
Abstract
Partial epoxidation of methyl linoleate was carried out at room temperature (30 °C) using a methyltrioxorhenium catalyst in the presence of pyridine and urea-hydrogen peroxide. Full epoxidation of methyl linoleate was carried out using the Prilezhaev method. The reactions were monitored using the oxirane oxygen content value. The products from partial and full epoxidation were analyzed using GC-FID, FTIR, NMR and GC–MS. Methyl 9,10-epoxy-12Z-octadecenoate and methyl 12,13-epoxy-9Z-octadecenoate were obtained as the major products from partial epoxidation, with a percent yield of 46 %. The product from full epoxidation afforded 97 % yield with methyl 9,10-12,13-diepoxyoctadecanoate as the major component. Physicochemical properties such as kinematic viscosity, viscosity index, crystallization temperature and oxidative stability were examined. Fully epoxidized methyl linoleate exhibits superior kinematic viscosity and oxidative stability due to the complete conversion of double bonds to epoxy groups. Partially epoxidized methyl linoleate exhibits intermediate kinematic viscosity, viscosity index, crystallization temperature and oxidative stability.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Vegetable oils and their derivatives have numerous advantages for various applications due to their availability from renewable resources, biodegradability and non-toxic natures. However, the use of vegetable oils is restricted to certain applications due to their inadequate oxidative stability, poor low-temperature properties and a narrow range at which viscosity is optimal [1–3]. Chemical modification of vegetable oils is an attractive way to overcome these shortcomings. Elimination of olefin groups by introducing additional functionalities could improve the oxidative stability of vegetable oils while, reducing structural uniformity to improve their low temperature properties [1, 4].
The physicochemical properties of vegetable oils and their derivative-based products can be enhanced by epoxidation reactions, which can eliminate olefin groups and add epoxy groups. For instance, epoxidized soybean oil showed maximum improvement of oxidative stability, followed by genetically modified high oleic soybean oil and crude soybean oil [5]. Fully and partially epoxidized oils and derivatives with different physicochemical properties can be applied in different domains. Low viscosities and melting points below room temperature are desirable features for reactive diluents, which may be obtained from homogenous oils comprising partially epoxidized oils, acids, or their esters [6]. Full epoxides are mainly applied as polyvinyl chloride-plasticizers and stabilizers [7]. In addition, full epoxides have high commercial importance as surface coatings, surfactants, lubricants and as intermediates in chemical reactions [8, 9].
There are several methods for producing partial and full epoxides from vegetable oils and their derivatives, including fatty acids and methyl ester. These methods depend on the type of feedstock, epoxidation reagent, catalyst and solvent used in the epoxidation reactions [10]. Conventionally, full epoxidation of unsaturated compounds is carried out using the Prilezhaev method [6], which uses peroxycarboxylic acids generated in situ or performed by reacting formic or acetic acid with aqueous hydrogen peroxide (H2O2). Enzymes such as lipase and immobilized oat seed peroxygenase can provide milder conditions for partial and full epoxidation, as has been recently reported [11, 12]. Nevertheless, these processes are time consuming and have low selectivity. To overcome these problems, a number of transition metal-based catalysts such as methyltrioxorhenium (MTO), meso-tetraphenylporphyrin manganese (III) chloride [Mn(TPP)Cl] and titanium catalyst continue to be investigated [13, 14].
Methyltrioxorhenium (MTO) is known as an effective transition metal-based catalyst for epoxidation of olefins. MTO is easily available, active at low concentrations and effective with a broad range of olefins. Moreover, it does not decompose H2O2 which acts as the oxidant during most of the MTO-catalyzed epoxidation into water and oxygen gas [15]. Previous studies have reported the possibility of tuning the degree of epoxidation by using MTO with aqueous H2O2 as an oxidant (MTO/H2O2) system [14, 16]. However, the MTO/H2O2 system retards the formation of epoxide with a high yield. The limitations include ring opening of the newly formed epoxide due to the acidic nature of the rhenium catalyst and facile decomposition of MTO to perrhenates [17]. For this reason, urea-hydrogen peroxide (UHP) rather than H2O2 was used in this study to avoid unwanted epoxide ring opening reactions. Furthermore, the addition of heterocyclic amine additives, such as pyridine, 3-cyanopyridine and pyrazole have been found to be successful in both suppressing such side reactions and substantially increasing the rate of epoxidation [17, 18].
An abundance of research related to epoxidation has been reported, but limited attention has been paid to partial epoxidation or to the physicochemical properties of the epoxidized products, such as their viscosity, oxidative stability and low temperature performance. In the present study, Jatropha curcas oil-based methyl linoleate was chosen as the substrate for partial and full epoxidation. We report the preparation of monoepoxidized methyl linoleate using the MTO/UHP system and the production of diepoxidized methyl linoleate using the Prilezhaev method. The physicochemical properties such as viscosity, viscosity index, crystallization temperature and oxidative stability of the partially epoxidized methyl linoleate (PEML) and fully epoxidized methyl linoleate (FEML) were determined and compared with those of methyl linoleate.
