
Abstract
The GRAS gene family performs a variety of functions in plant growth and development processes, and they also play essential roles in plant response to environmental stresses. Medicago truncatula is a diploid plant with a small genome used as a model organism. Despite the vital role of GRAS genes in plant growth regulation, few studies on these genes in M. truncatula have been conducted to date. Using the M. truncatula reference genome data, we identified 68 MtGRAS genes, which were classified into 16 groups by phylogenetic analysis, located on eight chromosomes. The structure analysis indicated that MtGRAS genes retained a relatively constant exon–intron composition during the evolution of the M. truncatula genome. Most of the closely related members in the phylogenetic tree had similar motif compositions. Different motifs distributed in different groups of the MtGRAS genes were the sources of their functional divergence. Twenty-eight MtGRAS genes were expressed in six tissues, namely root, bud, blade, seedpod, nodule, and flower tissues, suggesting their putative function in many aspects of plant growth and development. Nine MtGRAS genes were upregulated under cold, freezing, drought, ABA, and salt stress treatments, indicating that they play vital roles in the response to abiotic stress in M. truncatula. Our study provides valuable information that can be utilized to improve the quality and agronomic benefits of M. truncatula and other plants.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Transcription factors (TFs) are a vital part of functional genomics, which participate in various physiological processes and regulatory networks (Sun et al. 2012). The first TF was discovered in maize, and then a large number of TFs in vascular plants have been identified (Huang et al. 2015). GRAS family proteins are plant-specific TFs, which are named after the three functional members, GIBBERELLIC ACID INSENSITIVE (GAI), REPRESSOR OF GAI (RGA), and SCARECROW (SCR) (Sun et al. 2011). GRAS TFs typically consist of 400–770 amino acid residues and possess conserved motifs in their C-terminal region, including LHR I, VHIID, LHR II, PFYRE, and SAW (Bolle 2004). Other GRAS proteins are highly variable in N-terminal region, except that the DELLA subfamily contains conserved N-terminal domains of DELLA and TVHYNP (Tian et al. 2004; Lu et al. 2015). Based on the functional similarity and sequence homology, the GRAS protein family is divided into DELLA, SCR, PAT1, HAM, LS, SHR, and SCL9 protein subfamilies; each subfamily has distinct conserved domains and functions (Bolle 2004).
The GRAS family of putative transcriptional regulators has been identified in vascular plants including Arabidopsis, tomato, Prunus mume, rice, and Populus (Mayrose et al. 2006; Lee et al. 2008; Lu et al. 2015; Liu and Widmer 2014). These proteins play important role in plant growth and development, such as root and shoot development, phytochrome A signal transduction (PAT), gibberellic acid (GA) signal transduction, and disease resistance (Hirsch and Oldroyd 2009; Lu et al. 2015). PAT1 protein in Arabidopsis thaliana participates in phytochrome A signal transduction (Bolle et al. 2000), and Lateral suppressor (Ls) gene from tomato plays a significant role in the formation of lateral branches (Schumacher and Theres 1999). SHORT ROOT (SHR) is a TF essential for endodermis specification in the Arabidopsis root. SCR is a member of the GRAS protein family associated with radial patterning of both roots and shoots. Recently, it has been shown that BnSCL1, a SCARECROW-LIKE (SCL) protein from Brassica napus, is expressed predominantly not only in roots but also in shoots, suggesting its mode of action in the plant’s auxin response (Gao et al. 2004). SCL from Lilium longiflorum plays a transcriptional regulation role during microsporogenesis within the lily anther (Morohashi et al. 2003). Arbuscular Mycorrhizal 18 (OsAM18) is a GRAS protein in rice, which likely affects the colonization process and functionality of arbuscular mycorrhiza, and the systemic arbuscular mycorrhizal symbiosis (Fiorilli et al. 2015). A class of GRAS proteins, the DELLA proteins, regulates plant growth through hormone signaling. DELLA mutants are partially insensitive to gene induction in methyl-jasmonate treatment, whereas the constitutively active dominant DELLA mutant gai is sensitized for jasmonate; these results indicated that DELLA proteins are involved in the perception of jasmonate signaling (Navarro et al. 2008). Helianthus annuus GRAS-like gene (Ha-GRASL) lacking the DELLA motif belongs to the SCL4/7 subfamily. The metabolic flow of gibberellins was reduced in overexpressed Ha-GRASL Arabidopsis, and this modification could be relevant in axillary meristems development (Fambrini et al. 2015). In a recent study, the Jumonji-C domain-containing gene JMJ524 was not only upregulated by GA treatment in tomato, but it also responded to circadian rhythms. Moreover, JMJ524 altered GA responses by stem elongation or by regulating SlGLD1, a GRAS protein lacking DELLA domain, at least partially (Li et al. 2015). The GRAS proteins NODULATION SIGNALING PATHWAY 1 (NSP1) and NSP2 from Medicago truncatula directly bind to the promoter of a nodulation-associated gene, highlighting the importance of GRAS proteins for efficient nodulation in legume plants (Hirsch et al. 2009; Kaló et al. 2005). In Lotus japonicus, the NSP1 protein is involved in bacterial release, infection, and normal bacteroid formation in nodule cells (Heckmann et al. 2006).
Plant-specific GRAS TFs also play diverse roles in response to biotic and abiotic stress conditions. In a recent study, PeSCL7 was highly induced by high-salt and drought treatment but repressed by GA treatment in poplar leaves. Overexpressing PeSCL7 from poplar improved drought and salt tolerance in Arabidopsis (Ma 2010). Overexpressed OsGRAS23 in rice enhanced drought tolerance by reducing H2O2 accumulation in cells (Xu et al. 2015). Triticum aestivum TaSCL14 was expressed in various wheat organs by high light stress, with high levels detected in stems and roots. The study on silencing TaSCL14 in wheat revealed that TaSCL14 may act as a regulator involved in plant growth, photosynthesis, and tolerance to oxidative stress (Chen et al. 2015). The increased expression of the GRAS gene family in tomato (Solanum lycopersicum), which responds to disease stress, in part depends on jasmonic acid signaling. Expression of SlGRAS6 is suppressed by virus-induced gene silencing, impairing tomato resistance to Pseudomonas syringae (Mayrose et al. 2006).
