green fluorescence under UV light, which allowed for discrimination against wild-type plants. We selected 10 transplastomic plants emitting green fluorescence and conducted PCR to determine whether the transgenes had been incorporated properly into the plastid genome using the FI and RA primers that bind to the trnI gene and trnA gene, respectively (Table 1; Fig. 1). PCR analysis showed that the aadA and gfp transgenes, under the control of the rrn promoter and psbA terminator derived from the tobacco plastome, were incorporated correctly between trnI and trnA in the plastome of transplastomic plant lines 1, 2, 3, 5, 8, 9, and 10 (Table 1; Fig. 1b). However, total DNA from the other three plants (lines 4, 6, and 7) generated no amplicon bands (Fig. 1b).
PCR with the FI and GR1 primers designed to amplify the sequence between trnI and gfp detected a single amplicon band from all 10 plants (Table 1; Fig. 1c), whereas PCR with the A5 and RA primers, designed for the amplification of the DNA fragment between aadA to trnA, generated a single amplicon band from the above seven plants, but no amplicon bands from the above three plants. These results showed that the transgenes were connected to the trnI gene in all 10 plants as intended, whereas the transgenes were not connected to the trnA gene in the three plants, in which homologous recombination was probably underway. Homologous recombination occurs via a multistep process, in which one end is integrated into the host DNA before the other end (Klaus et al. 2004).
Southern blot analysis using the aadA gene as a probe verified that the transgenes were incorporated correctly between trnI and trnA in the plastome of the soil-grown transplastomic plant (line 1 in Fig. 1) as intended, which was indicated by a 9.3 kb band in the BamHI digestion and a 6.5 kb band in the BglII digestion (Fig. 1e). From the BamHI-digested plastid DNAs, 9.3 and 4.3 kb bands were detected, whereas 6.5 and 9.1 kb bands were observed with the BglII-digested plasmid DNA. The 9.3 and 6.5 kb bands were major bands, whereas the 4.3 and 9.1 kb bands were minor bands. The two major bands showed that the plastome was transformed via homologous recombination between the trnI and trnA intergenic region as intended. However, the two minor bands were not the intended ones, which indicates that additional unintended recombination events may have occurred in the initial recombinant plastome.
Postulation of a possible mechanism to generate subgenomic circles from the initial recombinant plastome
The initial recombinant plastome retained two sets of the additional rrn promoter and the psbA terminator as a consequence of homologous recombination for the integration of the transgenes (because the transgenes were incorporated into the inverted repeat region of the plastome, the recombinant plastome would eventually contain additional two sets of the promoter and terminator). For simplicity’s sake, we proposed illustrations depicting one set of the promoter and terminator in the one side of the inverted repeat. Therefore, the psbA terminators could be lined up as shown in Fig. 1a, and subsequent intramolecular recombination would occur, thereby resulting in the excision of the initial recombinant plastome to two subgenomic circles (21 and 139 kb), as proposed in Fig. 2a. The 21 kb subgenomic circle was postulated to repeat further excision via intramolecular recombination between intra-element rrn promoters to generate 2.8 and 18.2 kb subgenomic circles, as shown in Fig. 2a. Alternative production of subgenomic circles was postulated in Supplementary Fig. 1A and 1B.

Postulation of intramolecular recombination to release the subgenomic circles in the transplastome of the transplastomic plants. a Generation of 21 and 139 kb subgenomic circles via intramolecular recombination between psbA terminators. b Generation of 2.8 and 18.2 kb subgenomic circles by intramolecular recombination between rrn promoters of 21 kb subgenomic circles
Generation of a 21 kb subgenomic circle via intramolecular recombination between two psbA terminators
In transplastomic plants, the transgene unit (Prrn-aadA-gfp-TpsbA) was inserted into the site between trnI and trnA in the Inverted repeat region of the plastome. The resultant recombinant plastome harbored additional rrn promoter and psbA terminator sequences as direct repeats, and an inverted repeat for the native rrn promoter and psbA terminator sequences, respectively. The rrn promoter and psbA terminator are frequently utilized to control transgene expression in plastid transformation (Jeong et al. 2004; Maliga 2004; Karimi et al. 2013; Roh et al. 2014).