Experimental Procedures
Materials
Methyl linoleate (97.3 %, iodine value = 167.91 g I2/100 g) with 2.7 % methyl oleate by urea complexation method [19] was prepared from Jatropha curcas oil. MTO and UHP were obtained from Aldrich. Pyridine, dichloromethane (CH2Cl2), formic acid (≥98 %) and aqueous H2O2 (30 wt%) were obtained from Systerm. Chemicals obtained from commercial vendors were used as received.
Partial Epoxidation of Methyl Linoleate Using MTO/UHP/Pyridine
CH2Cl2 (5 mL) was added along with MTO (0.06 g, 0.25 mmol), UHP (9.34 g, 99.24 mmol) and pyridine (0.24 g, 2.98 mmol) to a 50-mL Erlenmeyer flask. The amounts of MTO, UHP and pyridine were calculated according to the double bonds content of the methyl linoleate. The mixture was vigorously stirred (1,100 rpm) for 10 min at room temperature (30 °C) until the solution became yellow and remained unchanged throughout the reaction. A solution of methyl linoleate (5 g, 33.08 mmol of double bonds) in 10 mL of CH2Cl2 was added all at once to the mixture and stirred at room temperature for 2 h. After completion of the reaction (2 h), the mixture was diluted with CH2Cl2 and washed with distilled water. Excess H2O2 from UHP in the yellow organic phase was decomposed by adding a small amount of manganese dioxide (MnO2) until the yellow color totally disappeared. The organic phase was separated and washed with a saturated solution of sodium chloride and then dried over anhydrous sodium sulfate. The solvent was removed with a rotary evaporator. The overall yield of the reaction was 91 %. The product mixture, which contained unreacted methyl linoleate and diepoxide, was purified using silica gel column chromatography. The eluent of ether contained dichloromethane (ether: CH2Cl2 = 2:98) and was used to separate monoepoxides from diepoxide and unreacted methyl linoleate. Isolated yield: 46 % monoepoxides, 28 % diepoxide and 26 % unreacted methyl linoleate. IR(NaCl) for the separated monoepoxides (PEML): 3,513, 3,464, 3,010, 2,954, 2,929, 2,858, 1,740, 1,656, 1,460, 1,438, 1,379, 1,246, 1,198, 1,172, 1,113, 1,014, 881, 844, 826, 787, 725 cm−1. 1H NMR (400 MHz, CDCl3) for the separated monoepoxides (PEML): δ 0.76–0.80 (t, J = 6.6 Hz, 3H, CH 3–CH2–), 1.20–1.24 (m, 12H, –CH 2–), 1.39–1.42 (m, 4H, –CHOCH–CH 2–CH 2–), 1.48–1.53 (m, 2H, –CH 2–CH2–COOCH3), 1.91–1.96 (m, 2H, –CH=CH–CH 2–CH2–), 2.04–2.27 (m, 4H, 11–CH 2– and –CH 2–COOCH3), 2.80–3.01 (m, 2H, –CHOCH–), 3.55 (s, 3H, –COOCH 3), 5.22–5.41 ppm (m, 2H, –CH=CH–). 13C NMR (100 MHz, CDCl3) for the separated monoepoxides (PEML): δ 13.92 (CH3–CH2–), 22.52–33.91 (–CH2–), 51.29 (–COOCH3), 54.24–57.06 (–CHOCH–), 123.89–132.53 (–CH=CH–), 174.05 ppm (–COOCH3). EI-MS: Methyl 12,13-epoxy-9Z-octadecenoate (GCRT: 14.39 min): m/z 310 [M+], 292 [M −18], 279 [M −31], 235 [CH2CH(O)CHCH2CH=CH(CH2)7COOCH3 −18]+, 221 [CH2CH(O)CHCH2CH=CH(CH2)7COOCH3 −32]+, 207 [O≡CCH2CH=CH(CH2)7COOCH3 −18]+, 179 [CH2CH=CH(CH2)7COOCH3 −18]+, 164 [M −146, loss of CH3(CH2)4CH(O)CHCH3 and H2O], 147 [CH2CH=CH(CH2)7COOCH3 −50]+, 136 [CH3(CH2)4CH(O)CHCH2CH=CH2 −18]+. Methyl 9,10-epoxy-12Z-octadecenoate (GCRT: 14.43 min): m/z 310 [M+], 292 [M −18], 279 [M −31], 235 [CH2CH=CHCH2CH(O)CH(CH2)7COOCH3 −18]+, 221 [CH2CH=CHCH2CH(O)CH(CH2)7COOCH3 −32]+, 207 [CH2CH=CHCH2CH(O)CH(CH2)7COOCH3 −46]+, 200 [CH2(O)CH(CH2)7COOCH3]+, 199 [CH(O)CH(CH2)7COOCH3]+, 185 [O≡C(CH2)7COOCH3]+, 168 [CH2(O)CH(CH2)7COOCH3 −32]+, 155 [HO=CH(CH2)7COOCH3 −32]+, 143 [CH2(CH2)5COOCH3]+, 135 [CH3(CH2)4CH(O)CHCHCH=CH2 −18]+.