Medicago truncatula is a diploid plant with a small genome and undetermined growth of nodules (López et al. 2008). The high-efficiency genetic transformation makes it an excellent model organism in studies on legume plants. Characterization of GRAS proteins, which has been conducted in other plant species, in M. truncatula is focused on plant signal transduction, growth, and nodulation signaling of these proteins. In this study, 68 members from the MtGRAS gene family were analyzed comprehensively, and the analysis included phylogenetic analysis, gene structure, chromosomal location, and expression profiles under different abiotic stresses. The investigation of GRAS genes in the whole genome of M. truncatula in response to abiotic stresses is essential, because it will provide an essential foundation for functional study on GRAS proteins in M. truncatula.
Materials and methods
Identification and characteristics of the GRAS family
The whole genome sequence of M. truncatula and the summary of gene annotation were downloaded from the Phytozome database (http://phytozome.jgi.doe.gov/medicago.php) (Goodstein et al. 2012). The hidden Markov model (HMM) profiles of the GRAS domain PF03514 were downloaded from Pfam database (Punta et al. 2012). HMM search of the GRAS domain PF03514 profiles from M. truncatula protein database were conducted with a cutoff E value of 1.0 (Johnson et al. 2010). After determining the GRAS domain profile, the integrity of the GRAS domain was evaluated with the online program SMART with an E value <0.1 (http://smart.embl-heidelberg.de/) (Letunic et al. 2012).
Phylogenetic analysis and conserved motifs and intron/exon structure identification
To investigate the phylogenetic relationship of the GRAS protein families in M. truncatula, full-length GRAS protein sequences were retrieved from Phytozome, and all the candidate GRAS proteins were aligned to Arabidopsis GRAS proteins (Goodstein et al. 2012). GRAS proteins sequence information of A. thaliana previously reported was retrieved from the Arabidopsis Information Resource (Grimplet et al. 2016). GRAS TFs were aligned using BioEdit (http://www.mbio.ncsu.edu/bioedit/bioedit.html), and phylogenetic trees were constructed using neighbor-joining method implemented in MEGA5.1 program with 1000 replicates (Tamura et al. 2011). The resulting tree file was visualized by Tree View1.6 (Shen et al. 2015). Branches corresponding to partitions that received less than 50% bootstrap support were collapsed. Genes were classified according to the distance homology with Arabidopsis genes (Lee et al. 2008).
To identify the unknown conserved motifs using the online MEME (http://meme.ebi.edu.au/meme/intro.html) analysis (Bailey and Elkan 1994), the parameters were as following: 1) optimum motif width was set to ≥6 and ≤50 (inclusive); 2) identification of the maximum motif number was set to 25; 3) the distribution of a single motif occurrences among sequences was set to 0 or 1 occurrence per sequence (-mod zoops). The MEME motifs were annotated using the Pfam database (Bailey et al. 2015). Based on the M. truncatula genome, the DNA and cDNA sequences corresponding to each predicted gene and the information about MtGRAS intron distribution pattern were obtained from Phytozome (Goodstein et al. 2012).
Chromosomal location and gene duplication of GRAS genes
Based on gene annotation information, sequences of putative GRAS genes were extracted from the whole genome sequence using a Perl script. M. truncatula GRAS gene structures was displayed using Gene Structure Display Server program (http://gsds.cbi.pku.edu.cn/index.php) (Hu et al. 2015). GRAS family genes were subjected to a BLASTN search to explore gene duplication. If two genes shared more than 70% similarity, they were identified as tandem duplications or segmental duplications (Li et al. 2016). The chromosomal locations of GRAS genes in M. truncatula were plotted by Circos software (Krzywinski et al. 2009).
In silico expression analysis of GRAS genes from M. truncatula
Genome-wide transcriptome data from different development tissues in M. truncatula were downloaded from the NCBI database (Accession No.: SRX099057–SRX099062) (Liu et al. 2015). The transcriptome data were obtained from six tissue types: root, bud, nodule, seedpod, blade, and flower tissues. TopHat and Cufflinks were used to analyze per million mapped reads (Trapnell et al. 2009, 2010). The expression profiles of the MtGRAS genes were retrieved from these expression data, analyzed, clustered, and displayed by the ggplot2 package in R software (Version 3.1.0) (Wickham 2009).
Expression of GRAS protein under abiotic stress in M. truncatula
According to the M. truncatula transcriptome data downloaded from the NCBI database (Accession numbers: SRX1056987–SRX1056992), the molecular functions of MtGRAS genes were investigated under abiotic stress. All of the 8-week-old seedlings of M. truncatula ‘Jemalong A17’ were randomly divided into six groups for stress treatments, namely control (untreated) and treatment with cold (4 °C), freeze (−8 °C), salt (200 mM NaCl solution), drought (300 mM mannitol solution), and abscisic acid (ABA) solution (100 μM ABA). All the seedlings were harvested 3 h after the treatment, and five whole seedlings per group were bulked separately as described in Shu et al. (2016). The expression levels of GRAS TF genes were detected in transcriptome data, which included five abiotic stresses treatments (cold, freezing, salt, drought, and ABA stresses).