The presence of subgenomic circles was demonstrated via PCR, Southern blot analysis, and PCR product sequencing analysis. The 21 kb subgenomic circle would be generated via intramolecular recombination between intra-element psbA terminators (Fig. 2). Two minor bands of 4.3 and 9.1 kb observed in Southern blot analysis (Fig. 1e) seemed to indicate the presence of two subgenomic circles, which could subsequently be generated from the excision of the 21 kb subgenomic circle, as the proposed 21 kb circle harbors two BamHI sites with a 4.3 kb distance around the transgenes (One site is located on the rrn promoter and the other site between rpl23 and ycf2). The minor 9.1 kb band indicated the presence of the 21 kb subgenomic circle, because the 21 kb circle could generate the 9.1 kb fragment harboring the transgenes after BglII digestion (One site is located between rps12 and ORF70B and the other site on ycf2).
Two amplicon bands of 2.3 and 4.0 kb were detected via PCR with the FI/H and FI/RP23 primers in all analyzed transplastomic plants, whereas the H and RP23 primers bind to the trnH and rpl23 genes, respectively (Table 1; Fig. 3). These two amplicon bands could not be generated from the wild-type plastome, or if the plastome was transformed via homologous recombination, could be generated only in the trnI and trnA intergenic region. To characterize the manner in which these two amplicon bands were generated, the 2.3 kb amplicon band was sequenced, comprising trnI-Prrn-aadA-gfp-TpsbA-trnH (Fig. 3c). If only the intended homologous recombination event had occurred between trnI and trnA, the order of genes should have been trnI-Prrn-aadA-gfp-TpsbA-trnA (The map of the recombinant plastome is shown in Fig. 3a). If another homologous recombination had occurred between intra-element psbA terminators for the expression of the dicistronic transgenes (aadA and gfp) and the native gene, the 21 kb circular DNA molecule harboring the trnI-Prrn-aadA-gfp-TpsbA-trnH DNA sequence could be generated as a subgenomic circle. To confirm the presence of the 21 kb circular DNA, three primer sets were designed with inward (LIP and RIP), outward (LOP and ROP), and 17 bp overlapped (OvRtP) to LOP with a reverse orientation on the trnH gene, after which long-range PCR was conducted with a transplastomic plant (Table 1; Fig. 3d). PCR detected a 21 kb DNA fragment within all three sets of primers from a transplastomic plant, but no amplicon bands from a wild-type plant, thereby indicating that the 21 kb subgenome was circular. Overall results, including the results of PCR, Southern blot analysis, and PCR product sequencing analysis definitively supported the presence of the 21 kb DNA subgenomic molecules in the transplastomic plant, in accordance with our expectations.

Demonstration of the presence of a 21 kb subgenomic circle. a Diagram of the 21 kb subgenome and initial recombinant plastome. b PCR analysis of the 21 kb subgenome. trnI-trnH, PCR amplification using the FI and H primers; trnI-RP23, PCR amplification using the FI and rpl23 primers. c Sequence of PCR product amplified using the FI and H primers. d Long-range PCR analysis for 21 kb subgenomic circle using the LIP/RIP, LOP/OvRtP, and LOP/ROP primer sets; 1–10, 10 independent transplastomic plants (T0 generation); WT wild-type plant; T transplastomic plant line 1; H lambda HindIII DNA size marker
Generation of a 139 kb subgenomic circle via intramolecular recombination between two psbA terminators
If the 21 kb DNA molecules had been derived from the plastome initially transformed by pCtVG, a plastome 21 kb shorter than the initial recombinant plastome must have been observed (The shorter plastome should be 139 kb long) (Figs. 2, 4a). To determine whether such a plastome exists, PCR using primers (RA/PAR1 and RR23/MK) was conducted, in which PAR1, RR23, and MK bind to the psbA coding region, the rrn23 gene, and the matK gene, respectively (Table 1; Fig. 4). The sequence distance between trnA and psbA was 21 kb. If the plastids had harbored only the initial recombinant plastome and the 21 kb subgenomic circle, PCR using the RA and PAR1 primers should have generated 21 kb amplicon bands or no amplicon bands, because the 21 kb subgenomic circle is too long to be amplified via regular PCR. However, PCR generated 1.4 (using RA/PAR1 primers) and 3.5 kb amplicon bands (using RR23/MK primers) from seven of 10 transplastomic plants (Fig. 4a–c). To determine how these amplicon bands were generated, the 1.4 kb amplicon band was sequenced, revealing trnA-psbA (Fig. 4a, the diagram and DNA sequence). These results of PCR amplicon and sequencing supported that a 139 kb subgenome was present in T0 plants. If only the intended homologous recombination event had occurred, the gene order of trnA-psbA could not have been generated. Southern blot analysis using the trnA gene as a probe detected three bands at 2.5, 1.9, and 0.5 kb from the recombinant plastome digested with BamHI/BglII in the T1 generation of the transplastomic plant (Fig. 4d). The 2.5 kb band was a major band and the other bands were minor bands, indicating that the recombinant plastome was transformed in accordance with the intended homologous recombination in the trnI and trnA intergenic region (one BamHI site on the rrn promoter and the other BglII site on trnA in the initial recombinant plastome, Fig. 3a), and the 1.9 kb band supported the presence of a 139 kb circular DNA molecule (one BglII site on trnA and the other BglII site between psbA and matK in Fig. 4a). These results bolstered the notion that a 139 kb circular DNA molecule coexisted with the initial recombinant plastome and the 21 kb subgenomic circle in the plastids of the transplastomic plants. The minor 0.5 kb band was also detected in the wild-type plants, thus suggesting that these transplastomic plants retained the residual wild-type plastome.

