
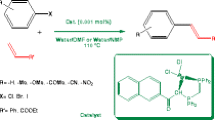
Abstract
We have achieved an efficient synthesis of C(3)-arylphthalides by coupling C(3)-bromophthalides and arylboronic acids under palladium catalysis. The C(sp3)-C(sp2) coupling worked well in the presence of water to provide products in a high yield.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The phthalide (the C(3)-substituted 3H-isobenzofuran-1-one and reduced product of phthalic anhydride; highlighted in 1, Fig. 1) structure is a medicinally privileged scaffold (Sadikogullari et al. 2022; Sharma et al. 2010). Molecules built on this scaffold display a wide range of pharmacological activities. The phthalide core is present in many natural products (Fig. 1) (Lin et al. 2005; Leon et al. 2017). Some of them exhibit extremely useful biological profiles (Beck and Chou 2007; Xioang et al. 2007; Mola et al. 2012a, b). For example, Isopestacin 1 isolated from the endophytic fungus Pestalotiopsis microspora is an antifungal agent and acts as an effective antioxidant towards both superoxide and hydroxide free radicals (Strobel et al. 2002; Meshram et al. 2018). Cryophonectric acid 2 isolated from the fungus Cryophonectria parasitica is its most abundant natural product, constituting more than 20% of the crude extract (Arnone et al. 2002). It exhibits profound antibacterial activity (Kukreti et al. 2015; Kaur et al. 2020). Chryocolide 3 isolated from the Japanese vegetable and garland flower-bearing plant, Chrysanthemum coronarium exhibits plant growth-inhibiting and insect anti-feeding activity (Tada et al. 1984). Typhaphthalide (a benzylphthalide) 4 isolated the rhizomes of Typha capensis is a part of the traditional medicines for venereal diseases, dysmenorrhea, diarrhoea, etc. (Shode et al. 2002). Corollosporine 5 isolated from the marine fungus corollospora maritima, shows antibacterial activity against Staphylococcus aureus and other microorganisms (Liberra et al. 1998; Ohzeki et al. 2001). In addition, several non-natural phthalides are emerging as medicinal drugs (Chen and Mori 2020; Luo et al. 2020). They found use in the treatment of circulatory disorders and heart diseases (Lei et al. 2019). Another important use of phthalides is their application as building blocks in organic synthesis (Awasthi et al. 2020; Karmakar et al. 2024; Mal and Pahari 2007Rathwell et al. 2007). They have been used in the synthesis of several functionalized naphthalenes, anthracenes, and naphthacenes, including some natural products having these structural motifs (Mousavi et al. 2023; Rathwell and Brimble 2007; Hernandez et al. 2007; Sankara et al. 2023; Zhang et al. 2023).

Biologically active compounds containing phthalide nucleus
During the last few decades, several methods (e.g. A-G, Scheme 1) have emerged for the synthesis of C(3)-arylphthalides (Shi and Li 2012). The majority of them (A-D) depend on metal catalysis to make the crucial C–C and C–O bonds during the benzofuran ring formation (Miura et al. 2018; Wang et al. 2021; Qiang et al. 2021; Ibraheem et al. 2021; Ye et al. 2010). Method E is a base-mediated condensation followed by lactone formation (Mola et al. 2012a; b). Method F is an example of photochemical Norrish type I cleavage, loss of carbon monoxide, and finally reductive alkylation (Mor et al. 2007; Roscini et al. 2008). Method G is an acid-mediated substitution of the labile C(3) hydroxyl group in phthalide with nucleophiles that include electron-rich aromatic compounds (Canonne et al. 1988; Ortega et al. 2022). However, most of them suffer from one or more drawbacks, like the need for expensive metal catalysts, stoichiometric or excessive amounts of acids or bases, harsh conditions, or low functional group tolerance. Developing a simple and efficient method for 3-arylphthalide remains a highly desirable goal. Enormous importance of phthalides in medicinal chemistry and the increasing awareness of environmentally benign chemical production created the need for the development of efficient, versatile, and scalable methods. In this context and in continuation of our efforts on the synthesis of phthalide mimics like isoindolinones (Mousavi et al. 2023) we have taken up the development of a new, facile, and scalable synthesis of C(3)-arylphthalides. We thought that readily available benzylic bromide 6 and arylboronic acids 7 would undergo palladium-catalyzed C(sp3)-C(sp2) Suzuki-type coupling to furnish C(3)-arylphthalides (method H, Scheme 1). We disclose details of the successful realization of this hypothesis towards several hitherto unknown phthalides. Most relevant to the present work are the findings of Zhang and Feng, who employed NiCl2.glyme catalysed (10 mol%) coupling of 6 and 7 in presence of the chiral ligands (12 mol%) under stringent condition to form enantiomerically enriched C(3) aryl substituted phthalides 8 (Xu et al. 2021). Although chemical yield of 8 were good, enantiomeric excess was moderate.