Quantitative real-time PCR analysis
The genes identified in the transcriptome analysis were validated and quantified by quantitative real-time PCR (qPCR). The primers were designed using Primer3plus 1.0.0 (Untergasser et al. 2012) based on transcriptome sequencing data; the primer pairs are listed in Table S1. According to manufacturer’s instructions, the total RNA from six groups alfalfa mentioned above was extracted using an RNAprep Pure Plant Kit (Tiangen, Beijing, China), and cDNA synthesized and qPCR analyzed were using a ReverTra Ace kit and SYBR Premix Ex TaqTM II (Toyobo, Shanghai, China), respectively. qPCR was performed in a Roche LightCycler 96 System (Roche, China). MtActin was used as an internal control to normalize the expression levels (Zhang et al. 2011). The details of qPCR analysis referenced to Song et al. (2016).
Results
Identification and phylogenetic analysis of GRAS proteins in M. truncatula
To identify the full complements of GRAS genes in M. truncatula, BLASTP searches were performed using the GRAS genes from Arabidopsis and the M. truncatula genome. Sixty-eight proteins were identified as predicted by GRAS genes in total. The length of amino acid sequences encoded by MtGRAS varied from 69 (MtGRAS26) to 805 (MtGRAS62) amino acids, the molecular mass ranged from 7939.1 (MtGRAS26) to 89030.2 (MtGRAS62) kDa, and the pI values of MtGRAS proteins varied from 4.72 (MtGRAS61) to 9.8 (MtGRAS48) (Table 1).
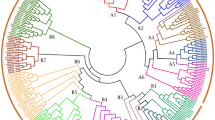
The evolutionary relationship between M. truncatula and Arabidopsis was assessed with respect to GRAS TFs and visualized with a neighbor-joining phylogenetic tree. As shown in Fig. 1, 16 groups were obtained according to the clade support values, topology of the tree, and classification of Arabidopsis. Among the GRAS proteins in M. truncatula, eight subfamilies, SCL3, HAM, SCL, SHR, PAT, SCR, DELLA, and LAS, were previously described in other plant species, and two newly detected subfamilies, GRAS8 and SCL26, were also found in Vitis vinifera. The remaining six subfamilies, labeled GRASM1–GRASM6, formed a clade with SCL3, SCL26, and SCL. The smallest subfamily was GRAS8, with only one member, and the largest subfamily was PAT, which contained 12 members. Regarding the PAT subfamily, 12 MtGRAS genes were clustered and homologous with genes from Arabidopsis and V. vinifera, indicating that they might be derived from gene duplications of the same gene locus during M. truncatula genome evolution. These results suggested that these paralogous genes may perform the same function in the life cycle of M. truncatula.

Phylogenetic analysis of Medicago truncatula and Arabidopsis GRAS genes. Sixteen subfamilies were identified in M. truncatula: the known SCL, SCL3, SCL26, SHR, SCR, HAM, PAT, LAS, DELLA, GRAS8, and six new subfamilies GRASM1-6
Structure and conserved motifs of GRAS family in M. truncatula
Exon–intron structural diversity is an important part of gene families’ evolution, and provided additional evidence to support phylogenetic groupings (Wei et al. 2016). To elucidate phylogenetic relationship and classification of MtGRAS, we employed the GRAS domain (PF03514) to search against these predicted GRAS genes. All predicted GRAS genes containing conserved motifs in the C-terminal and N-terminal region of their corresponding GRAS proteins were highly variable. The number of introns varied from one to seven, which was consistent with GRAS genes in M. truncatula (Fig. 2). The exon and intron number in M. truncatula GRAS genes ranged from 1 to 8 and 0 to 7, respectively. Among the 68 MtGRAS genes, 41 genes had introns, 34 genes had only one intron, and 27 genes had no introns. Group GRASM3 contained 0–7 introns, group GRASM6 contained 0–5 introns, group GRASM1 contained 1–4 introns, group PAT contained 0–2 introns, group SCL26 contained 0–4 introns, groups GRASM2, GRASM5, SCL, SCR, LAS, SCL26, and SHR contained 0–1 intron, groups GRASM4 and HAM contained only one intron, and groups GRAS8 and DELLA had no introns. Generally, MtGRAS genes within the same group showed similar exon–intron structure in the phylogenetic tree.

Exon–intron structure of GRAS genes in Medicago truncatula. Green boxes indicate exons, and black lines indicate introns. The scale represents 2500 bp. Thin lines indicated the length by the scale
The MEME analysis of GRAS family proteins in M. truncatula revealed 25 conserved motifs (designated motifs 1–25) (Fig. 3, Fig. S1; Table S2). Among those, motif 1 encoding the GRAS domain was found in all MtGRAS genes and was the most conserved motif in all MtGRAS proteins. A combination of motif 3 and other motifs (motif 2, 4, 13, 15, 17, and 19) was found in the C terminus, while the N terminus contained various motifs. The results indicated that most of the closely related members had similar motif composition in the phylogenetic tree, while among different groups or subgroups the motifs were divergent. For example, groups SHR, SCR, and DELLA had eight similar conserved motifs (motifs 9, 6, 5, 1, 8, 4, 14, and 3), groups SCL3, SCL26, and SCL had only one similar motif (motif 5), group HAM had four conserved motifs (motifs 12, 8, 4, and 3), and five motifs (motifs 3, 4, 1, 2, and 5) were present in groups GRASM2–5. While motif 19 was specific to group GRASM6, motif 25 was specific to groups SCL3 and GRASM1, and motifs 11, 15, 18, and 20 were specific to groups GRASM2–4. Based on the results, it indicated that different motifs distributed in different groups of the MtGRAS genes were the sources of their functional divergence.

