
Abstract
Thermostable DNA polymerases are being widely used in polymerase chain reaction for diagnosis, cloning and DNA sequencing purposes. We have cloned a gene from Pyrobaculum calidifontis encoding a thermostable DNA polymerase and expressed in Escherichia coli BL21(DE3). The gene product was produced in soluble and highly active form. However, the expression was low, which resulted in a poor yield of the recombinant enzyme. In order to get higher expression, we introduced a silent mutation at second codon by replacing the rare codon (AGG), for arginine, with an E. coli preferred codon (CGT). This resulted in nearly a 2-fold higher production of the recombinant protein. We further optimized the inducer concentration and time of cultivation after induction. Under optimized conditions nearly 3-fold higher production of the recombinant protein was observed. Furthermore, we changed the expression host to E. coli Rosetta DE3 cells. However, there was no increase in the expression level, rather it was slightly decreased. Such silent mutations and use of an alternate expression host can be used to enhance heterologous expression of other genes too.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
DNA polymerase is a vital enzyme for life as it copies DNA during replication. DNA polymerases have various applications including polymerase chain reaction (PCR), a method for amplification of desired DNA fragments (Kovarova and Draber 2000; Zhu et al. 2020). During PCR, the high temperature requires thermostable DNA polymerase. So, the DNA polymerases from thermophilic bacteria and archaea find utility in PCR. Taq DNA polymerase, an enzyme isolated from T. aquaticus, is the most commonly used enzyme for this purpose. However, this enzyme does not possess 3′ to 5′ exonuclease activity. Hence it has low fidelity during DNA replication. Furthermore, it lacks the ability to amplify longer DNA segments (Hamilton et al. 2001). These problems, can be overcome by using DNA polymerases of archaeal origin. Most of the DNA polymerases isolated from hyperthermophilic archaea belong to family B and possess 3′ to 5′ exonuclease activity (Lundberg et al., 1991; Takagi et al., 1997), thereby display high fidelity in DNA amplification. Several thermostable recombinant DNA polymerases have been characterized from euryarchaeota including Pyrococcus abyssi (Dietrich et al., 2002; Gueguen et al., 2001), Pyrococcus furiosus (Lundberg et al., 1991), Thermococcus litoralis (Kong et al., 1993), Thermococcus aggregans (Niehaus et al., 1997), Thermococcus gorgonarius (Hopfner et al. 1999), Thermococcus sp. 9_N-7 (Southworth et al., 1996), Thermococcus kodakarensis (Takagi et al., 1997; Yamashita et al. 2017), Thermococcus fumicolans (Cambon-Bonavita et al., 2000) and Thermococcus gammatolerans (Zhang et al. 2020). A few reports are also available from crenarchaeota (Feng et al., 2020) including a family B DNA polymerase from P. calidifontis (Ali et al., 2011; Guo et al., 2017). However, heterologous expression of DNA polymerase gene from P. calidifontis resulted in low yield of the recombinant enzyme.
Synonymous codon substitution, because of its apparent silent behavior, has long been considered inconsequential. However, this dogma has been refuted and significant impact on heterologous gene expression level has been observed by synonymous codon substitution (Bibi et al. 2017; Chamary et al. 2006; Saunders and Deane 2010; Plotkin and Kudla, 2011; Chen et al., 2020). Translational efficiency is considered to be influenced by the mRNA secondary structure. Expression level reduces significantly by strong hairpin loop formation centered at initiation codon (AUG) and Shine-Dalgarno ribosome binding site (Kubo and Imanaka, 1989). Synonymous codon substitution at the 5′-end can impact mRNA secondary structure and its stability, and thus the relative translation kinetics at initiation and elongation steps (Tuller et al., 2010). Furthermore, the speed of translation of a codon is also affected by its frequency. More frequent codon, in the genome of an organism, is translated faster because of abundance of its cognate tRNA (Sørensen and Pedersen, 1991; Sørensen et al., 1989; Victor et al., 2020). Through codon usage, the traffic of ribosomes on mRNA could be controlled. More frequent codons would give smooth fast traffic. The passage from a segment carrying rare codon to one carrying frequent codon would minimize the bottleneck for ribosome traffic and could reduce jamming, which in turn could increase the level of gene expression (Deana et al., 1998). We describe here the effect of: i) synonymous mutation from a rare codon to E. coli preferred codon, ii) reduction in hairpin loop formation in mRNA, iii) host cells, iv) inducer concentration, and v) time after induction, on expression level of a family B DNA polymerase gene from P. calidifontis.