Demonstration of the presence of a 139 kb subgenomic circle. a Diagram of a 139 kb subgenomic circle and sequence of PCR products amplified using the RA (for trnA) and PAR1 (for psbA) primers on the 139 kb subgenomic circle. b, c PCR analysis of transplastomic plants. trnA-psbA, PCR amplification using the RA and PAR1 primers; rrn23-matK, PCR amplification using the RR23 (for rrn23) and MK (for matK) primers. d Southern blot analysis of one transplastomic plant. 1–10, 10 independent transplastomic plants (T0 generation); WT wild-type plant; T1, T1 generation of transplastomic plant
Generation of 18.2 and 2.8 kb subgenomic circles via intramolecular recombination between two rrn promoters
Because the rrn promoter for the expression of the transgenes was also integrated into the recombinant plastome, another intramolecular recombination could occur between the two identical sequences derived from the vector and the native 16S rRNA gene within the initial recombinant plastome. As a result, we expected to generate another subgenomic circle including 2.8, 18.2, and 154 kb, as shown in the illustration in Fig. 2a, b and Supplementary Fig. 1B. To detect these subgenomes, PCR was conducted using various primer combinations (listed in Table 1). PCR detected one major amplicon band of 2.45 kb and one minor band of 5.25 kb using the 70B and H primers that bind to the orf70B gene and trnH genes, respectively, only from the transplastomic plants (T0 generation) (Fig. 5a). PCR also detected one major amplicon band of 1.1 kb and one minor band of 3.9 kb using the 70B and A3 primers for the aadA transgene (T0 generation) (Fig. 5b). The 5.25 and 3.9 kb amplicon bands can be amplified from the 21 kb subgenomic circle. However, the 2.5 and 1.1 kb amplicon bands cannot be generated from the proposed subgenomic circles of 21, 139, or 160 kb (initial recombinant plastome). These results demonstrate that the transgene cassette (Prrn-aadA-gfp-TpsbA) was rearranged in a different manner to generate the 21 and 139 kb subgenomes. These amplicon bands could be amplified if another homologous recombination event had occurred between the intra-element rrn promoters derived from the element for transgene expression and the native rrn gene in the plastome, the promoter region of the 16S rRNA gene, to generate a 18.2 kb subgenome (the 18.2 kb subgenome does not contain the 16S rRNA gene and trnI gene), which could be produced via intramolecular recombination between direct repeats of the rrn promoter sequences in the 21 kb subgenome (Figs. 2, 5c). DNA sequencing revealed that the 2.5 kb amplicon band was composed of orf70B-trnV-Prrn-aadA-gfp-TpsbA-trnH (without 16S rRNA gene and trnI gene) (Supplementary Fig. 2, instead of the expected gene order: orf70B-trnV-16S rRNA-trnI-Prrn-aadA-gfp-TpsbA-trnH, thereby indicating that another 18.2 kb subgenome without two 16S rRNA-trnI genes coexisted with other subgenomic DNA molecules within the plastids of transplastomic plants.