Different methods for the synthesis of C(3)-substituted phthalides
Results and discussion
To test the concept and to optimization of the reaction conditions, we started our investigation with 3-bromoisobenzofuran-1(3H)-one 6 (Qiang et al. 2017) and phenylboronic acid 7 as the reactants to form the C(3)-phenylphthalide 8 (Scheme 2, Table 1). From the series of experiments conducted with equi-milli molar amounts of 6 and 7, the one with PdCl2(PPh3)2 as the catalyst (1 mol%), Na2CO3 (1 equiv) as the base, a mixture (9:1) of water and THF as the solvent, and 70 °C as the bath temperature provided the best yield (94%) of the product 8 (entry 1, Table 1). Alternate reaction conditions invariably resulted in lower yields or there was no product even after 24 h. For example, the use of relatively inexpensive Pd(OAc)2 as the catalyst (1 mol%; 19%; entry 2) (Liu et al. 2005; Saikra et al. 2015) or Pd/C (no reaction; entry 3) (Schmidt and Riemer 2014; Tagata and Nishida 2003) under the standard Suzuki coupling conditions furnished in the product in low or no yield. Switching the base from Na2CO3 to the relatively stronger and better water-soluble K2CO3 (1 equiv; 34%; entry 4) or relatively milder and better water-soluble KHCO3 (1 equiv; 21%; entry 5) did not help. The reaction was significantly impacted by the choice of the solvent. We tried conducting the reaction in environmentally benign H2O (59%, entry 6), an equal proportion of H2O and THF (89%; entry 7), THF alone (68%, entry 8), 1,4-dioxane (53%, entry 9), or non-polar toluene (18%, entry 10), but the reactions did not give optimal yields. We concluded that the reaction requires water to proceed, however, water alone does not work to provide best yield of the product. The reaction was conducted as per entry 1 but at rt did not work (entry 11). Surprisingly, the yield of product 8 was lower when we employed a larger amount of the catalyst (10 mol%; 73% entry 12).

Synthesis of C(3)-arylphthalides
The workup of the reaction was simple. After completion of the reaction, the mixture of THF and water was recovered under reduced pressure. Filtration of the resulting viscous crude product as a solution of EtOAc (5 vol) through a pad of silica-gel followed by evaporation of the solvent and trituration by using n-pentane furnished the pure solid product with 94% yield. Notably, isolation of the product did not require column chromatography. We conducted this reaction on a deca-molar scale, and it worked without any event. The structure of the phthalide 8a was assigned based on the spectroscopic (IR, 1H NMR, 13C NMR, DEPT-135) and analytical (ESIMS HRMS) data.
After successfully unearthing optimal reaction conditions, we applied it to the reactions of 6 with six more arylboronic acids 7b-7 g to expand the scope towards the synthesis of six more C(3)-arylphthalides. As anticipated, the reaction of 6 with arylboronic acids 7b-7 g worked well under the optimized reaction conditions to provide the phthalides 8b-8 g as solids in excellent yields. Functional groups, such as methyl 8b, ethyl 8c, methoxy 8d, ethoxy 8e, naphthyl 8f, and fluro 8 g were tolerated in these conditions. The spectra IR and 1H, 13C, DEPT NMR, and HRMS of 8b-8 g supported the assigned structures and they matched with those of the parent 8a. In gist, we have demonstrated the generality of the palladium-catalyzed C(sp3)-C(sp2) Suzuki-type coupling to realize the C(3)-arylphthalides 8b-8 g in good to excellent yields. Notably, in all the cases, isolation of 8b-8 g did not require column chromatography for isolation.