Materials and methods
Chemicals, reagents, enzymes, plasmids and host cells
The chemicals used in this research work were of analytical grade and purchased either from Merck (Germany), Sigma (USA), Thermo Fisher Scientific (Leicestershire, UK) or Fluka (Buchs, Switzerland). The primers for gene amplification were synthesized by Macrogen (Korea). Taq DNA polymerase, T4 DNA ligase, dNTPs, restriction endonucleases, DNA and protein size markers were purchased from Thermo Fisher Scientific (USA).
E. coli propagation strain DH5α and expression host strains BL21 (DE3) and Rosetta (DE3) were from Novagen (Merck, Germany). Cloning vector pTZ57R/T was from Thermo Fisher Scientific (USA) and pET-21a(+) from Novagen (Merck, Germany). Luria-Bertani (LB) broth and agar were used as growth media of the host cells.
Cloning and expression of mutated DNA polymerase gene
A silent mutation immediately after initiation codon was introduced in the wild type gene. The low frequency codon in E. coli for arginine (AGG) was replaced by a high frequency codon (CGT).
Wild-type Gene: ATGAGGTTTTGGCCTCTAGACGCCACGTACTCTG……TAG.
Mutated Gene: ATGCGTTTTTGGCCTCTAGACGCCACGTACTCTG…….TAG.
For amplification of the mutant gene by polymerase chain reaction (PCR), a set of forward (Pol2N) and reverse (Pol2C) primers was used. Nucleotide sequence of the primers, their melting temperature (Tm) and GC contents are shown in Table 1. Genomic DNA of P. calidifontis strain VA1 was used as template for PCR amplification of the mutated gene. The PCR amplified gene was ligated in pTZ57R/T (Thermo Fisher Scientific) and the resulting construct was names pTZ-PolM. The DNA polymerase gene was liberated by digesting pTZ-PolM with NdeI and BamHI and ligated in pET-21a(+) digested with the same restriction enzymes. The resulting plasmid was named pET-PolM. Construction of expression plasmid for wild-type gene has been previously described (Ali et al., 2011). Escherichia coli BL21 (DE3) and Rosetta (DE3) were used as host cells for heterologous expression of the gene. Host cells carrying pET-PolM plasmid were cultivated at 37 °C and gene expression was induced with 0.12, 0.25, 0.5 and 1.0 mM of isopropyl β-D-1-thiogalactopyranoside (IPTG) when optical density of the culture at 600 nm was ~0.4. Cultivation was continued at the same temperature for 1, 2, 4, and 6 h after induction. The cells were harvested by centrifugation at 6000×g for 15 min and suspended in 50 mM Tris-Cl pH 8.5. The cells were lysed by sonication, using Vibra Cell 130 VC sonicator, with 50 pulses at 80% amplitude, 12 watts power, and 10 s of pulse with a pause of 1 min. The concentration of proteins in the lysate was measured by Bradford method (Bradford 1976). The samples were analyzed by 12% denaturing polyacrylamide gel electrophoresis (SDS-PAGE).
Purification of the recombinant protein
After cell lysis, soluble fraction was separated by centrifugation at 13,000×g and 4 °C for 15 min. Supernatant, containing the recombinant protein PolM, was heated at 85 °C for 20 min to denature the heat labile proteins of E. coli origin. The denatured and precipitated proteins were separated by centrifugation at 18,000×g for 25 min. The supernatant containing recombinant PolM was purified by using HiTrap Heparin (GE Healthcare) column. The proteins bound to the column were eluted using a linear gradient of 0 to 1 M NaCl. Fractions after column chromatography were analyzed by SDS-PAGE and enzyme activity assay.