Full Epoxidation of Methyl Linoleate Using the Prilezhaev Method
A 100-mL two-neck round-bottom flask was charged with methyl linoleate (5 g, 33.08 mmol of double bonds) and formic acid (3 g, 66.16 mmol). The mixture was heated under stirring until 40 °C was reached. Afterwards, H2O2 (30 wt%, 13.5 g, 396.96 mmol) was added drop by drop and the reaction mixture was heated at 47 °C under stirring for 2 h. After the reaction was complete (2 h), the mixture was cooled to room temperature (30 °C). The organic phase was separated and washed with a solution of sodium bicarbonate (5 wt%) several times to remove any traces of acid remaining in the reaction mixture. Lastly, the organic phase was washed with a solution of sodium chloride (5 wt%) and dried over anhydrous sodium sulfate. The solvent was removed from the final product using a rotary evaporator. The product was analyzed using oxirane oxygen content titration and instruments (GC-FID, FTIR, NMR and GC–MS) without further purification. The overall yield of the reaction was 97 %. IR (NaCl): 3,517, 3,463, 2,954, 2,929, 2,858, 1,740, 1,458, 1,437, 1,379, 1,248, 1,198, 1,172, 1,124, 1,110, 1,014, 881, 844, 824, 788, 728 cm−1. 1H NMR (400 MHz, CDCl3): δ 0.72–0.75 (t, J = 7.3 Hz, 3H, CH 3–CH2–), 1.18–1.37 (m, 14H, –CH 2–), 1.30–1.37 (m, 4H, 8–CH 2– and 14–CH 2–), 1.44–1.47 (m, 2H, –CH 2–CH2–COOCH3), 1.54–1.62 (m, 2H, 11–CH 2–), 2.11–2.15 (m, 2H, –CH 2–COOCH3), 2.80–2.96 (m, 4H, –CHOCH–), 3.49 ppm (s, 3H, –COOCH 3). 13C NMR (100 MHz, CDCl3): δ 13.77 (CH3–CH2–), 22.37–33.75 (–CH2–), 51.13 (–COOCH3), 53.92–56.70 (–CHOCH–), 173.83 ppm (–COOCH3). EI-MS: Methyl 9,10-12,13-diepoxyoctadecanoate (GCRT: 15.93 and 16.30 min) m/z 326 [M+], 308 [M −18], 277 [M −49], 255 [CH2CH(O)CHCH2CH(O)CH(CH2)7COOCH3 −14]+, 237 [CH2CH(O)CHCH2CH(O)CH(CH2)7COOCH3 −32]+, 211 [O=CHCH2CH(O)CH(CH2)7COOCH3 −31]+, 187 [HO=CH(CH2)7COOCH3]+, 164 [CH2CH(O)CH(CH2)7COOCH3 −49]+, 155 [HO=CH(CH2)7COOCH3 −32]+, 139 [CH3(CH2)4CH(O)CHCH2CH=OH −18]+, 124 [CH3(CH2)4CH(O)CHCH2CH=OH −33]+.
Oxirane Oxygen Content (OOC)
The evolution of the epoxidation reaction was monitored by measuring the OOC in accordance with the official and recommended practice according to AOCS Cd 9-57 [20]. Under the prescribed conditions of this method, the oxygen was titrated directly with hydrogen bromide in acetic acid. From the OOC measurement, the relative conversion to oxirane (RCO) value was calculated from Eq. 1:
where OOCex is the experimentally determined content of oxirane oxygen in 100 g of sample and OOCth is the theoretical maximum oxirane oxygen in 100 g of sample. The OOCth value was determined from Eq. 2 [21]:
where IVo is the initial iodine value of methyl linoleate (167.91 g I2/100 g) and Ai (126.9) and Ao (16.0) are the atomic weight of iodine and oxygen, respectively.
