
Abstract
Rapidly identifying protein complexes is significant to elucidate the mechanisms of macromolecular interactions and to further investigate the overlapping clinical manifestations of diseases. To date, existing computational methods majorly focus on developing unsupervised graph clustering algorithms, sometimes in combination with prior biological insights, to detect protein complexes from protein-protein interaction (PPI) networks. However, the outputs of these methods are potentially structural or functional modules within PPI networks. These modules do not necessarily correspond to the actual protein complexes that are formed via spatiotemporal aggregation of subunits. In this study, we propose a computational framework that combines supervised learning and dense subgraphs discovery to predict protein complexes. The proposed framework consists of two steps. The first step reconstructs genome-scale protein co-complex networks via training a supervised learning model of l2-regularized logistic regression on experimentally derived co-complexed protein pairs; and the second step infers hierarchical and balanced clusters as complexes from the co-complex networks via effective but computationally intensive k-clique graph clustering method or efficient maximum modularity clustering (MMC) algorithm. Empirical studies of cross validation and independent test show that both steps achieve encouraging performance. The proposed framework is fundamentally novel and excels over existing methods in that the complexes inferred from protein co-complex networks are more biologically relevant than those inferred from PPI networks, providing a new avenue for identifying novel protein complexes.
Article PDF
Similar content being viewed by others


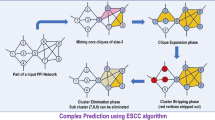
Avoid common mistakes on your manuscript.
References
Krogan N J, Peng W, Cagney G, Robinson M D, Haw R, Zhong G, et al. High-definition macromolecular composition of yeast RNA-processing complexes. Molecular Cell, 2004, 13(2): 225–239
Lage K, Karlberg E O, Størling Z M, Olason P I, Pedersen A G, Rigina O, et al. A human phenome-interactome network of protein complexes implicated in genetic disorders. Nature Biotechnology, 2007, 25(3): 309–316
Mewes H W, Amid C, Arnold R, Frishman D, Güldener U, Mannhaupt G, et al. MIPS: analysis and annotation of proteins from whole genomes. Nucleic Acids Research, 2004, 32(suppl_1): D41–D44
Ruepp A, Waegele B, Lechner M, Brauner B, Dunger-Kaltenbach, Fobo G, et al. CORUM: the comprehensive resource of mammalian protein complexes—2009. Nucleic Acids Research, 2010, 38(suppl_4): D497–D501
Keshava Prasad T S, Goel R, Kandasamy K, Keerthikumar S, Kumar S, Mathivanan S, et al. Human Protein Reference Database—2009 update. Nucleic Acids Research, 2009, 37(suppl_1): D767–D772
Li X, Wu M, Kwoh C K, Ng S K. Computational approaches for detecting protein complexes from protein interaction networks: a survey. BMC Genomics, 2010, 11(1): 1–19
Srihari S, Yong C H, Patil A, Wong L. Methods for protein complex prediction and their contributions towards understanding the organisation, function and dynamics of complexes. FEBS Letters, 2015, 589(19): 2590–2602
Zahiri J, Emamjomeh A, Bagheri S, Ivazeh A, Mahdevar G, Sepasi H, et al. Protein complex prediction: a survey. Genomics, 2020, 112(1): 174–183
Bron C, Kerbosch J. Finding all cliques of an undirected graph. Communications of the ACM, 1973, 16(9): 575–580
Bader G, Hogue C. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics, 2003, 4(1): 1–27
Van Dongen S. Graph clustering by flow simulation. University of Utrecht, 2000
Nepusz T, Yu H, Paccanaro A. Detecting overlapping protein complexes in protein-protein interaction networks. Nature Methods, 2012, 9(5): 471–472
