
Abstract
Behçet syndrome (BS) is a multi-systemic complex disorder with unknown etiology and a unique geographic distribution. It could not be possible to include it into specific classification schemes and it is certainly not a uniform disease. Several cluster and association studies revealed that it has been composed of multiple phenotypes ascribing the principal problem such as skin-mucosa, joint, eye, vascular, neurological and gastrointestinal involvement. Each phenotype has its own characteristic demographic and clinical features as such their management strategies and prognosis differ substantially. Actually, the concept of phenotyping has been well known for some time and is considered one of the basic elements of the still continuing debate whether to call this entity ‘disease’ or ‘syndrome’. Further supporting evidence comes from the observation of the geographical differences of disease expression. In this setting, BS resembles rather a construction made of several dynamic and interactive LEGO pieces of different shapes and colors. These pieces presenting phenotypes with their own disease mechanism have presumably different genetic determinants. The analysis of phenotyping could help us to identify this disorder and hence could contribute to find better ways of treatment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Behçet syndrome (BS) is a multi-systemic complex disorder with unknown etiology and a distinct geographical distribution [1]. The disease course characterized by exacerbations and spontaneous remissions, tends to be more active and severe during the first years [1, 2]. The usual onset is in the third decade. While the disease affects both genders equally, it runs a more severe course among males [1, 2]. Recurrent skin mucosa lesions and sight threatening panuveitis are the hallmark of the disease [1]. Veins or arteries of all sizes may be involved along with joints, gastrointestinal system and central nervous system [1, 2].
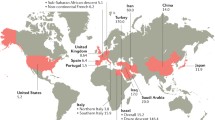
BS has a distinct geographical distribution including Mediterranean countries and Middle-East and Far East [3]. Turkey has the highest prevalence around the world with up to 420 per 100,000 adult population [4]. It has been considered as an orphan disease (≤ 50 per 100,000) in the rest of the world. However, there is evidence that its prevalence might be increasing in Europe especially when Asian and North African immigrant populations were evaluated as shown in a study done in a suburb of Paris about 10 years ago [5]. This is expected to be even higher especially after the European refugee crisis [6].
BS has been always in the limelight in both endemic and non-endemic places and attracted attention of physicians from several different specialties. It is one of the major causes of non-infectious uveitis [7] and one of the leading causes of Budd–Chiari syndrome [8]. The correct diagnosis and defining the main organ threat are of crucial importance for the proper management. All these efforts must be at the right time before the irreversible organ damage. Skin-mucosa lesions which are usually the presenting manifestations and considered as ‘innocent’ could be tip of the iceberg hiding an ensuing major organ involvement. It is well recognized that major organ involvements are responsible from serious morbidity and mortality [2], while skin-mucosa lesions may cause significant impact on quality of life [9, 10]. Moreover, work-related disability in BS is increased [10, 11]. Finally, in endemic places, BS exerts a significant economic burden [12].
The diagnosis of BS is clinical in that there are no diagnostic laboratory, histological, or radiological tests. International study group (ISG) criteria have been developed based on the incidence of clinical manifestations [13]. It has to be noted that these criteria should not be used to diagnose the individual case [14]. They ensure rather the uniformity of groups of patients for clinical and laboratory studies [14]. As seen in all biological events, BS is dynamic and clinical manifestations evolve over time [15]. Additionally and perhaps most important of all, it is not a single disease process which could complicate making a diagnosis [16].
In this review, I will describe common phenotypes and discuss difficulties with putting BS into a specific classification scheme. In addition, I will try to summarize geographical variances in disease expression and review the relevant literature.
Difficulties with classification
For the moment BS could be described best as a ‘multifactorial complex disorder’. As has been previously mentioned, it could not be possible to include it into a specific classification scheme [1, 16]. In particular:
-
a.
It is not a Mendelian disease yet ethnic predilection, well known HLA B51 and non-HLA associations, significantly increased sibling recurrence ratio and presumptive autosomal recessive mode of inheritance in the pediatric patient population are considered to support the role of the genetics in the ethiopathogenesis [16,17,18].
-
b.
It is certainly not an autoimmune disease since typical features of autoimmune diseases such as female dominance, increased B cell function or specifically impaired T cell activity, autoantibody production, association with auxiliary autoimmune conditions or features such as Raynaud phenomenon or premature atherosclerosis are either not increased or absent. Similarly, the association with organ-specific autoimmune disorders, as vitiligo or Hashimoto's disease are also unusual [1, 16, 19,20,21].
-
c.
BS has an episodic course and some clinical features such as oral ulcers and papulopustular lesions resemble to that seen in autoinflammatory diseases [17]. However, it rarely begins in the childhood, let alone not being a monogenic disease, its genetic associations are not so strong, fever episodes are rare and organ damage may occur more frequently than that observed in autoinflammatory conditions [22]. On the other hand, the response to anti-IL 1 treatment -a common therapeutic agent for many autoinflammatory conditions – is somewhat controversial. While there are several studies suggesting a potential role for anti-IL agents in ocular and muco-cutaneous phenotypes [23,24,25]; a few others report a less clear benefit [26, 27].
-
d.
It does not fit into a specific classification dedicated to ‘vasculitides’ either, since veins are the primary target in BS, which is rather unexpected for a typical vasculitis. Additionally, vasculitic changes are rarely found on the histopatologic examination of characteristic skin-mucosa lesions or Pathergy phenomenon [28]. Moreover, characteristic radiological or histological features ascribed to ‘vasculitis’ such as leukocytoclastic vasculitis, concentric thickening of aorta and its branches, granulomatous changes or necrotizing vasculitis are not features of BS. On the other hand, pulmonary artery aneurysms which are specific to BS, are unusual for ‘large vessel vasculitides’ [1, 28]. Having said that, BS is currently classified as a variable vessel vasculitis [29], which will be quite possibly subject to change in the imminent future.
Why do we need phenotyping?
Even scholars who are particularly focused on BS cannot decide whether to call it a ‘Behçet’s disease’ or ‘Behçet’s syndrome’. We prefer to describe it as a syndrome (a collection of signs and symptoms that frequently occur together) rather than a disease which usually describes a definite entity with known etiology and pathology [14]. For instance, a recent study showed that the prevalence of clinical manifestations (which may exceed 10 in total) has been estimated to be below 70% excluding that of skin-mucosa lesions, leading to a multitude variety of combinations of clinical presentation among patients [30].