Distribution of conserved motifs within GRAS genes family in Medicago truncatula
Chromosomal locations and gene duplication analysis of MtGRAS Genes
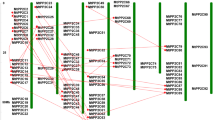
The physical locations of 68 GRAS TF genes were distributed on eight chromosomes of M. truncatula shown in Fig. 4. Each chromosome in M. truncatula contained between 1 and 16 GRAS genes. Chromosome 4 had the highest number of GRAS genes (16 genes) distributed throughout the chromosome. Chromosomes 2 and 3 contained 11 genes, distributed on both ends of the chromosome 2 or evenly distributed on chromosome 3. Moreover, chromosome 6 contained only one gene located in the middle of the chromosome. The GRAS genes were not randomly distributed on each chromosome, as is the case with some gene clusters on chromosomes forming “hot regions.” For example, chromosome 4 contained five GRAS genes (MtGRAS34–38) in a short chromosome region; similar gene clusters were observed in chromosomes 2, 3, and 5.

Chromosomal location of Medicago truncatula GRAS proteins. Similarity of segmental duplicate genes is linked by different lines. The red lines represent less than 95% similarity, the orange lines represent 90–95% similarity, the purple lines represent 85–90% similarity, the blue lines represent 80–85% similarity, and the green lines represent 75–80% similarity
MtGRAS gene clusters or hot regions were distributed in different chromosomes, such as clusters MtGRAS 34–38, MtGRAS 39–40, MtGRAS 41–42, MtGRAS 44–45, and MtGRAS 46–47 on chromosome 4; clusters MtGRAS 8–10 and MtGRAS 13–16 on chromosome 2; cluster MtGRAS 22–24 on chromosome 3; cluster MtGRAS 51–52 on chromosome 5. Many homologous GRAS genes on different chromosomes, which were produced by segmental duplication, will expand the number of MtGRAS genes from the same group. For example, as products of genome segmental duplication, MtGRAS4 and MtGRAS64 from group GRASM6 were distributed on chromosome 1 and chromosome 7, respectively, and MtGRAS6 and MtGRAS50 from group SCL3 were distributed on chromosome 1 and 5, respectively. In addition, 14 pairs of gene duplications that arose from tandem duplications and segmental duplications were identified in this study. MtGRAS gene clusters or hot regions were produced by tandem duplications, such as the cluster MtGRAS13–16 on chromosome 2 and cluster MtGRAS44–45 on chromosome 4.
Expression patterns analysis of GRAS genes in M. truncatula tissues
The MtGRAS gene expression profiles in six tissues (nodule, root, bud, flower, blade, and seedpod tissues) were investigated. The number of expressed genes in the six tissues was 51, 51, 45, 44, 42, and 31, respectively. The highest expression of MtGRAS genes was detected in nodule and root tissue (51/68, 75%), followed by bud (45/68, 66%), flower (44/68, 64.7%), blade (42/68, 61.8%), and seedpod (31/68, 45.6%) tissue. Of the 28 MtGRAS genes expressed in all tissues, 24 were upregulated, suggesting that the function of MtGRAS genes may affect plant growth and development, such as root system regulation, embryonic development, apical dominance, and shoot initiation and growth. The transcription patterns of MtGRAS genes were clustered across the six tissues (Fig. 5). All MtGRAS genes, except GRAS53, that were clustered in groups HAM, SCL, SCR, GRAS8, and DELLA were highly expressed across all the six tissues. A number of genes were expressed only in one tissue; for example, GRAS3 and GRAS67 were specifically expressed in the nodule, and GRAS10, GRAS22, and GRAS24 were specifically expressed in the root, suggesting that the expression of these genes is organ specific. Eleven GRAS genes (4, 23, 26, 27, 28, 39, 40, 52, 53, 58, and 67) were not expressed in any tissues. These results indicated that MtGRAS genes were expressed in a particular tissue or at certain developmental stages and thus were involved in the development of M. truncatula.

Expression profile of GRAS genes in tissues of Medicago truncatula. In the heat map, rows represent genes, while columns represent different tissues, including nodule, blade, flower, root, seedpod and bud. Red and light yellow color gradients indicate a decrease and increase in transcript abundance, respectively
Expression analysis of MtGRAS genes responses to abiotic stress
The expression levels of GRAS genes under stress treatments (cold, freezing, salt, drought, and ABA) were detected to explore the molecular functions of MtGRAS genes under abiotic stress (Accession numbers: SRX1056987–SRX1056992). Compared to the control library, of the 68 expressed MtGRAS genes, we identified 44 MtGRAS genes that were differentially expressed under five stress conditions (Fig. 6). In total, 48 MtGRAS genes were differentially expressed under freezing and drought stress, 47 MtGRAS genes were expressed under salt and cold treatment, and 45 MtGRAS genes were differentially expressed in ABA treatment. The expression of 22, 24, 25, 28, and 34 MtGRAS genes was downregulated under freezing, cold stress, drought, salt, and ABA stress, respectively. The other genes exhibited group-specific profiles; for example, the expression of MtGRAS genes clustered in groups GRASM4 and DELLA was highly upregulated under cold and freezing stress, and the expression of those clustered in groups GRASM2, GRASM4, and SCL was highly upregulated under drought stress. Notably, nine MtGRAS genes (MtGRAS11, MtGRAS13, MtGRAS18, MtGRAS19, MtGRAS29, MtGRAS35, MtGRAS37, MtGRAS38, and MtGRAS49) were upregulated under all stress conditions. In summary, the expression level of these genes under different stress conditions was as follows. Under cold stress: 24 up-, 26 downregulated; freeze stress: 26 up-, 24 downregulated; drought stress: 23 up-, 28 downregulated; salt stress: 20 up-, 31 downregulated; ABA: 16 up-, 33 downregulated.