Enzyme activity assay
Enzyme activity of recombinant PolM, stored at 4 °C in 10 mM Tris-Cl (pH 8.5) containing 10% glycerol, was examined by measuring the incorporation of TTP [methyl-3H] by using activated calf thymus DNA as a template. The reaction mixture contained: 25 mM Tris-Cl pH 8.5, 4 mM MgCl2, 100 μM each of dATP, dGTP, dCTP, dTTP, 0.5 μCi TTP [methyl-3H] (85 Ci/mmol), 5 μg activated calf thymus DNA, 0.2 mg/mL BSA and 0.1% Tween 20 in a total volume of 20 μL. The mixture was pre-warmed at 75 °C for 5 min before addition of the recombinant enzyme. After the addition of PolM, aliquots were taken at various intervals of time and spotted onto DE-81 filter paper discs (23 mm diameter, Whatman, Madison, UK). The filter paper discs were air dried at room temperature, washed three times in 0.5 M sodium phosphate buffer pH 7.0, followed by washing in 70% ethanol and air-dried. The radioactivity incorporated on the filter paper discs was measured in counts per second (cps) by using Raytest Malisa scintillation counter (Berlin, Germany).
Use of PolM in PCR
To evaluate the DNA amplification by the recombinant enzyme by PCR, DNA fragment cloned in pET-21a(+) was used as template. The primers, 522F and Pol2C, used for application of PolM in PCR are given in Table 1. Amplified product was obtained by using a set of gene specific primers. In addition to the primer-template set, the PCR mixture contained 10 mM Tris-Cl pH 8.5, 4 mM MgCl2, 200 μM each dNTP, 0.2 mg/mL BSA and 0.5 U recombinant PolM. Denaturation was done at 95 °C, annealing at 55 °C and extension at 72 °C for 25 cycles.
Results
Analysis of secondary structure of mRNA
Analysis of the secondary structure of 27 nucleotides from the 5′-end of the mRNA was done by using mfold program freely available at http://unafold.rna.albany.edu/?q=mfold. This indicated a hairpin loop formation between the codons of the second (AGG) and fifth (CCT) amino acids (Fig. 1A). Furthermore, AGG is not a preferred codon for E. coli. In order to avoid hairpin loop formation, we designed Pol2N primer for PCR amplification of the gene replacing AGG codon by CGT, a preferred codon in E. coli. When the secondary structure of mRNA of the mutated gene was analyzed, the hairpin loop in the structure was relaxed (Fig. 1B). Hence, this substitution exhibited a dual advantage.

Prediction of mRNA secondary structure of the 5′-end of the gene. A) mRNA secondary structure of the wild-type gene. B) mRNA secondary structure of the 5′-end of the synonymously mutated gene
Cloning of mutated DNA polymerase gene
PCR, using primers Pol2N, Pol2C and the wild-type gene as a template, resulted in amplification of a 2.4 kbp DNA fragment, matching the size of the DNA polymerase gene from P. calidifontis (Fig. 2A). Ligation of the PCR amplified DNA fragment in pTZ-57R/T resulted in a recombinant construct that was named pTZ-PolM. Digestion of pTZ-PolM with NdeI and BamHI liberated a 2.4 kbp DNA fragment, matching the size of PolM DNA polymerase gene (Fig. 2B). For expression of the gene, the liberated fragment was ligated in pET-21a(+) vector and the resulting plasmid was named pET-PolM. Cloning of PolM in pET-21a was confirmed by double digesting isolated pET-PolM with NdeI and BamHI. Digestion of pET-PolM resulted in liberation of a 2.4 kbp DNA fragment indicating the presence of the gene in pET-PolM (Fig. 2C). The presence of PolM gene in pET-PolM was further confirmed by digestion with HindIII, which resulted in liberation of a ~ 1.4 kbp DNA fragment (Fig. 2D), matching the expected fragment as the gene contains a HindIII recognition site at 1037 position.