In a typical BS cohort, each patient has their own individual complaint, sign and symptoms, treatment and outcome and yet all are classified as BS. Table 1 shows the detailed clinical history of six examplary patients with different dominant organ involvement. As if in theory we had been successful to dismantle their disorder, we would have find pieces similar to LEGO bricks that can be assembled and connected in many ways to construct individual syndromes. These pieces would have presumably different disease mechanisms and genetic determinants. Phenotyping analysis similar to what was done in other complex heterogeneous disorders could help us to decipher the pathogenesis and hence provide better management [31, 32]. Emerging developments on this field will eventually lead to individualized medicine for BS.
The concept of phenotyping BS is not a new concept, being introduced about 50 years ago by Lehner who had described a “spectral involvement or classification” of BS as stated by Barnes [14]. The concept has been further developed with the support of several cluster analysis mainly by Yazici and colleagues now became an essential element in our understanding disease mechanisms [1, 16, 33,34,35,36,37,38,39,40,41]. Our Japanese colleagues appear to have adapted it to routine practice already, but with another terminology: patients who have intestinal, vascular, or neurological involvement as a dominant clinical presentation are labeled as ‘variant types’ [42].
Cluster analysis and association studies
Several cluster analysis and other studies revealed important associations related to specific organ involvement. Table 2 summarizes the literature review of the prominent ones.
Two studies done 10 years apart in different BS populations studied organ associations with factor analysis and identified the same 4 clusters [34, 36]. These were: factor 1: oral ulcers, genital ulcers, and erythema nodosum; factor 2: superficial and deep vein thrombosis; factor 3: Uveitis; factor 4: papulopustular skin lesions and joint involvement. Another study from the same unit observed that enthesopathy was part of this association as well [39]. Importantly, the latter association was also shown in the family relatives of the patients with this specific cluster [36]. Recently, Kurosawa et al. identified subgroup classifications in BS using a correspondence analysis database of 2218 patients receiving financial aid for treatment between 2003 and 2014 [43]. Three distinct clusters were found in the data: group A (male, HLA-B51-positive, ocular inflammation, neurologic involvement), group B (female, onset age: < 30 years, HLA-B51-negative, genital ulcers, no ocular inflammation, no neurologic involvement), and group C (onset age: 30–39 years, skin lesions, arthritis) [43]. On the other hand, the clusters described were not observed in studies from Israel and Greece [44, 45].
Additionally, several studies using univariate analysis pointed out to significant positive and negative associations between organ systems [33, 35, 39,40,41,42,43, 46,47,48]. Positive associations could be listed as: cerebral venous sinus thrombosis (CVST) and peripheral major vessel disease; CVST and pulmonary artery involvement; Budd–Chiari syndrome and vena cava involvement/peripheral major vessel disease and finally, posterior uveitis and parenchymal neurological involvement. On the other hand, patients with ocular disease were found to be less likely to have genital ulceration and gastrointestinal and vascular involvement [47, 48]. Similarly, a negative association between vascular involvement and genital ulcers was also shown [42] supporting what was found in the cluster analysis [34, 36].
Phenotypes of Behçet’s syndrome
As revealed by cluster analyzes there are four predominant clinical phenotypes such as skin-mucosa involvement, uveitis, joint involvement and vascular disease (Table 3). In addition to these, gastrointestinal and central nervous system involvement could be other plausible candidates.
Phenotype 1: skin-mucosa involvement alone
One third of the patient population may present and continue for longtime with only recurrent skin-mucosa lesions without having any major organ involvement [2, 49, 50]. Recurrent oral and genital ulcerations defined as the hallmark of BS are the most common manifestations followed by papulo-pustular or nodular skin lesions. Clinical characteristics and differential diagnosis are summarized in Table 4.
Oral ulcers
Oral ulcers are usually the first and the most long lasting lesion [2, 49, 50]. They are easily differentiated from herpetic lesions because they are usually found on the mucous membranes of the lips, gingiva, cheeks, and tongue. On the other hand, it might be difficult to distinguish from recurrent aphthous stomatitis morphologically and pathologically. The increase in its frequency during the early years of the disease may predict the development of major organ involvement [51]. The recurrent episodes abate usually slowly after the fourth decade and oral ulcers are becoming fewer at each attack, less painful and less noticeable [2]. Fatigue or stress, some types of food and quitting smoking has been reported as triggering factors [52, 53]. It is also well known that menstruation may exacerbate oral ulcers as well as other skin mucosa lesions among female BS patients [54]. On the other hand, dental and periodontal therapies may decrease their number in longer follow-up indicating that, oral health is associated with disease severity [55].
Genital ulcers
Genital ulcers are the most pathognomonic lesion in BS. They usually occur on the scrotum or labiae and tend to form scars [56], with no urethritis or dysuria, unlike what is observed in reactive arthritis or venereal diseases [49].They are less frequent compared to oral ulcers and in about half of the patients they occur for only first few years then disappear. By definition, they can be considered as the first signals of BS onset.
Acne-like lesions or papulo-pustular lesions
Acne-like lesions or papulo-pustular lesions (PPL) are seen both at the usual acne sites as well as at uncommon sites such as extremities and are also indistinguishable from acne vulgaris by both in appearance and pathological examination [33, 39, 54, 57]. They may not be sterile [58]. Nevertheless, as found in a controlled survey, BS patients tend to have these lesions more on the lower extremities and continue to have PPL after the age of 50 [54, 57]. Additionally, acne-like lesions occur more frequently among patients with arthritis [33, 34, 36, 39] as mentioned earlier.
Nodular lesions
Erythemathous nodular lesions can be either due to panniculitis or superficial vein thrombosis [49]. Dermal ultrasonography could be helpful in their differentiation as well as other clinical features. While panniculitis are more likely to be seen among women, the latter often occur among men and in association with vascular involvement. The appearance of the nodular lesions due to panniculitis among BS patients is similar to that observed in idiopathic erythema nodosum. In about half of the cases neutrophilic vasculitis may be found in the histopathology [59].
Phenotype 2: joint involvement
Joint involvement has been reported in up to 50% in large case series [60]. The frequency can go up to 80% in some ethnicities [61]. However, it is estimated that only about 10% of the patient population suffers from solo joint involvement without any other organ involvements. The clinical presentation with mono or oligo-arthritis in the lower extremities may be similar to that seen in seronegative spondylarthropathies. Similarly, it is usually self-limited, resolves in 2–4 weeks; often leaves no deformity or erosions. Patients report no morning stiffness, and there is mild inflammation in the synovium [60, 61]. Patients with arthritis may have enthesopathy. However, different than SpA, sacroiliac joint involvement is not increased, spine is spared and HLA B27 association is weak.