A possible mechanism for the above Pd-catalyzed coupling reaction is given in Scheme 3. It is analogous to that of the earlier proposed mechanism for the Suzuki reaction. (Dalterio et al. 2021). The first step is the oxidative insertion of Pd(0) into C–Br of 6 to form 9. Since the reaction did not take place at rt, the bromide 6 remained as such at this temperature, and the reaction took place at 70 °C, we think that the oxidative insertion of Pdo into the C–Br bond is the rate-determining step. Transmetallation with concomitant elimination of B(OH)3 and HBr with the involvement of a molecule of water leads to the intermediate 10. Since the reaction worked best in H2O:THF (9:1) we think that this combination is suitable for the solubility of the reactants, catalyst, and base to afford good yield of the desired product 6. The in situ generated acid (HBr) is trapped by Na2CO3 to form sodium bromide which precipitated from the reaction medium. Transformation of the intermediate 10 through C(sp3)-C(sp2) coupling led to 8 and regeneration of the [Pd0] catalyst.

Proposed mechanism for the Pd-catalyzed C(sp3)-C(sp2) coupling
Conclusion
In conclusion, we have developed an efficient palladium-catalyzed C(sp3)-C(sp2) coupling for C(3)-arylphthalides from readily available C(3)-bromophthalides and arylboronic acids. The method offers several advantages such as easy handling, workup, environmentally benign conditions and excellent yields. The method allows the synthesis of a combinatorial library of medicinally important C(3)-arylphthalides.
Experimental
Materials and equipment
The progress of all reactions were monitored by TLC using a hexanes and ethyl acetate mixture as eluent. Column chromatography was performed on silica gel (100–200 mesh) using increasing percentages of ethyl acetate in hexanes. 1H NMR (400 MHz), 13C NMR (100 MHz), and DEPT-135 spectra were recorded for CDCl3) solutions on a Bruker Avance 400 spectrometer with TMS as the internal standard. Chemical shifts (in ppm) and coupling constants J are given in Hz. IR spectra were recorded as solid solutions in KBr on a Nicolet-6700 FT-IR spectrometer. High-resolution mass spectra (HRMS) were recorded on an Agilent 6350 B Q-TOF mass spectrometer using electrospray ionization mode. The melting points were recorded with an open-ended capillary tube with a VEEGO VMP-DS instrument and were uncorrected. Standard methods were used to dry organic solvents (Armarego and Chai 2003). Commercially obtained reagents were used after purification. Arylboronic acids, phthalic anhydride, and palladium salts were purchased from Sigma-Aldrich.
General procedure for the synthesis of 3-phenylisobenzofuran-1(3H)-one 8a

To the reaction tube with 3-bromoisobenzofuran-1(3H)-one (200 mg, 0.92 mmol), phenylboronic acid (114 mg, 0.92 mmol), Na2CO3 (100 mg, 0.92 mmol), PdCl2(PPh3)2 (6.4 mg, 0.009 mmol) a solution of 20 mL of H2O and THF (9:1, v/v). The reaction tube was kept stirring at 70 °C for 2 h. After the completion of the reaction, as monitored by TLC, the solvent was evaporated under reduced pressure. The residue was passed through a pad of silica gel with the help of EtOAc (50 mL) to give 3-phenylisobenzofuran-1(3H)-one 8a as a white solid (186 mg, 94% yield); mp 113 °C; IR (KBr, cm−1) 3340, 1762, 1603, 1499, 1356, 1282, 1193, 1092, 1008, 913, 749, 696, 638; 1H NMR (400 MHz, CDCl3) δ 7.96 (d, J = 8 Hz, 1H), 7.65 (td, J = 7.5, 1.1 Hz, 1H), 7.55 (t, J = 7.5 Hz, 1H), 7.40–7.35 (m, 3H), 7.33 (dd, J = 7.7, 0.8 Hz, 1H), 7.27 (dd, J = 7.0, 3.0 Hz, 2H), 6.41 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 170.7 (C), 149.7 (C), 136.4 (C), 134.4 (CH), 129.49 (CH), 129.43 (CH), 129.0 (CH), 127.0 (CH), 125.7 (CH), 125.6 (CH), 122.9 (C), 82.8 (CH); HRMS (ESI) calcd for C14H11O2 (M + H) 211.0759, found 211.0745.