Structural Analysis
The IR spectra were recorded on a Perkin Elmer Fourier transform infra-red (FTIR) spectrometer using the GX spectrum. Samples were prepared as thin films on sodium chloride (NaCl) plates. The scanning range was 700–4,000 cm−1 for 4 scans at a spectral resolution of 4 cm−1. The 1H- and 13C-NMR spectra were recorded using an NMR 400 MHz (JEOL–ECA series 400 MHz). A small amount of sample (0.2 g) was dissolved in 550 µL of CDCl3.
A gas chromatograph with flame ionization detector (GC-FID, Shimadzu, 17A series) equipped with a polar column BPX 70 (30 m × 0.25 mm i.d. × 0.25 μm film thickness; SGE) was used to determine the composition of the monoepoxides and diepoxides. The conditions of GC analysis were as follows: split injection with split ratio 29:1; rate 3 °C/min; FID temperature 280 °C; injector temperature 250 °C; initial oven temperature 120 °C with 1 min holding time and final oven temperature 250 °C with 15 min final holding time. Nitrogen was used as the carrier gas with flow rate of 0.4 mL/min.
Products were further characterized by GC with mass detection using an electron impact mass spectrometer (GC/EI-MS; Hewlett-Packard 5972 Series) equipped with a non-polar HP-5 ms column (30 m × 0.25 mm i.d × 0.25 µm film thickness; Agilent). The EI-MS detector was set to scan in the mass range of m/z 20–600 at 2 scans/s. The injector port temperature was 250 °C and the detector temperature was 290 °C. The initial oven temperature was set at 120 °C with 11 min holding time, and final oven temperature was 280 °C with 7 min holding time. Before 280 °C, the system was held for 4 min at 265 °C to ensure well separated peaks.
Determination of Kinematic Viscosity
The kinematic viscosities of the samples were measured at 40 and 100 °C on a Physica MCR 301 rheometer (Anton Paar, Germany). The rheometer was equipped with a P-PTD200 Lower plate Peltier temperature control system and a CP50-2 cone and plate measuring system shaft. The corresponding viscosity index was calculated according to ASTM D2270-04 [22].
Determination of Oxidative Stability
The experiments were carried out using a differential scanning calorimetry, DSC 1 (Mettler Tolero, Switzerland). Approximately 1.5 mg of sample was weighed in an aluminum pan closed with a pinhole lid to allow interaction of the sample with oxygen. The sealed aluminum pan was place in the DSC module. A film thickness of less than 1 mm was required to ensure proper oil–oxygen interaction and to eliminate any discrepancies in the results due to gas diffusion limitations [5]. The procedure involved heating the sample to 250 °C at 10 °C/min heating rate. Oxygen was pressurized in the module at a constant pressure of 3.5 mP. A flow rate of 50 mL/min was maintained throughout the experiment. Finally, the onset temperature (T o), which indicates the temperature for oxidative stability of the sample, was obtained by extrapolating the tangent drawn on the steepest slope of the exothermal reaction to the baseline in the thermogram.
Determination of Low Temperature Properties
Crystallization temperatures (T c) of the products were also determined by DSC1, where 10 mg of the sample was weighed in an open aluminum pan and placed in the DSC module. To homogenize the sample and especially waxy materials that could accelerate wax formation, the sample was rapidly heated to 50 °C and held under isothermal conditions for 10 min according to the procedure. The sample was then cooled to −70 °C at constant rate of 10 °C/min. Nitrogen gas was used as the cooling medium in this experiment. The T c was taken to be the onset temperature in the cooling process.
Results and Discussion
Synthesis
Preparation of monoepoxidized methyl linoleate from MTO-catalyzed partial epoxidation was carried out at room temperature (30 °C) in the presence of pyridine and UHP (Fig. 1). Partial epoxidation was initiated by the formation of monoperoxo- and diperoxorhenium complexes as a result of the interaction between MTO and UHP. The formation of peroxorhenium complexes were evident by the appearance of yellow color in the solution [23]. Upon addition of pyridine, the peroxorhenium complexes were transferred to the organic phase, where they reacted with the methyl linoleate. The urea that remained during the consumption of UHP modulated the pH of the solution and prevented acid-catalyzed ring opening [23]. As observed in Fig. 1, two of the monoepoxides formed were regioisomers. The epoxy group positioned between C12 and C13 only gave rise to compound 1 (methyl 12,13-epoxy-9Z-octadecenoate), which is a methyl ester of vernolic acid. Likewise, the epoxy group positioned between C9 and C10 gave rise to compound 2 (methyl 9,10-epoxy-12Z-octadecenoate), which is a methyl ester of coronaric acid. It is interesting to note that coronaric acid and vernolic acid are natural epoxy fatty acids [25]. The compositions of regioisomers represented by the ratio of 1 part of compound 1–1.2 parts of compound 2.