As indicated clearly in 2 factor analysis mentioned earlier and in several others, joint involvement is associated with acne or papulopustular lesions among BS patients [33, 34, 36, 39]. Diri et al. studied 44 patients with arthritis, 42 patients without arthritis along with 21 patients with rheumatoid arthritis and 33 healthy controls and found that the number of papules and pustules was significantly higher in the arthritis group than observed in the remaining three groups [33]. The presence of enthesopathy was investigated using ultrasonography among 35 patients with acne and arthritis, 38 patients without arthritis, 37 patients with ankylosing spondylitis (AS) and 25 patients with rheumatoid arthritis (RA) [39]. Patients with AS, followed by BS patients with acne and arthritis were found to have the significantly higher enthesopathy scores compared to the remaining study groups.
Phenotype 3: vascular involvement
Vascular disease develops in up to 40% with a definite male preponderance [62]. About half of the male patient population may be affected when followed prospectively for 20 years [2]. It is usually an early manifestation. Several lines of evidence suggest that the vascular involvement stands apart:
-
a.
The frequency of vascular involvement was not found to be high enough to be included among the listed clinical manifestations set for the formulation of International Criteria most probably due to geographical variances in disease expression [13]. The fact that it is not strongly associated with skin-mucosa and eye involvement could be also possible. Exclusion from the criteria was rightfully criticized later on by some experts [63].
-
b.
Some patients with typical vascular involvement may not fulfill the criteria at the onset and during the follow-up. In a large series of BS patients with vascular involvement including 882 patients, vascular involvement occurred in about 20% of the patients at the same time of disease onset and before fulfilling the ISG criteria in another 10% [40]. Similarly a literature review of 392 patients [64] and a large series of 47 patients with pulmonary artery involvement found that there were about 10% who did not fulfill the ISG criteria at presentation and during the follow-up [65].
-
c.
Two studies in separate cohorts and in different time lines identified vascular involvement as distinct cluster using factor analysis [34, 36].
-
d.
Furthermore, Hughes-Stovin syndrome, which is known as a rare condition characterized by aneurysms especially in pulmonary arteries and venous thrombosis of the lower extremities without the classical expected signs of BS is considered as the cardiovascular subtype of BS [66, 67]. There are around 50 cases reported in the literature. Hughes-Stovin syndrome supports the proposition that there is a weak association between vascular involvement and other BS stigmata.
Vascular involvement is associated with unique features and its presence plays a critical role in the differential diagnosis. These unique features can be summarized as thrombotic tendency, venous predilection and relapsing course. Moreover, it leads to increased morbidity and mortality.
Venous involvement is significantly more common than arterial disease and lower extremity deep vein thrombosis (DVT) is its most frequent manifestation [40]. Vena cava inferior and superior, upper extremity and jugular veins and also cerebral venous sinuses can be affected. Arterial disease involves mostly pulmonary arteries, followed by aorta and peripheral arteries and is manifested mostly in the form of aneurysms. It has to be noted here that anatomical and physiological characteristics of the pulmonary arteries bear striking resemblance to peripheral veins with thin walls, less elasticity and lower pressure. Arterial occlusions are rare. As a rule vascular involvement is almost always associated with intensive thrombosis and runs a relapsing course whether it affects veins or arteries. Thus far, no significant evidence related with thrombophilia was observed [68, 69].
In a retrospective study from our unit including 882 patients with vascular involvement, the cumulative risk of recurrent vascular events of all kinds was calculated to be 38% at 5 years [40]. When an inception cohort with DVT was prospectively followed, the rate of relapse reached 29%, 37% and 45%, at 6 months, first and second year, respectively [Ozguler, inpreparation for submission]. In a retrospective French study which included 296 patiens with venous thrombosis, 33.8% experienced at least 1 venous thrombosis relapse after a median follow-up of 5 years [70]. Similarly, relapses can be seen in roughly 20% of the patients with pulmonary artery involvement (PAI) or non-pulmonary arterial aneurysms who remained alive after a median of 5–7 years follow-up [65].
In vascular phenotype, several vascular associations can be seen such as superficial and deep vein thrombosis; BCS and inferior vena cava syndrome (IVCS); intracardiac thrombosis and PAI [40, 65]. Furthermore, it has to be noted that, CVST which is strongly associated with DVT and PAI is obviously a part of the vascular phenotype [35, 40]. The rare concomitant presence of CVST and parenchymal neurological also supports this contention. DVT is often concomitantly present in these associations. It is interesting that there are no definitive histopathologic features besides the presence of lymphocytic infiltration around the vaso vasorum. Concentric arterial thickening around the vessel wall, granuloma formation or fibrinoid necrosis is absent. Leukocytoclastic vasculitis is not a feature of BS either.
Vascular involvement is the major cause of mortality in BS. In 20 year outcome study which included 428 (286 M/ 142 F) patients with BS, large vessel involvement such as pulmonary artery aneurysms and Budd–Chiari syndrome was the leading cause of mortality (17/ 42; 40%) [2]. Similarly, in a French study, among 817 patients with BS, the main cause of death was major vessel disease (mainly arterial aneurysms and Budd–Chiari syndrome) (44%) [71]. Moreover, the French group reported that Budd–Chiari syndrome was associated with 9 times increase in the mortality rate [72]. On the other hand, DVT may lead to severe post-thrombotic syndrome in about half of the patients, and claudication in third [62, 73]. It is also associated with high rate of unemployement [73].
Phenotype 4: eye involvement
The prevalence of eye involvement is about 50% [2, 74, 75]. It is basically a non-granulomatous uveitis. Recurrent inflammatory attacks and spontaneous remission is a rule and most distinctive feature. It is usually bilateral; however the severity of involvement may not be equal in both eyes. While isolated anterior uveitis is rarely seen and mostly among females (5–10%), pan or posterior uveitis dominates the clinical picture [74]. Retinal vasculitis may be concomitantly present in about half of the cases with posterior uveitis and signifies severe involvement. Complications such as cataracts, glaucoma and eventually phthisis bulbi could be seen in treatment refractory cases. Loss of useful vision is estimated to be around 15–20% [1, 74]. Male gender, posterior involvement, having frequent attacks (> 3/year), strong vitreous opacity and macular edema are defined as poor prognostic factors [74,75,76].
Previous 2 factor analysis identified ‘uveitis’ as a separate cluster with its distinguishing features [34, 36]. Ocular involvement develops usually within the 2 years after fulfillment of diagnostic criteria and run its outmost severe course which could result in extensive damage during the first few years then abates [2]. Additionally, it may occur without the accompanying stigmata in up to 20% of the patients, similar to what was observed with the vascular involvement [75]. Results of a very recent study by Suwa et al. conducted in a large inception cohort (n = 3213; 1382 M/1831 F) with a very short disease duration (median 1 year [IQR 0–3]) in Japan support this finding [47]. A multivariate logistic regression analysis revealed that ocular involvement was not associated with genital ulcer or gastrointestinal involvement during the early course of the disease.