Demonstration of the presence of 2.8 and 18.2 kb subgenomic circles. a, b PCR analyses for subgenomic circles. 70B-trnH, PCR amplification using the 70B and H primers; 70B-aadA3, PCR amplification using the 70B and A3 primers. c Diagram for 2.8 kb subgenomic circle. d, e PCR analysis for a 2.8 kb subgenomic circle. trnIF3-rrn16R1, PCR amplification using the trnIF3 and rrn16R1 primers; rrn16, PCR amplification using the rrn16F5 and rrn16R1 primers with inward orientation; trnI, PCR amplification using the trnIF4 and trnIR7 primers with inward orientation. f Southern blot analysis for the presence of the 2.8 kb subgenome in T 0 and T 2 generations. WT wild-type plant; digested Southern blot with EcoRI-digested DNA; undigested Southern blot without prior restriction enzyme digestion
The 21 kb subgenome could be rearranged via intramolecular recombination at the direct repeats of rrn promoters of the transgenes and the native gene to release a 2.8 kb subgenomic circle harboring only the two 16S rRNA-trnI genes, leaving the 18.2 kb subgenomic circle missing these two genes (Figs. 2, 5c). PCR and Southern blot analysis were conducted to determine the presence of the 2.8 kb subgenomic circle in the transplastomic plant (T0 and T2 generations). PCR detected the 2 kb amplicon band using the trnIF3 and rrn16R1 primers that bind to the trnI and 16S rRNA genes, respectively (Fig. 5d). PCR with two sets of primers including rrn16R1/rrn16F5 and trnIR7/trnIF4, which bind to the 16S rRNA gene or the trnI gene with an inward orientation, respectively, verified the presence of the 2.8 kb subgenomic circle. A 3 kb amplicon band was detected only from transplastomic plants (Fig. 5e). These primer sets cannot amplify the specific amplicon bands from 160, 139, 21 kb, and other possible subgenomes, if any, with the exception of the 2.8 kb subgenomic circle. Sequencing of the 2 and 3 kb DNA fragments (Supplementary Fig. 2) showed that the trnI gene was followed by the 16S rRNA gene in the PCR product (The 16S rRNA gene is followed by trnI in the wild-type tobacco plastome). Southern blot analysis using the trnl gene as a probe detected several major bands and a minor band corresponding to 2.8 kb from the EcoRI-digested DNA of the T0 and T2 generations of transplastomic plants and other two minor bands between 1 and 3 kb from undigested DNA (Fig. 5f). These results indicate that a 2.8 kb circular DNA molecule coexists with other subgenomic molecules within the plastid. A similar intramolecular recombination event between direct repeat sequences of the Prrn promoter might also have occurred in the initial recombinant plastome (160 kb), which was subsequently excised to 2.8 and 154 kb subgenomic circles.
The overall results of this study demonstrated that intramolecular recombination occurred between direct repeats of the rrn promoter and inverted repeats of the psbA terminator, generating various subgenomic circles after initial homologous recombination between the plastid transformation vector and the plastome. All of these subgenomic circles were stably maintained in the absence of selection.
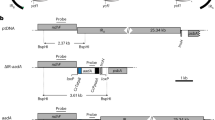
Construction of a new vector to prevent generation of subgenomic circles from the initial recombinant plastome
In this study, we demonstrated that the initial recombinant plastome generated by a conventional vector containing a native promoter and terminator for transgene expression cannot avoid subsequent intramolecular recombination between the intra-element promoters and between terminators. As a result, various subgenomic circles in the plastid were detected in all analyzed transplastomic plants, which is not the intent of plastid transformation. Transplastomic plants with such subgenomic circles would suffer from genetic instability owing to low levels of homoplasmy.
To prevent the generation of subgenomic circles from the initial recombinant plastome, we constructed a new plastid transformation vector by replacing the rrn promoter and psbA terminator for transgene expression in the pCtVG vector with the clp promoter from rice and the rrnB1B2 terminator of the rRNA gene from E. coli, respectively (Fig. 6a). The resultant recombinant plastome would contain no repeats of the rrn promoter and the psbA terminator, such that the generation of subgenomic circles via intramolecular recombination would be prevented. Accordingly, we generated transplastomic plants using the new vector. No extra bands were detected when soil-grown plants were subjected to Southern blot analysis using the aadA and trnA genes as probes (Fig. 6b, c), whereas two extra bands of 9.1 and 1.9 kb were detected from the transplastomic plants (T2 generation) generated using the pCtVG vector, which were assumed to represent the 21 kb and 139 kb subgenomic circles, respectively (lane T2 of Fig. 6b, c). To maximize the stringency of the detection of extra amplicon bands, if any, PCR was conducted using four sets of primers (FI/RA, FI/H, RR23/MK, and 70B/A3), resulting in no extra amplicon bands from transplastomic plants generated using the new vector, which were maintained in vitro in the presence of selection, whereas subgenomic circles were detected in control plants (pCtVG transformed T2 generation) (Fig. 6d). These results verified our hypothesis that subgenomic circles are generated from the initial recombinant plastome via intramolecular recombination between homologous promoters and/or between homologous terminators.