MTO-catalyzed partial epoxidation of methyl linoleate in the presence of UHP and pyridine
Preparation of diepoxidized methyl linoleate from full epoxidation was carried out using the Prilezhaev method (Fig. 2). Performic acid was generated in situ by combining H2O2 and formic acid in aqueous phase. An oxygen atom from performic acid was transferred to the olefin group for the formation of an oxirane ring in the organic phase. Owing to the water content in the 30 % H2O2 aqueous solution, the OOC values were used to closely monitor the epoxidation reaction to avoid undesired ring opening reactions. As shown in Fig. 2, the final product of full epoxidation was compound 3 (methyl 9,10-12,13-diepoxyoctadecanoate). Unlike compounds 1 and 2 (Fig. 1), the existence of compound 3 in nature is not yet reported.

Epoxidation of methyl linoleate by performic acid generated in situ using the Prilezhaev method
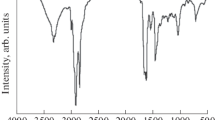
The methyl linoleate and products from partial and full epoxidation were analyzed by FTIR to determine the functional groups and to monitor the formation of epoxy group. The FTIR spectra of methyl linoleate, PEML and FEML from Fig. 3 show some common peaks at 3,467 (carbonyl C=O ester stretching overtone), 2,928 (methylene asymmetric C–H stretching), 2,856 (methylene symmetric C–H stretching), 1,743 (carbonyl C=O ester stretching), 1,456 (methylene asymmetric C–H bending), 1,380 (methyl symmetrical C–H bending) and 725 cm−1 (CH2 rocking). The additional peaks at 1,322, 1,245, 1,196, 1,172, 1,102, 1,018, 881 and 844 cm−1 are due to the stretching and rocking of the C–O group in the products.

Stacked FTIR spectra of methyl linoleate (ML), partially epoxidized methyl linoleate (PEML) and fully epoxidized methyl linoleate (FEML)
The alkene C–H vibration peak at 3,010 cm−1 gradually decreased from methyl linoleate to PEML and finally disappeared in FEML (Fig. 4a). The peaks at 1,660 (cis –CH=CH– stretching) and 914 cm−1 (=C–H out of plane bending) also showed a decreasing trend as 3,010 cm−1. The presence of new peaks in PEML at 826 cm−1 and in FEML at 824 cm−1 were ascribed to the epoxy group (Fig. 4b). This result is in agreement with a study of epoxidized soybean oil, which showed a characteristic peak at 823 cm−1 for the epoxy group [5]. These changes indicate the conversion of a C=C double bond in methyl linoleate to an epoxy group. Another peak that appeared at 787 cm−1 due to the ring vibration suggested the formation of an epoxide in cis configuration [26]. On these grounds, it seem reasonable to assume that the stereochemistry of the double bonds in methyl linoleate was retained in PEML and FEML.

Characteristic peaks of alkene C=C (a) and epoxide (b) in FTIR spectra for methyl linoleate (ML), partially epoxidized methyl linoleate (PEML) and fully epoxidized methyl linoleate (FEML)
Although the spectra obtained for PEML and FEML were found to have similar peaks, the peak intensity ratio for epoxide was different (Fig. 4). The intensity of the peak at 826 cm−1 for PEML was less than the peak at 824 cm−1 for FEML. This finding suggests that the double bonds of methyl linoleate were partially converted to epoxy group in PEML. This result was corroborated by the OOC titration method, in which the OOC values for PEML and FEML were 5.56 and 7.94 % respectively, and the RCO values were 58.06 and 82.98 % for the corresponding OOC. In addition to the C=O ester overtone characteristic peak at 3,467 cm−1, there was a shoulder band in the area of 3,517 cm−1 as a result of the intermolecular hydrogen bonded O–H, which involves an epoxy group. These results are obvious in the spectrum of FEML (Fig. 3) because of the formation of two epoxy groups.
The completion of the reaction was further confirmed by checking the 1H-NMR spectrum for the disappearance of protons attached to the C=C double bond at δ 5.22–5.41 ppm and the appearance of epoxide protons at δ 2.80–3.01 ppm. In the FEML 1H-NMR spectrum (Fig. 5a), the absence of alkene C=C peaks at δ 5.22–5.41 ppm indicated that the C=C double bonds from methyl linoleate were completely converted to epoxy group. Moreover, partial conversion of the C=C double bonds was verified by the simultaneous appearance of peaks at δ 5.22–5.41 and 2.80–3.01 ppm in the PEML 1H-NMR spectrum (Fig. 5b). The epoxy protons observed at the δ 2.80–2.96 ppm region further supported the cis configuration of the epoxide [14]. The presence of hydroxyl groups at δ 3.4 and 4.1 ppm were not detected by 1H-NMR spectroscopy in either case. Thus, no epoxy ring opening occurred in either epoxidation reaction.