Expression analysis of GRAS genes’ response to abiotic stress in Medicago truncatula. In the heat map, rows represent genes, while columns represent different abiotic stress, namely cold, freezing, salt, ABA and drought stress. Red and light yellow color gradients indicate a decrease and increase in transcript abundance, respectively
To further validate the reliability of the RNA-sequencing analysis, the expression of 16 subfamilies of GRAS proteins that were annotated by the NCBI Nr database was monitored by qPCR. Compared with the results of qPCR and RNA-Seq analysis, most of the MtGRAS gene expression patterns were basically the same, but the magnitude of the fold-changes was different between qPCR experiments and RNA-seq (Fig. 7). All of the above results indicated that MtGRAS genes play vital roles in response to abiotic stress, especially cold, freezing, and water deficit.

Validation of transcriptome sequencing data by qPCR analysis. Expression analysis of 15 GRAS genes from different subfamilies’ response to cold, freezing, salt, ABA and drought stress by qPCR. MtActin gene was used as internal control. The expression of the MtActin gene in non-treated controls was set to 1.0. The y-axis represents the relative expression, and the x-axis depicts the different abiotic stress. The mean values are calculated from three biological replicates. The error bars represented the standard deviation of the mean expression values
Discussion
With the development of bioinformatics analysis, various genomes with important information can be investigated to clarify the mechanisms about plant growth and development. Based on the M. truncatula genome database, 68 MtGRAS TF genes were identified and characterized in this study. The number of identified GRAS genes in M. truncatula is much higher than that in Arabidopsis (33), Carica papaya (42), Vitis vinifera (43), Prunus mume (46), and Oryza sativa (60), but lower than that in Musa acuminata (73), Populus trichocarpa (102), and Malus × domestica (127). Recent research on gene duplication events plays an essential role in the rapid expansion and evolution of gene families (Cannon et al. 2004; Lu et al. 2015). The considerably large GRAS gene family in Malus domestica is likely the result of a whole genome duplication event (Shu et al. 2016), whereas the GRAS gene family in Medicago truncatula has different characteristics and patterns of evolution.
Expansion of GRAS family in M. truncatula
The junction pattern analysis of the exon–intron can provide additional insights into gene families’ evolution (Huang et al. 2015). The exon–intron structures of MtGRAS genes vary significantly among the 16 groups. The number of introns and exons in all groups varied from 0 to 7 and from 1 to 8, respectively. Among the 68 identified MtGRAS genes, 24 genes had no introns. Meanwhile, the exon–intron analysis showed that 64.7% of GRAS genes lacked introns in M. truncatula (Fig. 2), compared to 82.2, 77.4, 67.6, 55, and 54.7% of genes that lacked introns in Prunus mume, tomato, Arabidopsis, rice, and Populus, respectively (Xin et al. 2016; Huang et al. 2015). This suggested that the structure of GRAS genes is species specific. Although, the groups had different intron structures, they had similar exon–intron structure, suggesting that the GRAS genes family retained a relatively constant exon–intron composition during the evolution of the M. truncatula genome. The different motifs were distributed in different groups of the MtGRAS genes and, therefore, were the source of their functional divergence.
Transcription regulators belonging to the same taxonomic group, exhibited a common evolutionary origins, and have conserved motifs associated with their molecular functions (Lu et al. 2015). A comparison of GRAS genes’ structure enhances our understanding of the roles of these TFs in M. truncatula. The results form phylogenetic analysis indicated that GRAS genes, which are involved in plant development, were clustered in 16 groups in M. truncatula. The MtGRAS genes were distributed on eight chromosomes, but most of them were present on chromosome 4. The distribution of MtGRAS genes showed that recent duplication events have occurred in this gene family. Segmental duplication, which expanded the number of MtGRAS genes from different groups, produced many homologous GRAS genes on different chromosomes. For example, MtGRAS28, 44, 45, 51, and 52 from group SHR are distributed on different chromosomes (MtGRAS28 on chromosome 3, MtGRAS44 and 45 on chromosome 4, MtGRAS51 and 52 on chromosome 5) and are products of genome segmental duplication. These results suggested that the expansion of MtGRAS gene family is the result of gene duplication events during the evolutionary process.
Variety in the expression patterns of GRAS family members in different tissues
Recent studies on functional and structural genomics in vascular plant model species revealed that GRAS TFs are involved in plant development in processes including GA signal transduction (DELLA), phytochrome signal transduction (PAT1, SCL21, and SCL13), axillary shoot meristem formation (HAM and MOC1), cell maintenance and proliferation (SCR and SHR), root radial pattering, and male gametogenesis (LlSCL) (Song et al. 2014). With tissue-specific expression in various plant species, the GRAS gene family may play essential roles in plant tissue development. The tissue expression analysis showed that 11 MtGRAS genes had no discernible expression level in any tissues/organs. These results suggest that those genes may have degenerated after gene duplication or undergone loss of function during the evolution of the gene family. Based on high-throughput sequencing data analysis, 58 MtGRAS genes were expressed in at least one tissue, and 28 MtGRAS genes were expressed in various tissues, root, blade, nodule, seedpod, bud, and flower tissues. These results suggested that these highly expressed genes may regulate plant growth and development. The groups HAM, SCL, SCR, GRAS8, and DELLA had the highest gene expression across all the six tissues during development. Notably, MtGRAS3 (homologous to At5g41920.1, At3g54220.1) and MtGRAS67 (homologous to At4g08250.1) were specifically expressed in nodule tissue, whereas MtGRAS10, 22, and 24 (not homologous to Arabidopsis) were expressed only in the roots. In the present study, the MtGRAS genes from SCR, SCL, and SHR groups were highly expressed in the root. SCR-regulated root meristem in Arabidopsis is located downstream of SHR. SCL13 regulates the expression of SIN1, which plays an essential role in lateral root elongation and root symbiosis and thereby nodulation in common bean (Mbe et al. 2014). These results suggested that the members from GRAS family in M. truncatula may have similar functions to those in Arabidopsis and grapevine. Many PAT genes, which regulate the phytochrome signal transduction pathway, might be involved in several developmental processes in Arabidopsis. A large number of genes from PAT subfamily in M. truncatula were highly expressed in the photosynthetic tissue, which was inconsistent with PAT expression in grapevine. These results indicated that PAT subfamily may regulate plant developmental processes, since various MtGRAS proteins were involved in diverse morphological features of plant development. In summary, the expression patterns of MtGRAS genes in different tissues may lay a foundation for further investigation of alfalfa development.