Production and analysis of transplastomic plants using a new plastid transformation vector. a Plastid transformation vector to prevent intramolecular recombination in transplastome. trnI, plastidic trnI gene of tobacco; rclpP, promoter region of the clp gene of rice; rrnB1B2, terminator region of the E. coli rrn gene; trnA, plastidic trnA gene of tobacco. b Southern blot analysis performed with aadA probe. c Southern blot analysis performed with trnA probe. d PCR analysis of transplastomic plants. Total DNA from soil-grown T0 transplastomic plants was digested with BglII for Southern blot hybridization. WT wild-type; 1–10 transplastomic plants generated by new vector; T 2 T2 generation of transplastomic plant generated by pCtVG vector; trnI-trnA PCR amplification using the FI and RA primers; trnI-trnH PCR amplification using the FI and H primers; rrn23-MatK, PCR amplification using the RR23 and MK primers; 70B-A3 PCR amplification using the 70B and A3 primers
Discussion
Extrachromosomal DNAs were detected in progeny
Extrachromosomal DNA molecules were also detected in the progeny of plastid-transformed tobacco plants. A 2.3 kb amplicon was generated by PCR amplification using trnI and trnH primers (lane T2 of Fig. 6d), indicating that the 21 kb subgenomic DNA sequence was present in the T2 progeny. PCR amplification with the RR23 and MK primers generated a 3.5 kb amplicon (lane T2 of Fig. 6d). Southern blot analysis indicated a 1.9 kb sequence from the recombinant plastome digested with BamHI/BglII in the T1 and T2 generations of the transplastomic plants (Figs. 4d, 6c), indicating that a 139 kb subgenomic DNA sequence was present in the progeny. PCR amplification with the trnIF/rrn16R and rrn16/trnI primers and Southern blot analysis detected 2.0 kb and 2.8 kb sequences and corresponding bands for a 2.8 kb subgenomic circle in the T2 generation (Fig. 5d–f). These data indicate that the 2.8 kb subgenomic DNA sequence was present in the progeny. PCR amplification with the 70B and A3 primers also generated a 1.1 kb amplicon in T2 progeny (lane T2 of Fig. 6d), indicating that an 18.2 or 154 kb subgenomic DNA sequence was present. Subgenomic circles were also detected in T3 generation (data not shown). Therefore, the extrachromosomal DNA molecules were likely generated via intramolecular recombination, regardless of the type of transgene. These results demonstrated that all these subgenomic circles were present in the progeny.
Extrachromosomal DNAs generated in the T0 generation were retained in the progeny
We demonstrated that all types of extrachromosomal DNAs found in the T0 generation were also detected in progeny through the T3 generation. Three different interpretations of these data are possible: (1) all types of extrachromosomal DNAs generated in the T0 generation were transmitted to the progeny; (2) only copies of the initially transformed plastid were transmitted to the progeny and all the extrachromosomal DNAs were newly generated in each generation; or (3) a combination of both events occurred. The second and third possibilities are supported by the finding that some of the extrachromosomal DNAs lacked the ori sequence, suggesting that some extrachromosomal DNAs were newly generated in progeny. Scharff and Koop (2007) reported that the oriA and/or oriB sequences are not essential for plastid DNA replication, implicating additional mechanisms of plastid DNA replication and/or additional origins of replication. To support this, recently, we found that episomal vector containing of putative replication origin of minicircle in plastid of marine dinoflagellate Heterocapsa triquetra was successfully replicated in the plastid of tobacco for 6 month (Min et al. 2015). Additionally, it would be difficult for all the extrachromosomal DNA molecules found in the T0 plants to replicate by themselves and be stably transmitted to progeny. Therefore, it is reasonable to hypothesize that all extrachromosomal DNA molecules detected in the progeny are newly generated in each generation from the initially transformed plastid DNA molecule transmitted to the progeny.
Previous studies indicate that the extrachromosomal DNA designated NICE1 was transmitted to progeny (Staub and Maliga 1994), but extrachromosomal DNA generated following CRE-lox recombination was not inherited by the progeny (Corneille et al. 2003). NICE1 contains the ori sequence, whereas the deleted genome following CRE-lox recombination lacked the ori sequence. Based on these and our findings, extrachromosomal DNAs lacking the ori sequence cannot be transferred to subsequent generations.