1H-NMR spectra of fully epoxidized methyl linoleate (FEML) (a) and partially epoxidized methyl linoleate (PEML) (b)
In the PEML 13C-NMR spectrum (Fig. 6b), an epoxy group was observed at δ 54.24–57.06 ppm and there were 4 peaks at δ 123.89–132.53 corresponding to the carbon of the C=C double bonds. Small numbers of C=C double bonds in the unreacted methyl linoleate were detected at δ 127.94 and δ 130.01 ppm. The peaks at δ 123.89 and δ 132.53 ppm were assigned to the C=C double bond located near the epoxy group. The differences in chemical shifts can be explained by the shielding effect of the epoxy group on the β olefinic carbon, causing the peak to appear farther upfield in the spectrum. On the contrary, the γ olefinic carbon shifted downfield as the result of the deshielding effect. Therefore, the olefinic carbon closer to the epoxy group had a lower chemical shift value (δ 123.89 ppm) than the other carbon on the same double bond. Small amounts of double bonds signals at δ 123.86 and 132.21 ppm were also detected in the FEML spectrum (Fig. 6a) due to trace amounts of monoepoxides. Furthermore, six peaks appeared in the FEML 13C-NMR spectrum (Fig. 6a) at δ 53.92–56.70 ppm, which were attributed to epoxide carbons. It seems possible that these peaks resulted from the formation of diepoxide in a mixture of anti and syn diastereomers [27].

13C-NMR spectra of fully epoxidized methyl linoleate (FEML) (a) and partially epoxidized methyl linoleate (PEML) (b)
The structures of the products formed upon partial and full epoxidation are difficult to determine by NMR alone due to the formation of different regioisomers. Hence, GC–MS was used to verify the molecular structure and especially the position of the epoxy groups. Larger fragments contain either oxygen or double bonds were used to differentiate between the regioisomers. The peak at the gas chromatography retention time (GCRT) 14.39 min was assigned to compound 1 with the molecular ion M+ of 310. The mass spectrum analysis showed mass to charge ratios (m/z) of 179, 164 and 147 as the characteristic peaks for compound 1 due to the epoxy group positioned between C12 and C13. On the other hand, compound 2 appeared at GCRT 14.43 min with the same molecular ion M+ of 310, indicating that the product was a regioisomer of compound 1. The characteristic peaks for compound 2 were m/z 185, 168 and 155 as a result of the epoxy group positioned between C9 and C10. It is noteworthy that both of the peaks at GCRT 15.93 and 16.30 min showed the same mass spectrum. These two peaks were assigned to compound 3 and were suggested to be anti and syn diastereomers, with melting points of 78 and 36 °C [27]. Compound 3 with the molecular ion m/z 326 showed characteristic peaks at m/z 255, 237, and 211 as a result of two epoxy groups at C9,10 and C12,13.
Before column chromatography separation, the product of partial epoxidation contained 16 % unreacted methyl linoleate, 74 % monoepoxidized methyl linoleate and 8 % diepoxidized methyl linoleate. The compositions of the product indicated that monoepoxides were further converted to diepoxides before methyl linoleate was entirely consumed. This finding is consistent with other research [14], which used the MTO/H2O2 system to epoxidize methyl linoleate. However, our study achieved higher composition of monoepoxides in milder conditions that used a lower amount of MTO and safer UHP. Monoepoxides were purified and separated from unreacted methyl linoleate and diepoxide by silica gel column chromatography. The unreacted methyl linoleate, which is less polar, eluted first in ether: dichloromethane (2:98). The diepoxide, which is more polar than monoepoxides, eluted last. GC-FID chromatograms indicated that the monoepoxides were well separated from the unreacted methyl linoleate and diepoxide. However, the regioisomers of monoepoxide were not separable in the silica gel column because the structural similarities of compounds 1 and 2 make them difficult to differentiate by column chromatography. The total composition of monoepoxides in PEML increased from 74 to 91 %, with 31 % of compound 1 and 60 % of compound 2. Although the monoepoxides were highly pure, the percentage yield was only 46 %. The low percentage yield was due to the tedious work-up procedure involving multiple extraction and adsorption steps to purify the monoepoxides. On the other hand, the percentage yield of FEML without purification was 97 %; however, this product was 87 % compound 3 and only 13 % monoepoxides.