GRAS family putatively involved in biotic and abiotic stress
TF families are involved in the regulation of defense responses to environmental stresses in plants. Because of the functional diversity of GRAS genes, the GRAS TF family in M. truncatula need to be further studied. The expression patterns analysis of MtGRAS genes could be useful in assessing their possible functions under various abiotic stress conditions. Compared to control samples, there were 44 MtGRAS genes differentially expressed under abiotic stress treatments (shown in Fig. 6). Among these, MtGRAS genes from groups GRASM2–4 and PAT were induced in response to all the tested abiotic stress conditions. Overexpression of VaPAT1, which belongs to the GRAS family, increases the cold, high-salinity, and drought tolerance in transgenic Arabidopsis (Yuan et al. 2016). Meanwhile, we found that 54 genes were responsive to freezing or cold treatment. Interestingly, 22 MtGRAS genes were highly upregulated under both freezing and cold stresses, including the members from the groups PAT, GRAS8, and GRASM2–4. MtGRAS genes that belong to the SCL subfamily respond to drought stress, and overexpression of the poplar GRAS protein SCL7 enhances the tolerance of drought in Arabidopsis (Ma 2010). Two poplar homologs of SCL were putatively associated with salt tolerance according to the single nucleotide polymorphism (SNP) method (Galovic et al. 2015). Overall, the above results suggested that not only different MtGRAS gene but also a single GRAS gene may participate in multiple signaling and stress processes. Furthermore, most of the MtGRAS genes can be induced quickly and significantly by cold, freezing, and drought stresses, indicating that GRAS genes play vital roles in the regulation of M. truncatula response to abiotic stress.
Conclusions
In summary, 68 GRAS genes were identified from the M. truncatula genome sequence and their classification, structure, evolution, and tissue-specific expression were investigated. The results in this study revealed that MtGRAS genes are broadly involved in the regulation of plant tissue development, in which a large number of tissue-specific expression. Meanwhile, we identified a number of candidate MtGRAS genes that participate in nodulation process of alfalfa; these were also differentially expressed by freezing and/or cold treatment. In addition, the results of the qPCR analysis were corroborated by the transcriptome analysis. These results indicated that nodulation might critically contribute to freezing tolerance of alfalfa. Furthermore, the expression levels of GRAS genes under abiotic stresses indicated that they had a comprehensive response to salt, freezing, cold, ABA, and drought stresses, thereby implying that GRAS may represent convergence points among different signaling pathways. In summary, this study would provide a solid foundation for further functional analysis of GRAS genes to be used for transgenic applications.
Author contribution statement
CG and LS conceived and designed the experiments; CG helped perform the analysis with constructive discussions. LS performed the experiments and wrote the manuscript. LT and HC contributed to treatment the materials. LL helped perform the analysis with the structure and chromosomal location of GRAS genes, and the expression of GRAS genes in different tissues. All authors read and approved the manuscript.
Abbreviations
- GA:
-
Gibberellic acid
- GAI:
-
Gibberellic acid insensitive
- HAM:
-
Hairy meristem
- Ls:
-
Lateral suppressor
- NSP1:
-
Nodulation signaling pathway 1
- NSP2:
-
Nodulation signaling pathway 2
- PAT:
-
Phytochrome A signal transduction
- RGA:
-
Repressor of GAI
- SCR:
-
Scarecrow
- SCL:
-
Scarecrow-like
- SHR:
-
Short root
- TF:
-
Transcription factor
References
Bailey TL, Elkan C (1994) Fitting a mixture model by expectation maximization to discover motifs in biopolymers. In: International conference on intelligent systems for molecular biology, pp 28–36
Bailey TL, Johnson J, Grant CE, Noble WS (2015) The MEME suite. Nucl Acids Res 43(1):39–49. doi:10.1093/nar/gkv416
Bolle C (2004) The role of GRAS proteins in plant signal transduction and development. Planta 218(5):683–692. doi:10.1007/s00425-004-1203-z
Bolle C, Koncz C, Chua NH (2000) PAT I, a new member of the GRAS family, is involved in phytochrome A signal transduction, Genes Dev 14, 1269-1278. Genes Dev 14(10):1269–1278. doi:10.1101/gad.14.10.1269Genes&Dev.2000
Cannon SB, Mitra A, Baumgarten A, Young ND, May G (2004) The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol 4(1):10–30. doi:10.1186/1471-2229-4-10
Chen K, Li H, Chen Y, Qi Z, Li B, Li Z (2015) TaSCL14, a Novel Wheat (Triticum aestivum L.) GRAS gene, regulates plant growth, photosynthesis, tolerance to photooxidative stress, and senescence. J Genet Genom 42(1):21–32. doi:10.1016/j.jgg.2014.11.002
Fambrini M, Mariotti L, Parlanti S, Salvini M, Pugliesi C (2015) A GRAS-like gene of sunflower (Helianthus annuus L.) alters the gibberellin content and the axillary meristem outgrowth in transgenic Arabidopsis plants. Plant Biol 17(6):1123–1134. doi:10.1111/plb.12358