In addition, extrachromosomal DNA with the ori sequence should be replicated more rapidly than a longer one. However, no extrachromosomal DNAs were considerably more prominent than the others in the progeny. These data further support that extrachromosomal DNAs could be continuously generated from the initially transformed plastid DNA retained in the progeny.
Extrachromosomal DNAs were circular
We demonstrated using inward, outward, and overlap extension PCR that the extrachromosomal DNAs were circular. However, it cannot be ruled out that the extrachromosomal DNAs were also present in forms other than the circular wild-type plastid chromosome (Lilly et al. 2001). In addition, as circular DNA molecules, two or more extrachromosomal DNAs derived from the initially transformed DNA could recombine with the initial plastid DNA by recombination between the same repeat sequences in the extrachromosomal DNAs as proposed in the subgenomic molecules and the master circle of the mitochondrial genome (Small et al. 1989).
In conclusion, this study demonstrated that extrachromosomal DNAs generated from the initially transformed plastid DNA via intramolecular recombination were found in progeny. Some of the extrachromosomal DNAs detected in the progeny lacked the ori sequence, suggesting that the initially transformed plastid DNA was transmitted to the progeny that generated new extrachromosomal DNAs.
References
Corneille S, Lutz KA, Azhagiri AK, Maliga P (2003) Identification of functional lox sites in the plastid genome. Plant J 35:753–762
Gray BN, Ahner BA, Hanson MR (2009) Extensive homologous recombination between introduced and native regulatory plastid DNA elements in transplastomic plants. Transgenic Res 18:559–572
Guda C, Lee SB, Daniell H (2000) Stable expression of biodegradable protein base polymer in tobacco chloroplasts. Plant Cell Rep 19:257–262
Karimi F, Mousavi A, Salmanian AH, Alizadeh H, Rafati S (2013) Immunogenicity of EIT chimeric protein expressed in transplastomic tobacco plants towards development of an oral vaccine against Escherichia coli O157:H7. Plant Biotechnol Rep 7:535–546
Klaus SMJ, Huang FC, Golds TJ, Koop HU (2004) Generation of marker-free plastid transformants using a transiently cointegrated selection gene. Nat Biotechnol 22:225–229
Lilly JW, Havey MJ, Jackson SA, Jiang J (2001) Cytogenomic analyses reveal the structural plasticity of the chloroplast genome in higher plants. Plant Cell 13:245–254
Maliga P (2004) Plastid transformation in higher plants. Ann Rev Plant Biol 55:289–313
Min SR, Davarpanah SJ, Jung SH, Park YI, Liu JR, Jeong WJ (2015) An episomal vector system for plastid transformation in higher plants. Plant Biotechnol Rep (Submitted)
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol Plant 15:473–497
Roh KH, Choi SB, Kwak B-K, Seo S-C, Lee S-B (2014) A single cupredoxin azurin production in transplastomic tobacco. Plant Biotechnol Rep 8:421–429
Scharff LB, Koop HU (2007) Targeted inactivation of the tobacco plastome origins of replication A and B. Plant J 50:782–794
Small ID, Suffolk R, Leaver CJ (1989) Evolution of plant mitochondrial genomes via substoichiometric intermediates. Cell 58:69–76
Staub JM, Maliga P (1992) Long regions of homologous DNA are incorporated into the tobacco plastid genome by transformation. Plant Cell 4:39–45
Staub JM, Maliga P (1994) Extrachromosomal elements in tobacco plastids. Proc Natl Acad Sci USA 91:7468–7472
Acknowledgments
This work was supported by a grant from the KRIBB Research Initiative program, a Grant from the Advanced Biomass R&D Center (ABC) of Korea Grant funded by the Ministry of Education, Science and Technology (ABC-2011-0031343), and a Grant from the Golden Seed Project, Ministry of Agriculture, Food, and Rural Affairs (MAFRA), the Ministry of Oceans and Fisheries (MOF), Rural Development Administration (RDA), and Korea Forest Service (KFS).
Author information
Authors and Affiliations
Corresponding authors
Additional information
J. R. Liu and W.-J. Jeong contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Min, S.R., Jung, S.H., Liu, J.R. et al. The fate of extrachromosomal DNAs in the progeny of plastid-transformed tobacco plants. Plant Biotechnol Rep 9, 431–442 (2015). https://doi.org/10.1007/s11816-015-0380-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11816-015-0380-5