Ethidium bromide stained 1% agarose gel demonstrating cloning of PolM gene. A) PCR amplified PolM gene. B) Cloning of PolM gene in pTZ57R/T. C) Cloning of PolM gene in pET-21a(+) expression vector. D) Confirmation of cloning of PolM gene in pET-21a(+) expression vector
Production of recombinant DNA polymerase in E.coli
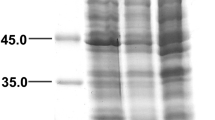
When the expression of the wild-type and the mutant genes was compared by inducing the host cells carrying pET-PolM construct with 0.5 mM IPTG, a 2-fold higher production of recombinant protein was observed in the cells harboring pET-PolM plasmid compared to the cells containing the wild-type gene in pET-Pol construct (Fig. 3A). This was further confirmed by analyzing the enzyme activity in the cell-free lysates of both the samples. Cells carrying pET-PolM plasmid (mutant gene) exhibited nearly 2-fold higher activity (~6150 U/L of the culture) compared to the cells containing pET-Pol (wild-type gene; ~3295 U/L of the culture). These results were in agreement with the production of the recombinant protein demonstrated by SDS-PAGE. We further replaced the expression host, E. coli BL21 (DE3) by E. coli Rosetta (DE3), however no increase in expression level was found (Fig. 3B). The time of cultivation, after induction, is an important factor for optimal expression. We analyzed the samples after various times after induction and found relative higher production of recombinant protein at 4 h after induction (Fig. 4A). In addition to the time after induction, concentration of the inducer also affects the production of recombinant proteins. When various concentrations of IPTG were used to induce the gene expression, relatively higher expression was observed at 0.12 mM (Fig. 4B). Under optimized expression conditions, ~9120 U of DNA polymerase activity was found per litre of the culture.

Coomassie brilliant blue stained 12% SDS-PAGE demonstrating the expression levels of wild-type and mutated genes. A) Comparison of expression of wild-type and mutated genes in E. coli BL21(DE3). Lane M, standard molecular weight marker (ThermoFisher Scientific # SM0661); lane TLM, total lysate of cells carrying mutated gene; lane TLW, total lysate of cells carrying wild-type gene. B) Comparison of heterologous expression in BL21(DE3) and Rosetta (DE3) cells of E. coli. Lane M, standard molecular weight marker (ThermoFisher Scientific # SM0661); lane BL, BL21(DE3) cells; lane Ros, Rosetta (DE3) cells

Coomassie brilliant blue stained 12% SDS-PAGE demonstrating the expression levels at various times after induction and induction with various concentrations of IPTG. A) Comparison of expression level at various time intervals. Lane M, standard molecular weight marker; lane 3 h, sample at 3 h after induction; lane 4 h, sample at 4 h after induction; lane 5 h, sample at 5 h after induction; and lane 6 h, sample at 6 h after induction. B) Comparison of expression level after induction with various concentrations of IPTG. Lane M, standard molecular weight marker; lane 0.0, sample without induction; lane 0.12, sample induced with 0.12 mM IPTG; lane 0.25, sample induced with 0.25 mM IPTG; lane 0.5, sample induced with 0.5 mM IPTG; and lane 1.0, sample induced with 1.0 mM IPTG
Partial purification and application in PCR
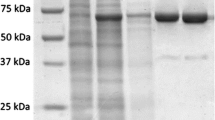
Thermostable recombinant PolM was partially purified by heat treatment and affinity column chromatography. Heat treatment of the clarified lysate resulted in precipitation of majority of the host-cell proteins. Recombinant PolM remained in the soluble fraction after heat treatment and was further purified by using HiTrap Heparin column (Fig. 5). Recombinant PolM eluted between 30 and 40% of 1 M NaCl. Concentration of NaCl at elution peak was 350 mM. The specific activities after heat treatment and column chromatography were 90 and 395 U/mg, respectively. The recombinant PolM was purified to ~6-fold with a yield of 60% (~23 mg). The partially purified recombinant PolM was successfully applied in PCR for amplification of 0.8 kbp gene fragment cloned in pET-21a(+) vector (Fig. 6).