The definition of eye lesions in the ISG criteria set includes non-specific terminology such as anterior uveitis, posterior uveitis, cells in the vitreous on slit-lamp examination, or retinal vasculitis observed by the ophthalmologist [75]. It is ophthalmologist’s responsibility to make the correct diagnosis which relies upon distinctive characteristics. However relapsing remitting course could not be easily recognized or could not be sufficient in some patients which necessitate additional clues that could be helpful in discrimination. For this purpose, Tugal-Tutkun et al. investigated the discriminative role of eye lesions in BS as presented on ocular clinical photographs [77]. Fourteen uveitis specialists, masked to demographic and clinical features of patients, independently labeled ocular photographs of 29 patients with BS and 30 patients with other diagnoses as “Behcet uveitis” or “non-Behcet uveitis”. While exact agreement with the correct diagnosis was found between 56 and 81%, 7 reviewers correctly labeled more than 70% of photographs. Smooth layered hypopyon, superficial retinal infiltrate with retinal hemorrhages, and branch retinal vein occlusion with vitreous haze were correctly recognized as Behcet uveitis by the majority of reviewers [77].
Phenotype 5: parenchymal neurological involvement
The prevalence of neurological involvement is approximately 5% with a significant male predilection. This prevalence may go up to 10% when followed prospectively for 2 decades [78,79,80]. Approximately 3/4th present with parenchymal central nervous system (CNS) involvement (p-NBS), while the remaining cases present with CVST which is considered as part of vascular phenotype (previous section) and considerably different than parenchymal type in terms of demographic and clinical characteristics, as well as disease course and prognosis. Parenchymal neurological involvement occurs late during the course of the disease after a mean of 5 years disease onset. Approximately, 6% of patients may present without fulfilling the ISG diagnostic criteria for BS. It usually affects the telencephalic–diencephalic junction, brainstem, and spinal cord. These patients present with a subacute (or rarely acute) onset of severe headache, cranial nerve palsy, dysarthria, ataxia, and hemiparesis. p-NBS is an important cause of morbidity and mortality [2, 78,79,80]. About 2 decades ago Akman–Demir studied the outcome of 200 patients with neurological involvement and found that among 110 patients who were followed for at least 3 years the rate of being death or severely disabled was found to be 60% in 10 years [78]. A recent study by Noel et al reported somewhat better but still severe outcome: in a cohort of 115 patients exclusively with parenchymal neurological involvement the mortality or severe disability rate was estimated to be 25% in 7 years [80].
Parenchymal neurological involvement can be associated with eye involvement
Cluster studies stated earlier did not include neurological involvement because of the very low prevalence [30, 32]. However, there are evidences indicating that parenchymal neurological involvement could be associated with eye disease. Bitik et al. found that patients with posterior uveitis had increased frequency of neurological involvement compared to those without (p < 0.001; odds ratio: 3.9; 95% CI 1.8–8.6) [46]. Moreover, a recent study including a considerably large cohort, identified a distinct cluster which is composed of eye disease, parenchymal neurological involvement in association with male gender and HLA B 51 positivity [43]. Additionally, a considerably high frequency of eye disease (up to 66%) was observed in large case series of patients with neurological involvement [78, 80].
Phenotype 6: gastrointestinal involvement
Prevalence of gastrointestinal involvement changes significantly across different ethnicities, reaching up to 50% in the Far-East to being negligeable in Turkey and in the majority of Middle-East. Both males and females are affected equally regarding prevalence and severity. The gastrointestinal manifestations may develop about 5–10 years after the onset of oral ulcers; however, it is also known to occur before the appearance of BS related symptoms. Due to the low prevalence, it was not studied in previous cluster analysis [34, 36], but studies show that it is less likely to be associated with eye involvement [42, 47]. Clinical presentation varies considerably; patients may have only mild abdominal discomfort or present with emergency conditions such as hemorrhage or perforation [81,82,83]. On the histopathology, chronic or active mucosal inflammation as well as neutrophilic phlebitis could be seen. Granuloma formation is again not a typical feature. Relapses are seen in about 20% of the affected patients [83].
Similarities with Crohn’s disease
Among all the phenotypes of BS, gastrointestinal involvement with its obvious similarity to Crohn’s disease (CD), constitutes the most strong evidence for the proposal of different disease mechanisms playing role in BS [16].
Both BS and CD typically occurs at a young age, have a chronic, relapsing remitting course and somewhat similar extraintestinal manifestations with a less frequency in CD [81, 82]. When patients with CD were screened formally for the presence of clinical features of BS, it was found that none of the CD patients fulfilled ISG criteria for BS or had major organ involvement such as eye, vascular or neurological disease [80]. Papulopustular lesions (24%) and oral ulcers (22%) were the most common lesions, followed by pathergy positivity (11%), genital ulcers (4%), arthritis (3%) and nodular lesions (2%). Moreover, apart from a few hints, clinical and endoscopic appearance of intestinal involvement could not be differentiated easily from CD [81, 82]. In BS typically terminal ileum is affected, while perianal and rectal area involvement are seldom seen. Ulcers tend to be single or less than five, are usually confined to the ileocecal area, more likely to be deep and round and prone to perforation [82]. On the other hand, this lingering debate over distinction seems in vain since the medical management of the two conditions is rather similar including their response to biological drugs. Interestingly exacerbations with secukinumab-an anti-IL 17 agent- have been reported among both patients with CD and BS [85, 86]. Conversely, secukinumab is known be quiet effective and licensed for ankylosing spondylitis, psoriasis and psoriatic arthritis [87].
Geographical differences in disease expression
There is a significant variation in frequency and prevalence of clinical manifestations in different parts of the world. Table 5 highlights the main results of major studies focusing on disease expression in several ethnicities.
Gastrointestinal disease is the prototype as mentioned previously being more frequent in the Far-East Asia [81, 82]; by contrast, vascular disease is less common in this region [88]. In a literature review looking at specifically published articles around the world on pulmonary artery involvement due to BS, the number of articles from Japan was significantly lower while that from Turkey was significantly higher than expected [89]. Interestingly, evidence shows that this could not be specific to BS since there is tendency to see low frequency of vein thrombosis in Asian populations. On the other hand aortic valve involvement has been reported mainly from Japan and Korea [90]. Krause et al. showed that patients from North Africa had higher rates of arthritis, overall vascular disease, deep vein thrombosis, and neuro-Behçet in contrast to patients with origins in Iran, Iraq and Turkey [91]. Central nervous system involvement has been reported with higher prevalence in Tunisian patients especially in males compared with Italian and Spanish [92, 93]. Similarly, in a French multiethnic country, sub Saharan African patients exhibited the highest mortality rate [94]. They displayed a higher frequency of CNS involvement and a trend toward higher cardiovascular involvement compared to those from Europe or North Africa [94].