Fiorilli V, Volpe V, Zanini S, Vallino M, Abbà S, Bonfante P (2015) A rice GRAS gene has an impact on the success of arbuscular mycorrhizal colonization. Am J Plant Sci 6(12):1905–1915. doi:10.4236/ajps.2015.612191
Galovic V, Orlovic S, Fladung M (2015) Characterization of two poplar homologs of the GRAS/SCL gene, which encodes a transcription factor putatively associated with salt tolerance. iForest Biogeosci For 8(6):780–786. doi:10.3832/ifor1330-008
Gao MJ, Parkin I, Lydiate D, Hannoufa A (2004) An auxin-responsive SCARECROW-like transcriptional activator interacts with histone deacetylase. Plant Mol Biol 55(3):417–431. doi:10.1007/s11103-004-0892-9
Goodstein DM, Shu S, Howson R, Neupane R, Hayes RD, Fazo J, Mitros T, Dirks W, Hellsten U, Putnam N (2012) Phytozome: a comparative platform for green plant genomics. Nucl Acids Res 40:1178–1186. doi:10.1093/nar/gkr944
Grimplet J, Agudelo-Romero P, Teixeira RT, Martinez-Zapater JM, Fortes AM (2016) Structural and functional analysis of the GRAS gene family in grapevine indicates a role of GRAS proteins in the control of development and stress responses. Front Plant Sci 7:353–374. doi:10.3389/fpls.2016.00353
Heckmann AB, Lombardo F, Miwa H, Perry JA, Bunnewell S, Parniske M, Wang TL, Downie JA (2006) Lotus japonicus nodulation requires two GRAS domain regulators, one of which is functionally conserved in a non-legume. Plant Physiol 142(4):1739–1750. doi:10.1104/pp.106.089508
Hirsch S, Oldroyd GE (2009) GRAS-domain transcription factors that regulate plant development. Plant Signal Behav 4(8):698–700. doi:10.4161/psb.4.8.9176
Hirsch S, Kim J, Muñoz A, Heckmann AB, Downie JA, Oldroyd GED (2009) GRAS proteins form a DNA binding complex to induce gene expression during nodulation signaling in Medicago truncatula. Plant Cell 21(21):545–557. doi:10.1105/tpc.108.064501
Hu B, Jin J, Guo AY, Zhang H, Luo J, Gao G (2015) GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics 31(8):1296–1297. doi:10.1093/bioinformatics/btu817
Huang W, Xian Z, Xia K, Tang N, Li Z (2015) Genome-wide identification, phylogeny and expression analysis of GRAS gene family in tomato. BMC Plant Biol 15(1):209–226. doi:10.1186/s12870-015-0590-6
Johnson LS, Eddy SR, Portugaly E (2010) Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinf 11:431–438. doi:10.1186/1471-2105-11-431
Kaló P, Gleason C, Edwards A, Marsh J, Mitra RM, Hirsch S, Jakab J, Sims S, Long SR, Rogers J (2005) Nodulation signaling in legumes requires NSP2, a member of the GRAS family of transcriptional regulators. Science 308(5729):1786–1789. doi:10.1126/science.110951
Krzywinski M, Schein J, Birol İ, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA (2009) Circos: an information aesthetic for comparative genomics. Genome Res 19(9):1639–1645. doi:10.1101/gr.092759.109
Lee MH, Kim B, Song SK, Heo JO, Yu NI, Lee SA, Kim M, Dong GK, Sohn SO, Lim CE (2008) Large-scale analysis of the GRAS gene family in Arabidopsis thaliana. Plant Mol Biol 67(6):659–670. doi:10.1007/s11103-008-9345-1
Letunic I, Doerks T, Bork P (2012) SMART 7: recent updates to the protein domain annotation resource. Nucl Acids Res 40:302–305. doi:10.1093/nar/gkr931
Li J, Yu C, Hua W, Luo Z, Bo O, Long C, Zhang J, Ye Z (2015) Knockdown of a JmjC domain-containing gene JMJ524 confers altered gibberellin responses by transcriptional regulation of GRAS protein lacking the DELLA domain genes in tomato. J Exp Bot 66(5):1413–1426. doi:10.1093/jxb/eru493
Li W, Xu H, Liu Y, Song L, Guo C, Shu Y (2016) Bioinformatics analysis of MAPKKK family genes in Medicago truncatula. Genes 7(4):13–29. doi:10.3390/genes7040013
Liu X, Widmer A (2014) Genome-wide comparative analysis of the GRAS gene family in populus, Arabidopsis and rice. Plant Mol Biol Rep 32(6):1129–1145. doi:10.1007/s11105-014-0721-5
Liu Y, Zhang J, Li W, Guo C, Shu Y (2015) In silico identification, phylogeny and expression analysis of expansin superfamily in Medicago truncatula. Biotechnol Biotechnol Equip 1:1–7. doi:10.1080/13102818.2015.1093919
López M, Herrera-Cervera JA, Iribarne C, Tejera NA, Lluch C (2008) Growth and nitrogen fixation in Lotus japonicus and Medicago truncatula under NaCl stress: nodule carbon metabolism. J Plant Physiol 165(6):641–650. doi:10.1016/j.jplph.2007.05.009
Lu J, Wang T, Xu Z, Sun L, Zhang Q (2015) Genome-wide analysis of the GRAS gene family in Prunus mume. Mol Genet Genom 290(1):303–317. doi:10.1007/s00438-014-0918-1
Ma HS (2010) The salt- and drought-inducible poplar GRAS protein SCL7 confers salt and drought tolerance in Arabidopsis thaliana. J Exp Bot 61(14):4011–4019. doi:10.1093/jxb/erq217
Mayrose M, Ekengren SK, Melech-Bonfil S, Martin GB, Sessa G (2006) A novel link between tomato GRAS genes, plant disease resistance and mechanical stress response. Mol Plant Pathol 7(6):593–604. doi:10.1111/j.1364-3703.2006.00364.x
Mbe ESK, Fotsin HB, Kengne J, Woafo P (2014) A nuclear factor Y interacting protein of the GRAS family is required for nodule organogenesis, infection thread progression, and lateral root growth. Plant Physiol 164(3):1430–1442. doi:10.1104/pp.113.230896