Pellegrini M, Baglioni M, Geraci F. Protein complex prediction for large protein protein interaction networks with the Core&Peel method. BMC Bioinformatics, 2016, 17(12): 37–58
Hernandez C, Mella C, Navarro G, Olivera-Nappa A, Araya J. Protein complex prediction via dense subgraphs and false positive analysis. PLoS ONE, 2017, 12: e0183460
Wu M, Xie Z, Li X, Kwoh C K, Zheng J. Identifying protein complexes from heterogeneous biological data. Proteins, 2013, 81(11): 2023–2033
Gavin A C, Aloy P, Grandi P, Krause R, Boesche M, Marzioch M, et al. Proteome survey reveals modularity of the yeast cell machinery. Nature 2006, 440(7084): 631–636
Geva G, Sharan R. Identification of protein complexes from coimmunoprecipitation data. Bioinformatics, 2011, 27(1): 111–117
Krogan N J, Cagney G, Yu H, Zhong G, Guo X, Ignatchenko A, et al. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature, 2006, 440(7084): 637–643
Qi Y, Balem F, Faloutsos C, Klein-Seetharaman J, Bar-Joseph Z. Protein complex identification by supervised graph local clustering. Bioinformatics, 2008, 24(13): i250–i268
Fabregat A, Sidiropoulos K, Garapati P, Gillespie M, Hausmann K, Haw R, et al. The Reactome pathway Knowledgebase. Nucleic Acids Research, 2016, 44(D1): D481–D487
Wu G, Feng X, Stein L. A human functional protein interaction network and its application to cancer data analysis. Genome Biology, 2010, 11(5): 1–23
Chatr-Aryamontri A, Breitkreutz B J, Oughtred R, Boucher L, Heinicke S, Chen D, et al. The BioGRID interaction database: 2015 update. Nucleic Acids Research, 2015, 43(D1): D470–D478
Orchard S, Ammari M, Aranda B, Breuza L, Briganti L, Broackes-Carter F, et al. The MIntAct project–IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Research, 2014, 42(D1): D358–D363
Collins S R, Kemmeren P, Zhao X C, Greenblatt J F, Spencer F, Holstege F C, et al. Toward a comprehensive atlas of the physical interactome of Saccharomyces cerevisiae. Molecular & Cellular Proteomics, 2007, 6(3): 439–450
Yu H, Braun P, Yildirim M A, Lemmens I, Venkatesan K, Sahalie J, et al. High-quality binary protein interaction map of the yeast interactome network. Science, 2008, 322(5898): 104–110
Ito T, Chiba T, Ozawa R, Yoshida M, Hattori M, Sakaki Y. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proceedings of the National Academy of Sciences of The United States of America, 2001, 98(8): 4569–4574
Uetz P, Giot L, Cagney G, Mansfield T A, Judson R S, Knight J R, et al. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature, 2000, 403(6770): 623–627
Pu S, Wong J, Turner B, Cho E, Wodak S J. Up-to-date catalogues of yeast protein complexes. Nucleic Acids Research, 2009, 37(3): 825–831
Maetschke S, Simonsen M, Davis M, Ragan M A. Gene ontology-driven inference of protein-protein interactions using inducers. Bioinformatics, 2012, 28(1): 69–75
Qi Y, Tastan O, Carbonell J G, Klein-Seetharaman J, Weston J. Semi-supervised multi-task learning for predicting interactions between HIV-1 and human proteins. Bioinformatics, 2010, 26(18): i645–i652
Mei S, Zhu H. A novel one-class SVM based negative data sampling method for reconstructing proteome-wide HTLV-human protein interaction networks. Scientific Reports, 2015, 5: 8034
Mei S. In silico enhancing M. tuberculosis protein interaction networks in STRING to predict drug-resistance pathways and pharmacological risks. Journal of Proteome Research, 2018, 17(5): 1749–1760
Mei S, Flemington E K, Zhang K. Transferring knowledge of bacterial protein interaction networks to predict pathogen targeted human genes and immune signaling pathways: a case study on M. tuberculosis. BMC Genomics, 2018, 19(1): 1–21