In general, series from non-endemic geographies, include more female patients and have a milder course and outcome with less severe organ involvement [93, 95,96,97,98]. However, studies which do not favor a significant ethnic influence or end up with even contradictory results are also present [5, 99,100,101,102]. For example a recent study revealed that eye and vascular disease were similar in frequency in both USA and Turkish patients [99]. However, more gastrointestinal and neurologic disease was seen in the USA [99]. Addionally, associations between major organs or clusters as described in Turkish BS patients have not been found among Israeli and Greek patients [44, 45].
Conclusions
BS is still an enigma; we could not define nor classify. It should be considered as an ensemble of phenotypes with different clinical characteristics, outcome and treatment modalities, similar to LEGO pieces of different shapes and colors. It is possible that these pieces with their own disease mechanism have different genetic determinants. In this context, geographical differences of phenotypic expression are regarded as further supportive evidence. Defining the phenotypes could help us to decipher this enigma.
References
Yazici H, Seyahi E, Hatemi G, Yazici Y. Behçet syndrome: a contemporary view. Nat Rev Rheumatol. 2018 14:107–119 (Erratum in: Nat Rev Rheumatol. 2018 14 119)
Kural-Seyahi E, Fresko I, Seyahi N, Ozyazgan Y, Mat C, Hamuryudan V, Yurdakul S, Yazici H (2003) The long-term mortality and morbidity of Behçet syndrome: a 2-decade outcome survey of 387 patients followed at a dedicated center. Medicine (Baltimore). 82:60–76
Yurdakul S, Yazici Y (2010) Epidemiology of Behçet’s syndrome and regional differences in disease expression. In: Yazici Y, Yazici H (eds) Behçet’s Syndrome, 1st edn. Springer, New York, pp 35–52
Azizlerli G, Köse AA, Sarica R, Gül A, Tutkun IT, Kulaç M, Tunç R, Urgancioğlu M, Dişçi R (2003) Prevalence of Behçet's disease in Istanbul. Turkey. Int J Dermatol. 42:803–806
Mahr A, Belarbi L, Wechsler B, Jeanneret D, Dhote R, Fain O, Lhote F, Ramanoelina J, Coste J, Guillevin L (2008) Population-based prevalence study of Behçet's disease: differences by ethnic origin and low variation by age at immigration. Arthritis Rheum. 58:3951–3959
Yalçındaǧ FN, Özdal PC, Özyazgan Y, Batıoǧlu F, Tugal-Tutkun I (2018) Demographic and clinical characteristics of Uveitis in Turkey: the first national registry report. Ocul Immunol Inflamm. 26:17–26
Bayraktar Y, Balkanci F, Bayraktar M, Calguneri M (1997) Budd-Chiari syndrome: a common complication of Behçet's disease. Am J Gastroenterol. 92:858–862
Fabiani C, Vitale A, Orlando I, Sota J, Capozzoli M, Franceschini R, Galeazzi M, Tosi GM, Frediani B, Cantarini L (2017) Quality of life impairment in Behçet's disease and relationship with disease activity: a prospective study. Intern Emerg Med. 12:947–955
Senusi AA, Ola D, Mather J, Mather J, Fortune F. Behçet's syndrome and health-related quality of life: influence of symptoms, lifestyle and employment status. Clin Exp Rheumatol. 2017;35 Suppl 108(6):43-50.
Mehta P, Ambrose N, Haskard DO (2014) Work-related disability in Behçet's syndrome: a British case series. Clin Exp Rheumatol. 32(4 Suppl 84):S173–S174
Sut N, Seyahi E, Yurdakul S, Senocak M, Yazici H (2007) A cost analysis of Behcet's syndrome in Turkey. Rheumatology (Oxford) 46:678–682
International Study Group for Behçet’s Disease (1990) Criteria for diagnosis of Behçet’s disease. Lancet 335:1078–1080
Barnes Colin G (2010) History and diagnosis. In: Yazici Y, Yazici H (eds) Behçet’s Syndrome, 1st edn. Springer, New York, pp 7–34
Ideguchi H, Suda A, Takeno M, Ueda A, Ohno S, Ishigatsubo Y (2011) Behçet disease: evolution of clinical manifestations. Medicine (Baltimore). 90:125–132
Yazici H, Ugurlu S, Seyahi E (2012) Behçet syndrome: is it one condition? Clin Rev Allergy Immunol. 43:275–280
Gül A (2015) Pathogenesis of Behçet's disease: autoinflammatory features and beyond. Semin Immunopathol. 37:413–418
Gül A (2014) Genetics of Behçet's disease: lessons learned from genomewide association studies. Curr Opin Rheumatol. 26:56–63
Direskeneli H, Saruhan-Direskeneli S (2010) Disease mechanisms. In: Yazici Y, Yazici H (eds) Behçet’s syndrome, 1st edn. Springer, New York, pp 243–264
Oran M, Hatemi G, Tasli L, Garip F, Kadioglu P, Mat C, Yazici H (2008) Behçet's syndrome is not associated with vitiligo. Clin Exp Rheumatol. 26(Suppl 50):S107–S109
Seyahi E, Ugurlu S, Cumali R, Balci H, Ozdemir O, Melikoglu M, Hatemi G, Fresko I, Hamuryudan V, Yurdakul S, Yazici H (2008) Atherosclerosis in Behçet's syndrome. Semin Arthritis Rheum. 38:1–12
Yazici H, Fresko I (2005) Behçet's disease and other autoinflammatory conditions: what's in a name? Clin Exp Rheumatol. 23(4 Suppl 38):S1–2