References
Armarego WLF, Chai CLL (2003) Purification of Laboratory Chemicals, 5th edn. Elsevier, Oxford, UK
Arnone A, Assante G, Nasini G et al (2002) Cryphonectric acid and other minor metabolites from a hypovirulent strain of Cryphonectria Parasitica. J Nat Prod 65:48–50. https://doi.org/10.1021/np0103012
Awasthi A, Singh M, Rathee G et al (2020) Recent advancements in synthetic methodologies of 3-substituted phthalides and their application in the total synthesis of biologically active natural products. RSC Adv 10:12626–12652. https://doi.org/10.1039/D0RA00701C
Beck JJ, Chou SC (2007) The structural diversity of phthalides from the Apiaceae. J Nat Prod 70:891–900. https://doi.org/10.1021/np0605586
Canonne P, Plamondon J et al (1988) Reactions Selectives des organonagnesiens avec les lactols et les lactones. synthese des diols primaires secondaires. Tetrahedron 44:2903–2912. https://doi.org/10.1016/S0040-4020(88)90027-0
Chen LZ, Wu J, Li K et al (2020) Novel phthalide derivatives: synthesis and anti-inflammatory activity in vitro and in vivo. Eur J Med Chem 206:112722. https://doi.org/10.1016/j.ejmech.2020.112722
DAlterio MC, Casals-Cruañas E, Tzouras NV et al (2021) Mechanistic aspects of the palladium-catalyzed Suzuki-Miyaura cross-coupling reaction. Chem Eur J 27:13481–13493. https://doi.org/10.1002/chem.202101880
DiMola A, Croce G, More V et al (2012a) Active methylene compounds in a very effective approach to 3-substituted isobenzofuranones through tandem aldol/lactonization Reactions. Tetrahedron 68:6146–6151. https://doi.org/10.1016/j.tet.2012.05.079
DiMola A, Palombi L, Massa A (2012b) Active methylene compounds in the synthesis of 3-substituted isobenzofuranones, isoindolinones and related compounds. Curr Org Chem 16:2302–2320. https://doi.org/10.2174/138527212803520254
Hernández E, Vélez JM, Vlaar CP (2007) Synthesis of 1, 4-dihydro-benzo [d][1, 3] oxazin-2- ones from phthalides via an aminolysis-hofmann rearrangement protocol. Tetrahedron Lett 48:8972–8975
Ibraheem W, Wils Q, Camiade E et al (2021) Synthesis and antibacterial activity of racemic paecilocin a and its derivatives against methicillin-sensitive and-resistant Staphylococcus aureus. Tetrahedron Lett 67:152888. https://doi.org/10.1016/j.tetlet.2021.152888
Karmakar R, Pahari P, Mal D (2024) Phthalides and phthalans: synthetic methodologies and their applications in the total synthesis. Chem Rev 114:6213–6284. https://doi.org/10.1021/cr400524q
Kaur P, Kaur G (2020) Microbial endophytes: an untapped resource with antitumor and anti-microbial properties. Appl Biol Chem 1:9–20. https://doi.org/10.52679/tabcj.2020.0003
Kukreti V (2015) Synthesis of some new bio active phthalides and their antibacterial activity. Acta Ciencia Indica 3:139
Lei W, Deng YF et al (2019) Phthalides, senkyunolide A and ligustilide, showimmuno modulatory effect in improving atherosclerosis, through inhibiting AP-1 and NF-κB expression. Biomed Pharmacother 117:109074. https://doi.org/10.1016/j.biopha.2019.109074
León A, Del-Angel M, Avila JL et al (2017) Phthalides: distribution in nature, chemical reactivity, synthesis, and biological activity. Prog Chem Org Nat Prod 127:246. https://doi.org/10.1007/978-3-319-45618-8_2
Liberra K, Jansen R, Lindequist U (1998) Corollosporine, a new phthalide derivative from the marine fungus Corollospora maritima. Pharmazie 53:578–581