Coomassie brilliant blue stained 12% SDS-PAGE demonstrating partially purified PolM DNA polymerase. Lane M, standard molecular weight marker (ThermoFisher Scientific # SM0671); lane 1, partially purified PolM

Ethidium bromide stained 1% agarose gel demonstrating amplification of 0.8 kb DNA fragment by PCR using PolM DNA polymerase. Lane M, standard molecular weight marker (ThermoFisher Scientific # SM0331); lane 1, PCR amplified product
Discussion
E. coli expression system is one of the most popular expression systems. Usually high levels of recombinant proteins are produced by this system. However, sometimes no or very low production of recombinant proteins is reported (Rosano and Ceccarelli 2014). This could be due to a hairpin loop formation in the mRNA structure or codon bias of the host organism. Similar was the case with the DNA polymerase gene from P. calidifontis. When this gene was expressed in E. coli, a low level of recombinant protein was observed. Analysis of the secondary structure of the mRNA, using mfold web server, revealed the formation of a hairpin loop. In order to remove this loop, AGG codon for arginine, at position 2, was replaced by CGT, which also codes for arginine. This silent mutation resulted in removal of the hairpin loop formation at this position and ultimately resulted in a higher expression of the gene. Hairpin loop formation in mRNA can also be avoided by altering the expression vector. A gene encoding glyceraldehyde-3-phosphate dehydrogenase from Trypanosoma brucei was cloned in two different vectors, pET-3a and pET-28a(+), and expressed in E. coli BL21(DE3) cells. There was two-fold increase in the production of recombinant protein from pET-28a(+) vector as compared to pET-3a (Hannaert et al. 1995). In the present study, we too cloned the DNA polymerase gene in two different vectors, pET-21a(+) and pET-28a(+), and expressed in E. coli. However there was no significant difference in production of the recombinant protein from both the vectors (data not shown). In addition to the vector, expression host is one of the factors which affect the level of the gene expression. It has been suggested that E. coli Rosetta (DE3) strain is advantageous for producing the recombinant proteins that are difficult to produce in E. coli BL21(DE3) (Tegel et al., 2010). In our case the results were opposite. There was a relatively higher expression in BL21(DE3) compared to the Rosetta (DE3).
Optical density of the culture at the time of induction and cultivation time after induction play a crucial role in the production of a recombinant protein. Inducing the expression at early log phase or early mid-log phase is beneficial in most of the cases. There was notable decrease in recombinant protein production when induction was carried out after mid-log phase (Berrow et al., 2006). We, therefore induced the culture in early mid-log phase to get optimal expression. In addition to culture density, heterologous expression level is usually related to the amount of the inducer. A very low level of inducer results in low expression while a very high induction level is toxic to the cells (Lopes et al., 2019). Therefore, we examined the expression at various concentrations of the inducer and found 0.12 mM as optimal concentration.
In conclusion, the results obtained in this study indicated that mRNA secondary structure, codon bias, expression host and inducer concentration contribute to the heterologous expression of a gene. Silent mutations in the gene to avoid hairpin loop formation, use of suitable expression host and utilization of codons preferred by the host can be exploited to enhance heterologous expression of foreign genes.
Abbreviations
- PCR:
-
polymerase chain reaction
- Taq :
-
Thermus aquaticus
- tRNA:
-
transfer RNA
- mRNA:
-
messenger RNA
- dNTP:
-
deoxynucleoside triphosphate
- dATP:
-
deoxyadenosine triphosphate
- dGTP:
-
deoxyguanosine triphosphate
- dCTP:
-
deoxycytidine triphosphate
- dTTP:
-
deoxythymidine triphosphate
- SDS:
-
sodium dodecyl sulphate
- PAGE:
-
polyacrylamide gel electrophoresis
- Tm:
-
melting temperature
- IPTG:
-
isopropyl β-D-1-thiogalactopyranoside.
References