Fabiani C, Vitale A, Rigante D, Emmi G, Lopalco G, Di Scala G, Sota J, Orlando I, Franceschini R, Frediani B, Galeazzi M, Iannone F, Tosi GM, Cantarini L (2018) The Presence of Uveitis Is Associated with a Sustained Response to the Interleukin (IL)-1 Inhibitors Anakinra and Canakinumab in Behçet's Disease. Ocul Immunol Inflamm. 27:1–7
Fabiani C, Vitale A, Emmi G, Lopalco G, Vannozzi L, Guerriero S, Gentileschi S, Bacherini D, Franceschini R, Frediani B, Galeazzi M, Iannone F, Tosi GM, Cantarini L (2017) Interleukin (IL)-1 inhibition with anakinra and canakinumab in Behçet's disease-related uveitis: a multicenter retrospective observational study. Clin Rheumatol. 36(1):191–197
Grayson PC, Yazici Y, Merideth M, Sen HN, Davis M, Novakovich E, Joyal E, Goldbach-Mansky R, Sibley CH (2017) Treatment of mucocutaneous manifestations in Behçet's disease with anakinra: a pilot open-label study. Arthritis Res Ther. 19(1):69
Tugal-Tutkun I, Pavesio C, De Cordoue A, Bernard-Poenaru O, Gül A (2018) Use of Gevokizumab in patients with behçet's disease uveitis: an international, randomized, double-masked, placebo-controlled study and open-label extension study. Ocul Immunol Inflamm. 26(7):1023–1033
Cantarini L, Vitale A, Scalini P, Dinarello CA, Rigante D, Franceschini R, Simonini G, Borsari G, Caso F, Lucherini OM, Frediani B, Bertoldi I, Punzi L, Galeazzi M, Cimaz R (2015) Anakinra treatment in drug-resistant Behcet's disease: a case series. Clin Rheumatol. 34(7):1293–1301
Melikoglu M, Kural-Seyahi E, Tascilar K, Yazici H (2008) The unique features of vasculitis in Behçet's syndrome. Clin Rev Allergy Immunol. 35:40–46
Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F et al (2013) 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 65:1–11
Ishido T, Horita N, Takeuchi M, Kawagoe T, Shibuya E, Yamane T, Hayashi T, Meguro A, Ishido M, Minegishi K, Yoshimi R, Kirino Y, Kato S, Arimoto J, Ishigatsubo Y, Takeno M, Kurosawa M, Kaneko T, Mizuki N (2017) Clinical manifestations of Behçet's disease depending on sex and age: results from Japanese nationwide registration. Rheumatology (Oxford) 56:1918–1927
Chu JH, Hersh CP, Castaldi PJ, Cho MH, Raby BA, Laird N, Bowler R, Rennard S, Loscalzo J, Quackenbush J, Silverman EK (2014) Analyzing networks of phenotypes in complex diseases: methodology and applications in COPD. BMC Syst Biol. 8:78
Brito-Zerón P, Retamozo S, Ramos-Casals M (2018) Phenotyping Sjögren's syndrome: towards a personalised management of the disease. Clin Exp Rheumatol. 36(Suppl 112):198–209
Diri E, Mat C, Hamuryudan V, Yurdakul S, Hizli N, Yazici H (2001) Papulopustular skin lesions are seen more frequently in patients with Behçet's syndrome who have arthritis: a controlled and masked study. Ann Rheum Dis. 60:1074–1076
Tunc R, Keyman E, Melikoglu M, Fresko I, Yazici H (2002) Target organ associations in Turkish patients with Behçet's disease: a cross sectional study by exploratory factor analysis. J Rheumatol. 29:2393–2396
Tunc R, Saip S, Siva A, Yazici H (2004) Cerebral venous thrombosis is associated with major vessel disease in BS's syndrome. Ann Rheum Dis. 63:1693–1694
Karaca M, Hatemi G, Sut N, Yazici H (2012) The papulopustular lesion/arthritis cluster of Behçet's syndrome also clusters in families. Rheumatology (Oxford) 51:1053–1060
Yazici H. The lumps and bumps of Behcet's syndrome. Autoimmun Rev 2004; Suppl 1:S53-4.
Yazıcı H, Seyahi E (2016) Behçet syndrome: the vascular cluster. Turk J Med Sci. 46:1277–1280
Hatemi G, Fresko I, Tascilar K, Yazici H (2008) Enthesopathy is increased among Behçet’s syndrome patients with acne and arthritis: an ultrasonographic study. Arthritis Rheum 58:1539–1545
Tascilar K, Melikoglu M, Ugurlu S, Sut N, Caglar E, Yazici H (2014) Vascular involvement in Behçet's syndrome: a retrospective analysis of associations and the time course. Rheumatology (Oxford) 53:2018–2022
Seyahi E, Caglar E, Ugurlu S, Kantarci F, Hamuryudan V, Sonsuz A, Melikoglu M, Yurdakul S, Yazici H (2015) An outcome survey of 43 patients with Budd-Chiari syndrome due to Behçet's syndrome followed up at a single, dedicated center. Semin Arthritis Rheum. 44:602–609
Suzuki T, Horita N, Takeuchi M et al (2018) Clinical features of early-stage possible Behçet's disease patients with a variant-type major organ involvement in Japan. Mod Rheumatol. 28:1–18
Kurosawa M, Takeno Y, Kirino Y, Soejima N, Mizuki M. Subgroup classification of Behçet's disease using clinical information: analysis of a clinical database of patients receiving financial aid for treatment. ISBD conference 2018, Rotterdam.
Krause I, Leibovici L, Guedj D, Molad Y, Uziel Y, Weinberger A (1999) Disease patterns of patients with Behçet's disease demonstrated by factor analysis. Clin Exp Rheumatol. 17:347–350
Arida A, Vaiopoulos G, Markomichelakis N, Kaklamanis P, Sfikakis PP. Are clusters of patients with distinct clinical expression present in Behçet's disease? Clin Exp Rheumatol. 2009;27(Suppl 53):S48–51.