Lin G, Chan SSK, Chung HS et al (2005) Chemistry and biological activities of naturally occurring phthalides. Stud Nat Prod Chem 32:611–669. https://doi.org/10.1016/S1572-5995(05)80065-1
Liu L, Zhang Y, Wang Y et al (2005) Phosphine-free palladium acetate catalyzed suzuki reaction in water. J Org Chem 70:6122–6125. https://doi.org/10.1021/jo050724z
Luo L, Song Q, Li Y et al (2020) Design, synthesis and evaluation of phthalide alkyl tertiary amine derivatives as promising acetylcholinesterase inhibitors with high potency and selectivity against Alzheimer’s disease. Bioorg Med Chem 28:115400. https://doi.org/10.1016/j.bmc.2020.115400
Mal D, Pahari P (2007) Recent Advances in the Hauser Annulation. Chem Rev 107:1892–1918. https://doi.org/10.1021/cr068398q
Meshram V, Uppal K, Gupta M (2018) Endophytes: a Gold mine of enzyme inhibitors. Microbial Bioprospect Sustain Dev 61:92. https://doi.org/10.1007/978-981-13-0053-0_4
Miura H, Terajima S, Shishido T (2018) Carboxylate-directed addition of aromatic C-H bond to aromatic aldehydes under ruthenium catalysis. ACS Catal 8:6246–6254. https://doi.org/10.1021/acscatal.8b00680
Mor S, Dhawan SN, Kumar D et al (2007) Photochemical synthesis of isomeric (E/Z)-3-alkylidene-3H-isobenzofuranones. Tetrahedron 63:594–597. https://doi.org/10.1016/j.tet.2006.11.021
Mousavi MS, Di Mola A, Massa A (2023) Multifaceted behaviour of 2-cyanobenzaldehyde and 2-acylbenzonitriles in the synthesis of isoindolinones, phthalides and related heterocycles. Eur J Org Chem 26:202300289. https://doi.org/10.1002/ejoc.202300289
Ohzeki T, Mori K (2001) Synthesis of corollosporine, an antibacterial metabolite of the marine fungus Corollospora maritima. Biosci Biotechnol Biochem 65:172–175. https://doi.org/10.1271/bbb.65.172
Ortega MJ, Parra-Torrejon B, Cano-Cano F et al (2022) Synthesis and antioxidant/anti-inflammatory activity of 3-arylphthalides. Pharmaceuticals 15:588. https://doi.org/10.3390/ph15050588
Qiang X, Li Y, Yang X et al (2017) DL-3-N-Butylphthalide-edaravone hybrids as novel dual inhibitors of amyloid-β aggregation and monoamine oxidases with high antioxidant potency for alzheimer’s therapy. Bioorg Med Chem Lett 27:718–722. https://doi.org/10.1016/j.bmcl.2017.01.050
Qiang Q, Liu F, Rong ZQ (2021) Direct and selective synthesis of 3-arylphthalides via nickel-catalyzed aryl addition/intramolecular esterification. Tetrahedron 89:132162. https://doi.org/10.1016/j.tet.2021.132162
Rathwell K, Brimble MA (2007) Use of stabilized phthalide anion annulation reactions in synthesis: an update. Synthesis 05:643–662. https://doi.org/10.1055/s-2007-965915
Roscini C, Davies DME, Berry M et al (2008) Product selection through photon flux: laser-specific lactone synthesis. Angew Chem Int Ed 47:2283–2286. https://doi.org/10.1002/ange.200704816
Sadikogullari BC, Senel P, Cini N et al (2022) An overview of natural and synthetic phthalides involved in cancer studies: past, present, and future. ChemistrySelect 7:e202202004. https://doi.org/10.1002/slct.202202004
Saikia B, Boruah PR, Ali AA et al (2015) Simple and efficient phosphine-free Pd(OAc)2 catalyzed urea accelerated suzuki–miyaura cross-coupling reactions in iPrOH-H2O at room temperature. Tetrahedron Lett 56:633–635. https://doi.org/10.1016/j.tetlet.2014.12.061