Ali SF, Rashid N, Imanaka T, Akhtar M (2011) Family B DNA polymerase from a hyperthermophilic archaeon Pyrobaculum calidifontis: cloning, characterization and PCR application. J BiosciBioeng 2:118–123. https://doi.org/10.1016/j.jbiosc.2011.03.018
Berrow NS, Büssow K, Coutard B, Diprose J, Ekberg M, Folkers GE, Levy N, Lieu V, Owens RJ, Peleg Y, Pinaglia C, Quevillon-Cheruel S, Salim L, Scheich C, Vincentelli R, Busso D (2006) Recombinant protein expression and solubility screening in Escherichia coli: a comparative study. Acta Crystallogr D Biol Crystallogr 62:1218–1226. https://doi.org/10.1107/S0907444906031337
Bibi T, Ali M, Rashid N, Muhammad MA, Akhtar M (2017) Enhancement of gene expression in Escherichia coli and characterization of highly stable ATP-dependent glucokinase from Pyrobaculum calidifontis. Extremophiles 22(2):247–257. https://doi.org/10.1007/s00792-017-0993-4
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72(1):248–254. https://doi.org/10.1006/abio.1976.9999
Cambon-Bonavita MA, Schmitt P, Zeiger M, Flaman JM, Lesongeur F, Raguenes G et al (2000) Cloning, expression and characterization of DNA polymerase I from the hyperthermophilic archaeon Thermococcus fumicolans. Extremophiles 4:215–225. https://doi.org/10.1007/pl00010714
Chamary JV, Parmley JL, Hurst LD (2006) Hearing silence: non-neutral evolution at synonymous sites in mammals. Nat Rev Genet 7(2):98–108. https://doi.org/10.1038/nrg1770
Chen K, Guo R, Wei C (2020) Synonymous mutation rs2515641 affects CYP2E1 mRNA and protein expression and susceptibility to drug-induced liver injury. Pharmacogenomics 21(7):459–470. https://doi.org/10.2217/pgs-2019-0151
Deana A, Ehrlich R, Reiss C (1998) Silent mutations in the Escherichia coli ompA leader peptide region strongly affect transcription and translation in vivo. Nucleic Acids Res 26(20):4778–4782. https://doi.org/10.1093/nar/26.20.4778
Dietrich J, Schmitt P, Zeiger M, Preve B, Rolland JL, Chaabihi H et al (2002) PCR performance of the highly thermostable proof-reading B-type DNA polymerase from Pyrococcus abyssi. FEMS Microbiol Lett 217:89–94. https://doi.org/10.1111/j.1574-6968.2002.tb11460.x
Feng X, Liu X, Xu R, Zhao R, Feng W, Liao J, Han W, She Q (2020) A unique B-family DNA polymerase facilitating error-prone DNA damage tolerance in crenarchaeota. Front Microbiol 11:1585. https://doi.org/10.3389/fmicb.2020.01585
Gueguen Y, Rolland JL, Lecompte O, Azam P, Le Romancer G, Flament D et al (2001) Characterization of two DNA polymerases from the hyperthermophilic euyarchaeon Pyrococcus abyssi. Eur J Biochem 268:5961–5969. https://doi.org/10.1046/j.0014-2956.2001.02550.x
Guo J, Zhang W, Coker AR, Wood SP, Cooper JB, Ahmad S, Ali S, Rashid N, Akhtar M (2017) Structure of the family B DNA polymerase from the hyperthermophilic archaeon Pyrobaculum calidifontis. Acta Crystallogr D Struct Biol 73:420–427. https://doi.org/10.1107/S2059798317004090
Hamilton SC, Farchaus JW, Davis MC (2001) DNA polymerases as engines for biotechnology. Biotechniques 31:370–383. https://doi.org/10.2144/01312rv01
Hannaert V, Opperdoes FR, Michels PA (1995) Glycosomal glyceraldehyde-3-phosphate dehydrogenase of Trypanosoma brucei and Trypanosoma cruzi: expression in Escherichia coli, purification, and characterization of the enzymes. Protein Expr Purif 6(3):244–250. https://doi.org/10.1006/prep.1995.1031
Hopfner KP, Eichinger A, Engh RA, Laue F, Ankenbauer W, Huber R, Angerer B (1999) Crystal structure of a thermostable type B DNA polymerase from Thermococcus gorgonarius. Proc Natl Acad Sci U S A 96(7):3600–3605. https://doi.org/10.1073/pnas.96.7.3600
Kong H, Kucera RB, Jack WE (1993) Characterization of a DNA polymerase from the hyperthermophile archaea Thermococcus litoralis. Vent DNA polymerase, steady state kinetics, thermal stability, processivity, strand displacement and exonuclease activities. J Biol Chem 268:1965–1975
Kovarova M, Draber P (2000) New specificity and yield enhancer of polymerase chain reactions. Nucleic Acids Res 28(13):E70. https://doi.org/10.1093/nar/28.13.e70
Kubo M, Imanaka T (1989) mRNA secondary structure in an open reading frame reduces translation efficiency in Bacillus subtilis. J Bacteriol 171(7):4080–4082. https://doi.org/10.1128/jb.171.7.4080-4082