Bitik B, Tufan A, Sahin K, Sucullu Karadag Y, Can Sandikci S, Mercan R, Ak F, Karaaslan Y, Ozturk MA, Goker B, Haznedaroglu S (2016) The association between the parenchymal neurological involvement and posterior uveitis in Behçet's syndrome. Clin Exp Rheumatol. 34(6 Suppl 102):82–85
Suwa A, Horita N, Ishido T, Takeuchi M, Kawagoe T, Shibuya E, Yamane T, Hayashi T, Meguro A, Ishido M, Minegishi K, Yoshimi R, Kirino Y, Kato S, Arimoto J, Fukumoto T, Ishigatsubo Y, Kurosawa M, Kaneko T, Takeno M, Mizuki N (2018) The ocular involvement did not accompany with the genital ulcer or the gastrointestinal symptoms at the early stage of Behçet's disease. Mod Rheumatol. 15:1–6
Hussein MA, Eissa IM, Dahab AA (2018) Vision-Threatening Behcet's Disease: Severity of Ocular Involvement Predictors. J Ophthalmol. 2018:9518065
Mat C, Yurdakul S, Sevim A, Özyazgan Y, Tüzün Y (2013) Behçet's syndrome: facts and controversies. Clin Dermatol. 31:352–361
Alibaz-Oner F, Mumcu G, Kubilay Z, Ozen G, Celik G, Karadeniz A, Can M, Oner SY, Inanc N, Atagunduz P, Ergun T, Direskeneli H (2014) Unmet need in Behcet's disease: most patients in routine follow-up continue to have oral ulcers. Clin Rheumatol. 33:1773–1776
Hamuryudan V, Hatemi G, Sut N, Ugurlu S, Yurdakul S, Yazici H (2012) Frequent oral ulceration during early disease may predict a severe disease course in males with Behçet's syndrome. Clin Exp Rheumatol. 30(3 Suppl 72):S32–S34
Volle G, Fraison JB, Gobert D, Goulenok T, Dhote R, Fain O, Gonzalez-Chiappe S, Lhote F, Papo T, Thuillier A, Rivière S, Mahr A (2017) Dietary and nondietary triggers of oral ulcer recurrences in Behçet's disease. Arthritis Care Res (Hoboken). 69:1429–1436
Soy M, Erken E, Konca K, Ozbek S (2000) Smoking and Behçet's disease. Clin Rheumatol. 19:508–509
Guzelant G, Ozguler Y, Esatoglu SN, Karatemiz G, Ozdogan H, Yurdakul S, Yazici H, Seyahi E (2017) Exacerbation of Behçet's syndrome and familial Mediterranean fever with menstruation. Clin Exp Rheumatol. 35(6):95–99
Mumcu G, Ergun T, Inanc N, Fresko I, Atalay T, Hayran O, Direskeneli H (2004) Oral health is impaired in Behçet's disease and is associated with disease severity. Rheumatology (Oxford) 43:1028–1033
Mat C, Göksugur N, Engin B et al (2006) The frequency of scarring after genital ulcers in Behçet’s syndrome: a prospective study. Int J Dermatol 45:554–556
Kutlubay Z, Mat CM, Aydin Ö, Demirkesen C, Calay Ö, Engin B, Tüzün Y, Yazici H (2015) Histopathological and clinical evaluation of papulopustular lesions in Behçet's disease. Clin Exp Rheumatol. 33(6 Suppl 94):S101–S106
Hatemi G, Bahar H, Uysal S, Mat C, Gogus F, Masatlioglu S, Altas K, Yazici H (2004) The pustular skin lesions in Behcet's syndrome are not sterile. Ann Rheum Dis. 63:1450–1452
Demirkesen C, Tüzüner N, Mat C, Senocak M, Büyükbabani N, Tüzün Y, Yazici H (2001) Clinicopathologic evaluation of nodular cutaneous lesions of Behçet syndrome. Am J Clin Pathol. 116(3):341–346
Yurdakul S, Yazici H, Tuzun Y et al (1983) The arthritis of Behçet’s disease: a prospective study. Ann Rheum Dis 42:505–515
Yurdakul S, Hatemi G (2010) Locomotor System Disease in Behçet’s Syndrome. In: Yazici Y, Yazici H (eds) Behçet’s syndrome, 1st edn. Springer, New York, pp 149–164
Seyahi E (2016) Behçet's disease: how to diagnose and treat vascular involvement. Best Pract Res Clin Rheumatol. 30:279–295
Schirmer M, Calamia KT, O'Duffy JD (1999) Is there a place for large vessel disease in the diagnostic criteria of Behçet's disease? J Rheumatol. 26:2511–2512
Uzun O, Akpolat T, Erkan L (2005) Pulmonary vasculitis in behcet disease: a cumulative analysis. Chest 127:2243–2253
Seyahi E, Melikoglu M, Akman C, Hamuryudan V, Ozer H, Hatemi G, Yurdakul S, Tuzun H, Oz B, Yazici H (2012) Pulmonary artery involvement and associated lung disease in Behçet disease: a series of 47 patients. Medicine (Baltimore). 91:35–48
Erkan D, Yazici Y, Sanders A, Trost D, Yazici H (2004) Is Hughes-Stovin syndrome Behçet's disease? Clin Exp Rheumatol. 22(4 Suppl 34):S64–S68
Bennji SM, du Preez L, Griffith-Richards S, Smit DP, Rigby J, Koegelenberg CFN, Irusen EM, Allwood BW (2017) Recurrent pulmonary aneurysms: Hughes–Stovin syndrome on the spectrum of Behçet disease. Chest 152:e99–e103
Seyahi E, Yazici H (2016) To anticoagulate or not to anticoagulate vascular thrombosis in Behçet's syndrome: an enduring question. Clin Exp Rheumatol. 34(Suppl 95):S3–S4
Leiba M, Seligsohn U, Sidi Y et al (2004) Thrombophilic factors are not the leading cause of thrombosis in Behcet's disease. Ann Rheum Dis 63:1445–1449
Desbois AC, Wechsler B, Resche-Rigon M, Piette JC, Huong Dle T, Amoura Z, Koskas F, Desseaux K, Cacoub P, Saadoun D (2012) Immunosuppressants reduce venous thrombosis relapse in Behçet's disease. Arthritis Rheum. 64:2753–2760
Saadoun D, Wechsler B, Desseaux K, Le Thi Huong D, Amoura Z, Resche-Rigon M, Cacoub P (2010) Mortality in Behçet's disease. Arthritis Rheum. 62:2806–2812
Desbois AC, Rautou PE, Biard L, Belmatoug N, Wechsler B, Resche-Rigon M, Zarrouk V, Fantin B, de Chambrun MP, Cacoub P, Valla D, Saadoun D, Plessier A (2014) Behcet's disease in Budd–Chiari syndrome. Orphanet J Rare Dis. 9:104
Ugurlu S, Seyahi E, Oktay V, Kantarci F, Tuzun H, Yigit Z, Arslan C, Yazici H (2015) Venous claudication in Behçet's disease. J Vasc Surg. 62(3):698–703.e1
Tugal-Tutkun I, Onal S, Altan-Yaycioglu R et al (2004) Uveitis in Behçet disease: an analysis of 880 patients. Am J Ophthalmol 138:373–380
Tugal-Tutkun I, Gupta V, Cunningham ET (2013) Differential diagnosis of behçet uveitis. Ocul Immunol Inflamm. 21:337–350
Takeuchi M, Hokama H, Tsukahara R, Kezuka T, Goto H, Sakai J, Usui M (2005) Risk and prognostic factors of poor visual outcome in Behcet's disease with ocular involvement. Graefes Arch Clin Exp Ophthalmol. 243:1147–1152
Tugal-Tutkun I, Onal S, Ozyazgan Y, Soylu M, Akman M (2014) Validity and agreement of uveitis experts in interpretation of ocular photographs for diagnosis of Behçet uveitis. Ocul Immunol Inflamm. 22:461–468
Akman-Demir G, Serdaroglu P, Tasci B (1999) Clinical patterns of neurological involvement in Behçet’s disease: evaluation of 200 patients The Neuro-Behcet Study Group. Brain 122:2171–2182
Siva A, Kantarci OH, Saip S et al (2001) Behçet’s disease: diagnostic and prognostic aspects of neurological involvement. J Neurology 248:95–103
Noel N, Bernard R, Wechsler B, Resche-Rigon M, Depaz R, Le Thi Huong Boutin D, Piette JC, Drier A, Dormont D, Cacoub P, Saadoun D. Long-term outcome of neuro-Behçet's disease. Arthritis Rheumatol. 2014;66:1306-14.