Sankara CS, Gaikwad SP, Namboothiri IN (2023) Synthesis of natural products, carbocycles, and heterocycles by hauser–kraus annulation. Synlett. https://doi.org/10.1055/a-2068-7126
Schmidt B, Riemer M (2014) Suzuki–miyaura coupling of halophenols and phenol boronic acids: systematic investigation of positional isomer effects and conclusions for the synthesis of phytoalexins from pyrinae. J Org Chem 79:4104–4118. https://doi.org/10.1021/jo500675a
Sharma U, Kumar P, Kumar N et al (2010) Recent advances in the chemistry of phthalimide analogues and their therapeutic potential. Mini-Rev Med Chem 10:678–704. https://doi.org/10.2174/138955710791572442
Shi X, Li CJ (2012) A novel rhodium-catalyzed cascade cyclization: direct synthesis of 3-substituted phthalides from aldehydes and aromatic acids. Adv Synth Catal 354:2933–2938. https://doi.org/10.1002/adsc.201200690
Shode FO, Mahomed AS, Rogers CB (2002) Typhaphthalide and typharin, two phenolic compounds from Typha Capensis. Phytochemistry 61:955–957. https://doi.org/10.1016/S0031-9422(02)00439-9
Strobel G, Ford E, Worapong J et al (2002) Isopestacin, an isobenzofuranone from Pestalotiopsis microspora, possessing antifungal and antioxidant activities. Phytochemistry 60:179–183. https://doi.org/10.1016/S0031-9422(02)00062-6
Tada M, Chiba K (1984) Novel plant growth inhibitors and an insect antifeedant from Chrysanthemum coronarium. Agric Biol Chem 48:1367–1369. https://doi.org/10.1271/bbb1961.48.1367
Tagata T, Nishida M (2003) Palladium charcoal-catalyzed suzuki − miyaura coupling to obtain arylpyridines and arylquinolines. J Org Chem 68:9412–9415. https://doi.org/10.1021/jo034970r
Wang S, Miao E, Wang H et al (2021) Rh-Catalyzed cascade C-H activation/C-C cleavage/cyclization of carboxylic acids with cyclopropanols. Chem Commun 57:5929–5932. https://doi.org/10.1039/D1CC01778K
Xiong MJ, Li ZH (2007) Progress in syntheses of 3-n-butylphthalide and its analogues. Curr Org Chem 11:833–844. https://doi.org/10.2174/138527207780831738
Xu SY, Zhang R, Zhang SS et al (2021) Enantioselective Synthesis of 3-arylphthalides through a nickel-catalyzed stereoconvergent cross-coupling reaction. Org Biomol Chem 19:4492–4496. https://doi.org/10.1039/D1OB00487E
Ye Z, Qian P, Lv G et al (2010) Palladium-catalyzed cascade aryl addition/intramolecular lactonization of phthalaldehyde to access 3-aryl-and alkenylphthalides. J Org Chem 75:6043–6045. https://doi.org/10.1021/jo101203b
Zhang Y, Zhang Z, Xia Y et al (2023) TfOH-Catalyzed cascade reaction: metal-free access to 3, 3-disubstituted phthalides from o-alkynylbenzoic acids. J Org Chem. https://doi.org/10.1021/acs.joc.3c00760
Acknowledgements
HSPR thanks Pondicherry University (PU) for the financial support to carry out the work described here. We thank DST-FIST and UGC-SAP for the instrumentals given to the Department of Chemistry. JP thanks UGC-RGNF for the fellowship. We thank the Central Instrumentation Facility, PU for spectral and analytical data.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Rao, H.S.P., Prabhakaran, J. Palladium-catalyzed C(sp3)-C(sp2) coupling: synthesis of C(3)-arylphthalides. Chem. Pap. 78, 5985–5991 (2024). https://doi.org/10.1007/s11696-024-03520-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11696-024-03520-4