Altschul S F, Madden T L, Schäffer A A, Zhang J, Zhang Z. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research, 1997, 25(17): 3389–3402
Boeckmann B, Bairoch A, Apweiler R, Blatter M C, Estreicher A, Gasteiger E, et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Research, 2003, 31(1): 365–370
Barrell D, Dimmer E, Huntley R P, Binns D, O’Donovan C, Apweiler R, et al. The GOA database in 2009–an integrated gene ontology annotation resource. Nucleic Acids Research, 2009, 37(D1): D396–D403
Yu F, Huang F, Lin C. Dual coordinate descent methods for logistic regression and maximum entropy models. Machine Learning, 2011, 85: 41–75
Fan R, Chang K, Hsieh C, Wang X, Lin C. LIBLINEAR: a library for large linear classification. Machine Learning Research, 2008, 9(2): 1871–1874
Palla G, Derényi I, Farkas I, Vicsek T. Uncovering the overlapping community structure of complex networks in nature and society. Nature, 2005, 435(7043): 814–818
Adamcsek B, Palla G, Farkas I J, Derényi I, Vicsek T. CFinder: locating cliques and overlapping modules in biological networks. Bioinformatics, 2006, 22(8): 1021–1023
Noack A, Rotta R. Multi-level algorithms for modularity clustering. In: Proceedings of the 8th International Symposium on Experimental Algorithms. 2009, 257–268
Rossi F, Villa-Vialaneix N. Représentation d’un grand réseau à partir d’une classification hiérarchique de ses sommets. Journal de la Société Française de Statistique, 2011, 152: 34–65
Newman M E. Finding community structure in networks using the eigenvectors of matrices. Physical Review E, 2006, 74: 036104
Zhang L V, Wong S L, King O D, Roth F P. Predicting co-complexed protein pairs using genomic and proteomic data integration. BMC Bioinformatics, 2004, 5(1): 1–15
Qiu J, Noble W S. Predicting co-complexed protein pairs from heterogeneous data. PLoS Computational Biology, 2008, 4(4): e1000054
Kikugawa S, Nishikata K, Murakami K, Sato Y, Suzuki M, Altaf-Ul-Amin M, et al. PCDq: human protein complex database with quality index which summarizes different levels of evidences of protein complexes predicted from H-Invitational protein-protein interactions integrative dataset. BMC Systems Biology, 2012, 6(Suppl 2): S7
Romero-Molina S, Ruiz-Blanco Y B, Harms M, Münch J, Sanchez-Garcia E. PPI-Detect: a support vector machine model for sequence-based prediction of protein-protein interactions. Journal of Computational Chemistry, 2019, 40(11): 1233–1242
Chen M, Ju C J, Zhou G, Chen X, Zhang T, Chang K W, et al. Multifaceted protein-protein interaction prediction based on Siamese residual RCNN. Bioinformatics, 2019, 35(14): i305–i314
Author information
Authors and Affiliations
Corresponding author
Additional information
Supporting information
The supporting information is available online at journal.hep.com.cn and springerlink.bibliotecabuap.elogim.com..
Suyu Mei received his PhD in computer science from Fudan University, China. His research fields cover machine learning and bioinformatics. He further conducted postdoctoral research of computational biology in Southern Medical University, China. His research topics focused on studying pathogen-host signaling cross-talks and systems pharmacology. He has published more than 20 first-authored papers in international peer-review journals. His current research topics cover the studies of plant and soil microbiome, microbial ecology and human microbiome-associated diseases via microbiomics and machine learning approaches.
Electronic Supplementary Material
Rights and permissions
About this article

Cite this article
Mei, S. A framework combines supervised learning and dense subgraphs discovery to predict protein complexes. Front. Comput. Sci. 16, 161901 (2022). https://doi.org/10.1007/s11704-021-0476-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11704-021-0476-8