Cheon JH, Kim WH (2015) An update on the diagnosis, treatment, and prognosis of intestinal Behçet's disease. Curr Opin Rheumatol. 27:24–31
Hatemi I, Hatemi G, Çelik AF (2018) Gastrointestinal Involvement in Behçet Disease. Rheum Dis Clin North Am. 44:45–64
Hatemi I, Esatoglu SN, Hatemi G, Erzin Y, Yazici H, Celik AF (2016) Characteristics, treatment, and long-term outcome of gastrointestinal involvement in Behcet's syndrome: a strobe-compliant observational study from a dedicated multidisciplinary center. Medicine (Baltimore). 95:e3348
Hatemi I, Hatemi G, Celik AF, Melikoglu M, Arzuhal N, Mat C, Ozyazgan Y, Yazici H (2008) Frequency of pathergy phenomenon and other features of Behçet's syndrome among patients with inflammatory bowel disease. Clin Exp Rheumatol. 26(4 Suppl 50):S91–S95
Hueber W, Sands BE, Lewitzky S, Vandemeulebroecke M, Reinisch W, Higgins PD et al (2012) Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn's disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut 61:1693–1700
Dick AD, Tugal-Tutkun I, Foster S, Zierhut M, Melissa Liew SH, Bezlyak V et al (2013) Secukinumab in the treatment of noninfectious uveitis: results of three randomized, controlled clinical trials. Ophthalmology 120(4):777–787
Baeten D, Sieper J, Braun J, Baraliakos X, Dougados M, Emery P et al (2015) Secukinumab, an Interleukin-17A inhibitor, in ankylosing spondylitis. N Engl J Med. 373:2534–2548
Ideguchi H, Suda A, Takeno M, Ueda A, Ohno S, Ishigatsubo Y (2011) Characteristics of vascular involvement in Behçet's disease in Japan: a retrospective cohort study. Clin Exp Rheumatol. 29(4 Suppl 67):S47–53
Celik S, Yazici Y, Sut N, Yazici H (2015) Pulmonary artery aneurysms in Behçet's syndrome: a review of the literature with emphasis on geographical differences. Clin Exp Rheumatol. 33(6 Suppl 94):S54–S59
Han JK, Kim HK, Kim YJ, Cho GY, Kim MA, Sohn DW, Park YB (2009) Behçet's disease as a frequently unrecognized cause of aortic regurgitation: suggestive and misleading echocardiography findings. J Am Soc Echocardiogr. 22:1269–1274
Krause I, Mader R, Sulkes J, Paul M, Uziel Y, Adawi M, Weinberger A (2001) Behçet's disease in Israel: the influence of ethnic origin on disease expression and severity. J Rheumatol. 28(5):1033–1036
Houman MH, Neffati H, Braham A, Harzallah O, Khanfir M, Miled M, Hamzaoui K. Behçet's disease in Tunisia. Demographic, clinical and genetic aspects in 260 patients. Clin Exp Rheumatol. 2007;25(Suppl 45):S58–64.
Leccese P, Yazici Y, Olivieri I (2017) Behcet's syndrome in nonendemic regions. Curr Opin Rheumatol. 29:12–16
Savey L, Resche-Rigon M, Wechsler B, Comarmond C, Piette JC, Cacoub P, Saadoun D (2014) Ethnicity and association with disease manifestations and mortality in Behçet's disease. Orphanet J Rare Dis. 9:42
Zouboulis CC, Kotter I, Djawari D, Kirch W, Kohl PK, Ochsendorf FR, Keitel W, Stadler R, Wollina U, Proksch E, Söhnchen R, Weber H, Gollnick HP, Hölzle E, Fritz K, Licht T, Orfanos CE (1997) Epidemiological features of Adamantiades Behcet's disease in Germany and in Europe. Yonsei Med J 38:411–422
Mok CC, Cheung TC, Ho CT, Lee KW, Lau CS, Wong RW (2002) Behçet's disease in southern Chinese patients. J Rheumatol. 29:1689–1693
Muhaya M, Lightman S, Ikeda E, Mochizuki M, Shaer B, McCluskey P, Towler HM (2000) Behçet's disease in Japan and in Great Britain: a comparative study. Ocul Immunol Inflamm. 8:141–148
Rozenbaum M, Boulman N, Slobodin G, Zisman D, Mader R, Yankevitch A, Weinberger A, Rosner I (2007) Behcet disease in adult Druzes in north Israel: the influence of ethnic origin on disease expression and severity. J Clin Rheumatol. 13:124–127
Sibley C, Yazici Y, Tascilar K, Khan N, Bata Y, Yazici H, Goldbach-Mansky R, Hatemi G (2014) Behçet syndrome manifestations and activity in the United States versus Turkey—a cross-sectional cohort comparison. J Rheumatol. 41:1379–1384
Mohammad A, Mandl T, Sturfelt G, Segelmark M (2013) Incidence, prevalence and clinical characteristics of Behcet's disease in southern Sweden. Rheumatology (Oxford) 52:304–310
Kötter I, Vonthein R, Müller CA, Günaydin I, Zierhut M, Stübiger N (2004) Behçet's disease in patients of German and Turkish origin living in Germany: a comparative analysis. J Rheumatol. 31:133–139
Kappen JH, van Dijk EH, Baak-Dijkstra M, van Daele PL, Lam-Tse WK, van Hagen PM, van Laar JA (2015) Behçet's disease, hospital-based prevalence and manifestations in the Rotterdam area. Neth J Med. 73:471–477
Funding
None.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Statements on human and animal rights
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
No informed consent was necessary for the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The article is part of the Topical Collection: Behcet disease.
Rights and permissions
About this article

Cite this article
Seyahi, E. Phenotypes in Behçet’s syndrome. Intern Emerg Med 14, 677–689 (2019). https://doi.org/10.1007/s11739-019-02046-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11739-019-02046-y