Physicochemical Properties
The physicochemical properties of methyl linoleate, PEML and FEML are presented in Table 1. The results showed that viscosity of the methyl linoleate increased gradually with increased degrees of epoxidation. FEML exhibited the highest kinematic viscosity at 40 and 100 °C due to the stronger intermolecular interactions at two oxygens in the diepoxide compared with the π electrons of the double bonds [27]. Stronger interactions between molecules restrict the fluidity of the liquid, thereby increasing the viscosity. As expected, the viscosity of methyl linoleate, PEML and FEML decreased as the temperature increased due to high temperatures weakening intermolecular interactions. This factor could be responsible for the significantly larger kinematic viscosity of methyl linoleate, PEML and FEML at 40 °C than at 100 °C. The changes in viscosity with the changes in temperature are expressed by the viscosity index, which depends on the rate of change in viscosity. A low viscosity index indicates a great variation of viscosity with a change in temperature; a high viscosity index indicates less variation of viscosity with the same changes in temperature. Although FEML exhibited the highest kinematic viscosity, the viscosity index was lowest compared to methyl linoleate and PEML due to the higher epoxy ring opening degradation in the temperature range. The degradation of epoxidized methyl linoleate can be accelerated when the compound is exposed to heat and light [16].
The low temperature properties of methyl linoleate, PEML and FEML were determined using thermal analysis (DSC). The onset crystallization temperature (T c) in the DSC thermogram indicates the highest temperature at which the substance still remains in the liquid state. This temperature is correlated to the pour point, as it is the lowest temperature at which a substance can flow without freezing. Among methyl linoleate, PEML and FEML (Table 1), methyl linoleate showed the lowest T c whereas FEML showed the highest T c. PEML was found to have an intermediate T c of −56 °C. Crystallization requires not only intermolecular interaction but also good molecular packing to reach an adequate entropy level [27]. The presence of olefinic groups with cis configurations in the methyl linoleate and PEML afforded the spatial arrangements that disrupt the packing efficiency and resist formation of macro crystals, thus lowering the T c. Furthermore, stronger intermolecular interactions in FEML due to hydrogen bonds promote nucleation, causing relatively high T c [4].
The oxidative stabilities of methyl linoleate, PEML and FEML were determined using DSC. The onset temperatures (T o) are shown in Table 1. T o is the temperature at which a rapid increase in the rate of oxidation is observed in a DSC thermogram. A high T o suggests a high oxidation stability of the oil. Methylene interrupted polyunsaturation is the main factor that causes the low oxidative stability of products as a result of the rapid breakdown of the double bond, leading to oxidative polymerization [1]. The oxidative stability of oil decreases as the degree of unsaturation increases. Therefore, methyl linoleate showed the lowest oxidative stability at 157 °C while FEML exhibited the highest oxidative stability at 194 °C. Owing to the structure of one double bond and one epoxy group in the PEML, it had an intermediate oxidative stability at 165 °C.
Conclusions
In conclusion, we have shown that MTO-catalyzed partial epoxidation of methyl linoleate using UHP and pyridine is an efficient method for the synthesis of high purity monoepoxidized methyl linoleate containing two regioisomers. The important finding was that monoepoxides were further converted to diepoxides before methyl linoleate was entirely consumed. Full epoxidation of methyl linoleate using the Prilezhaev method also successfully synthesized high purity diepoxide, and no by-products were formed by epoxy ring opening. The complete removal of unsaturation by converting double bonds to epoxy groups in FEML improved both viscosity and oxidative stability but weakened the low temperature properties compared to methyl linoleate. PEML demonstrated intermediate kinematic viscosity, viscosity index, crystallization temperature and oxidative stability as a result of the functional groups of one double bond and one epoxy group in the structure. Therefore, PEML not only improved the viscosity and oxidative stability of methyl linoleate but also had better low temperature properties compared to FEML. In general, PEML and FEML exhibit different properties and could have interesting industrial applications in different fields.