Lopes C, Dos Santos NV, Dupont J, Pedrolli DB, Valentini SR, de Carvalho S-EV, Pereira JFB (2019) Improving the cost effectiveness of enhanced green fluorescent protein production using recombinant Escherichia coli BL21(DE3): decreasing the expression inducer concentration. Biotechnol Appl Biochem 66(4):527–536. https://doi.org/10.1002/bab.1749
Lundberg KS, Shoemaker DD, Adams MW, Short JM, Soge JA, Mathur EJ (1991) High-fidelity amplification using a thermostable DNA polymerase isolated from Pyrococcus furiosus. Gene 108:1–6. https://doi.org/10.1016/0378-1119(91)90480-y
Niehaus F, Frey B, Antranikian G (1997) Cloning and characterization of a thermostable alpha-DNA polymerase from the hyperthermophilic archaeon Thermococcus sp. TY Gene 204:153–158. https://doi.org/10.1016/s0378-1119(97)00536-2
Plotkin JB, Kudla G (2011) Synonymous but not the same: the causes and consequences of codon bias. Nat Rev Genet 12(1):32–42. https://doi.org/10.1038/nrg2899
Rosano GL, Ceccarelli EA (2014) Recombinant protein expression in Escherichia coli: advances and challenges. Front Microbiol 5:172. https://doi.org/10.3389/fmicb.2014.00172
Saunders R, Deane CM (2010) Synonymous codon usage influences the local protein structure observed. Nucleic Acids Res 38(19):6719–6728. https://doi.org/10.1093/nar/gkq495
Sørensen MA, Kurland CG, Pedersen S (1989) Codon usage determines translation rate in Escherichia coli. J Mol Biol 207(2):365–377. https://doi.org/10.1016/0022-2836(89)90260-x
Sørensen MA, Pedersen S (1991) Absolute in vivo translation rates of individual codons in Escherichia coli. The two glutamic acid codons GAA and GAG are translated with a threefold difference in rate. J Mol Biol 222(2):265–280. doi: https://doi.org/10.1016/0022-2836(91)90211-n
Southworth MW, Kong H, Kucera RB, Ware J, Jannasch HW, Perler FB (1996) Cloning of thermostable DNA polymerases from hyperthermophilic marine archaea with emphasis on Thermococcus sp. 9_ N-7 and mutations affecting 3′–5′ exonuclease activity. Proc Natl Acad Sci U S A 93:5281–5285. https://doi.org/10.1073/pnas.93.11.5281
Takagi M, Nishioka M, Kakihara H, Kitabayashi M, Inoue H, Kawakami B et al (1997) Characterization of DNA polymerase from Pyrococcus sp. strain KOD1 and its application to PCR. Appl Environ Microbiol 63:4504–4510. https://doi.org/10.1128/aem.63.11
Tegel H, Tourle S, Ottosson J, Persson A (2010) Increased levels of recombinant human proteins with the Escherichia coli strain Rosetta(DE3). Protein Expr Purif 69(2):159–167. https://doi.org/10.1016/j.pep.2009.08.017
Tuller T, Carmi A, Vestsigian K, Navon S, Dorfan Y, Zaborske J, Pan T, Dahan O, Furman I, Pilpel Y (2010) An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell 141(2):344–354. https://doi.org/10.1016/j.cell.2010.03.031
Victor MP, Acharya D, Chakraborty S, Ghosh TC (2020) The combined influence of codon composition and tRNA copy number regulates translational efficiency by influencing synonymous nucleotide substitution. Gene 745:144640. https://doi.org/10.1016/j.gene.2020.144640
Yamashita M, Xu J, Morokuma D, Hirata K, Hino M, Mon H, Takahashi M, Hamdan SM, Sakashita K, Iiyama K, Banno Y, Kusakabe T, Lee JM (2017) Characterization of recombinant Thermococcus kodakaraensis (KOD) DNA polymerases produced using Silkworm-Baculovirus expression vector system. Mol Biotechnol 59:221–233. https://doi.org/10.1007/s12033-017-0008-9
Zhang L, Jiang D, Shi H, Wu M, Gan Q, Yang Z, Oger P (2020) Characterization and application of a family B DNA polymerase from the hyperthermophilic and radioresistant euryarchaeon Thermococcus gammatolerans. Int J Biol Macromol 156:217–224. https://doi.org/10.1016/j.ijbiomac.2020.03.204
Zhu H, Zhang H, Xu Y, Laššáková S, Korabečná M, Neužil P (2020) PCR past, present and future. Biotechniques 69:317–325. https://doi.org/10.2144/btn-2020-0057
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ahmad, S., Ali, S.F., Azim, N. et al. Studies on enhancement of production of recombinant DNA polymerase originated from Pyrobaculum calidifontis. Biologia 76, 3579–3586 (2021). https://doi.org/10.1007/s11756-021-00887-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11756-021-00887-7