Morohashi K, Minami M, Takase H, Hotta Y, Hiratsuka K (2003) Isolation and characterization of a novel GRAS gene that regulates meiosis-associated gene expression. J Biol Chem 278(23):20865–20873. doi:10.1074/jbc.M301712200
Navarro L, Bari R, Achard P, Lisón P, Nemri A, Harberd NP, Jones JD (2008) DELLAs control plant immune responses by modulating the balance of jasmonic acid and salicylic acid signaling. Curr Biol 18(9):650–655. doi:10.1016/j.cub.2008.03.060
Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, Heger A, Holm L, Sonnhammer Erik L L, Eddy SR, Bateman A, Finn RD (2012) The Pfam protein families database. Nucl Acids Res 40:290–301. doi:10.1093/nar/gkr1065
Schumacher K, Theres K (1999) The Lateral suppressor (Ls) gene of tomato encodes a new member of the VHIID protein family. Proc Natl Acad Sci USA 96(1):290–295
Shen C, Yue R, Sun T, Zhang L, Xu L, Tie S, Wang H, Yang Y (2015) Genome-wide identification and expression analysis of auxin response factor gene family in Medicago truncatula. Front Plant Sci 6(11):73–84. doi:10.3389/fpls.2015.00073
Shu Y, Liu Y, Zhang J, Song L, Guo C (2016) Genome-wide analysis of the AP2/ERF superfamily genes and their responses to abiotic stress in Medicago truncatula. Front Plant Sci 6(676):1247–1263. doi:10.3389/fpls.2015.01247
Song XM, Liu TK, Duan WK, Ma QH, Ren J, Wang Z, Li Y, Hou XL (2014) Genome-wide analysis of the GRAS gene family in Chinese cabbage (Brassica rapa ssp. pekinensis). Genomics 103(1):135–146. doi:10.1016/j.ygeno.2013.12.004
Song L, Jiang L, Chen Y, Shu Y, Bai Y, Guo C (2016) Deep-sequencing transcriptome analysis of field-grown Medicago sativa L. crown buds acclimated to freezing stress. Funct Integr Genom 16(5):495–511. doi:10.1007/s10142-016-0500-5
Sun X, Xue B, Jones WT, Rikkerink E, Dunker AK, Uversky VN (2011) A functionally required unfoldome from the plant kingdom: intrinsically disordered N-terminal domains of GRAS proteins are involved in molecular recognition during plant development. Plant Mol Biol 77(3):205–223. doi:10.1007/s11103-011-9803-z
Sun X, Jones WT, Rikkerink EH (2012) GRAS proteins: the versatile roles of intrinsically disordered proteins in plant signalling. Biochem J 442(1):1–12. doi:10.1042/BJ20111766
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28(10):2731–2739. doi:10.1093/molbev/msr121
Tian C, Ping W, Sun S, Li J, Chen M (2004) Genome-wide analysis of the GRAS gene family in rice and Arabidopsis. Plant Mol Biol 54(4):519–532. doi:10.1023/B:PLAN.0000038256.89809.57
Trapnell C, Pachter L, Salzberg SL (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25(9):1105–1111. doi:10.1093/bioinformatics/btp120
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, Baren MJV, Salzberg SL, Wold BJ, Pachter L (2010) Transcript assembly and abundance estimation from RNA-Seq reveals thousands of new transcripts and switching among isoforms. Nat Biotechnol 28(5):511–515. doi:10.1038/nbt.1621
Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG (2012) Primer3—new capabilities and interfaces. Nucl Acids Res 40(15):115–126. doi:10.1093/nar/gks596
Wei Y, Shi H, Xia Z, Tie W, Ding Z, Yan Y, Wang W, Hu W, Li K (2016) Genome-wide identification and expression analysis of the WRKY gene family in cassava. Front Plant Sci 7:211–228. doi:10.3389/fpls.2016.00025
Wickham H (2009) ggplot2: elegant graphics for data analysis. Springer Publishing Company, Berlin
Xin S, Xie Z, Cheng Z, Wu W, Wang B, Fang J (2016) A characterization of grapevine of GRAS domain transcription factor gene family. Funct Integr Genom 97(4):541–548. doi:10.1007/s10142-016-0479-y
Xu K, Chen S, Li T, Ma X, Liang X, Ding X, Liu H, Luo L (2015) OsGRAS23, a rice GRAS transcription factor gene, is involved in drought stress response through regulating expression of stress-responsive genes. BMC Plant Biol 15(1):141. doi:10.1186/s12870-015-0532-3
Yuan Y, Fang L, Karungo SK, Zhang L, Gao Y, Li S, Xin H (2016) Overexpression of VaPAT1, a GRAS transcription factor from Vitis amurensis, confers abiotic stress tolerance in Arabidopsis. Plant Cell Rep 35(3):655–666. doi:10.1007/s00299-015-1910-x
Zhang LL, Zhao MG, Tian QY, Zhang WH (2011) Comparative studies on tolerance of Medicago truncatula and Medicago falcata to freezing. Planta 234(3):445–457. doi:10.1007/s00425-011-1416-x
Acknowledgements
This work was supported by the MOST 863 project (2013AA102607-5); Key Scientific and Technological Project of Heilongjiang Province of China (GA15B105-1); the Natural and Science Foundation of China (No. 31470571); the Graduate Innovation Fund of Harbin Normal University (HSDBSCX2014-04); the National Major Project for Cultivation of Transgenic Crops (#2011ZX08004-002-003).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by M. Hajduch.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

{kind=link}
Cite this article
Song, L., Tao, L., Cui, H. et al. Genome-wide identification and expression analysis of the GRAS family proteins in Medicago truncatula . Acta Physiol Plant 39, 93 (2017). https://doi.org/10.1007/s11738-017-2393-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11738-017-2393-x