References
Erhan SZ, Asadauskas S (2000) Lubricant basestocks from vegetable oils. Ind Crops Prod 11:277–282
Adhvaryu A, Erhan SZ, Perez JM (2003) Wax appearance temperatures of vegetable oils determined by differential scanning calorimetry: effect of triacylglycerol structure and its modification. Thermochim Acta 395:191–200
Fox NJ, Stachowiak GW (2007) Vegetable oil-based lubricants—a review of oxidation. Tribol Int 40:1035–1046
Hwang HS, Erhan SZ (2001) Modification of epoxidized soybean oil for lubricant formulations with improved oxidative stability and low pour point. J Am Oil Chem Soc 78:1179–1184
Adhvaryu A, Erhan SZ (2002) Epoxidized soybean oil as a potential source of high-temperature lubricants. Ind Crops Prod 15:247–254
Klaas MR, Warwel S (1999) Complete and partial epoxidation of plant oils by lipase-catalyzed perhydrolysis. Ind Crops Prod 9:125–132
Gan LH, Ooi KS, Goh SH, Gan LM, Leong YC (1995) Epoxidized esters of palm olein as plasticizers for poly(vinyl chloride). Eur Polym J 31:719–724
Doll KM, Erhan SZ (2006) Synthesis and performance of surfactants based on epoxidized methyl oleate and glycerol. J Surfactants Deterg 9:377–383
Salimon J, Salih N, Yousif E (2011) Chemically modified biolubricant basestocks from epoxidized oleic acid: improved low temperature properties and oxidative stability. J Saudi Chem Soc 15:195–201
Goud VV, Patwardhan AV, Dinda S, Pradhan NC (2007) Kinetics of epoxidation of jatropha oil with peroxyacetic and peroxyformic acid catalysed by acidic ion exchange resin. Chem Eng Sci 62:4065–4076
Piazza GJ, Nuñez A, Foglia TA (2003) Epoxidation of fatty acids, fatty methyl esters, and alkenes by immobilized oat seed peroxygenase. J Mol Catal B Enzym 21:143–151
Sun S, Yang G, Bi Y, Liang H (2011) Enzymatic epoxidation of corn oil by perstearic acid. J Am Oil Chem Soc 88:1567–1571
Arends IWCE, Sheldon RA (2002) Recent developments in selective catalytic epoxidations with H2O2. Top Catal 19:133–141
Du G, Tekin A, Hammond EG, Woo LK (2004) Catalytic epoxidation of methyl linoleate. J Am Oil Chem Soc 81:477–480
Kühn FE, Scherbaum A, Herrmann WA (2004) Methyltrioxorhenium and its applications in olefin oxidation, metathesis and aldehyde olefination. J Organomet Chem 689:4149–4164
Gerbase AE, Gregório JR, Martinelli M, Brasil MC, Mendes NF (2002) Epoxidation of soybean oil by methyltrioxorhenium–CH2Cl2/H2O2 catalytic biphasic system. J Am Oil Chem Soc 79:179–181
Wang W, Espenson JH (1998) Effects of pyridine and its derivatives on the equilibria and kinetics pertaining to epoxidation reactions catalyzed by methyltrioxorhenium. J Am Chem Soc 120:11335–11341
Adolfsson H, Converso A, Sharpless KB (1999) Comparison of amine additives most effective in the new methyltrioxorhenium-catalyzed epoxidation process. Tetrahedron Lett 40:3991–3994
Wanasundara UN, Shahidi F (1999) Concentration of omega 3-polyunsaturated fatty acids of seal blubber oil by urea complexation: optimization of reaction conditions. Food Chem 65:41–49
American Oil Chemists’ Society (1998) Official Methods Cd 9-57. Official methods and recommended practices of the American Oil Chemists’ Society, 5th edn AOCS Press, Champaign
Petrovic ZS, Zlatanic A, Lava CC, Sinadinovic-Fiser S (2002) Epoxidation of soybean oil in toluene with peroxyacetic and peroxyformic acids- kinetics and side reactions. Eur J Lipid Sci Technol 104:293–299
American Society for Testing Materials (2006) D 2270—04. Standard practice for calculating viscosity index from kinematic viscosity at 40 and 100 °C. ASTM International, West Conshohocken, PA
Owens GS, Abu-Omar MM (2000) Methyltrioxorhenium-catalyzed epoxidations in ionic liquids. Chem Commun 13:1165–1166
Padley FB, Gunstone FD, Harwood JR (1994) Occurrence and characteristics of oils and fats. In: Gunstone FD, Harwood JL, Padley FB (eds) The lipid handbook, 2nd edn. Chapman and Hall, London, p 50
Socrates G (2001) Infrared and Raman characteristic group frequencies: tables and charts, 3rd edn. Wiley, Chichester
Orellana-Coca C, Adlercreutz D, Andersson MM, Mattiasson B, Hatti-Kaul R (2005) Analysis of fatty acid epoxidation by high performance liquid chromatography coupled with evaporative light scattering detection and mass spectrometry. Chem Phys Lipids 135:189–199
Rodrigues JDA, Cardoso FDP, Lachter ER, Estevão LRM, Lima E, Nascrimento RSV (2006) Correlating chemical structure and physical properties of vegetable oil esters. J Am Oil Chem Soc 83:353–357
Acknowledgments
We would like to thank Universiti Kebangsaan Malaysia for financial support via research grants UKM-AP-2011-017 and DPP-2013-054 and the supporting staff of the School of Chemical Sciences and Food Technology, who contributed greatly to our research.
Author information
Authors and Affiliations
Corresponding author
About this article

Cite this article
Lye, Y.N., Salimon, J. Synthesis and Physicochemical Properties of Partially and Fully Epoxidized Methyl Linoleate Derived from Jatropha curcas Oil. J Am Oil Chem Soc 92, 257–266 (2015). https://doi.org/10.1007/s11746-014-2584-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11746-014-2584-1