
Abstract
The coumarin nucleus is a simple privileged scaffold distributed in many plants. It has recently gained attention for its diverse biological activities and interactions with enzymes and receptors. The vascular endothelial growth factor receptor-2 (VEGFR-2), a receptor tyrosine kinase, is a crucial cancer target as it is involved in angiogenesis. This study employs virtual screening, molecular docking, and molecular simulation studies to identify potential coumarin candidates against VEGFR-2 from the COCONUT database. After thorough docking studies, CNP0056360, CNP0340213, and CNP0366287 were identified as final hits. Molecular dynamics simulation studies revealed strong stability and better binding energies for CNP0056360 and CNP0340213, outperforming lenvatinib; CNP0366287 showed comparable behaviour. The identified coumarins exhibited good in-silico pharmacokinetics and demonstrated low toxicity.
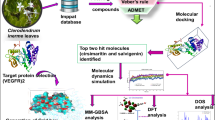
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Coumarins are a class of secondary metabolites obtained extensively from plants, but also present in microorganisms and fungi (Annunziata et al. 2020). The coumarin molecule gets its name from ‘coumarou,’ a French word for Tonka beans (seeds of Dipteryx odorata or Coumarouna odorata) from which they were first isolated in 1820 (Maria João et al. 2015). More than 1300 different coumarins have been reported from more than 150 plant species belonging principally to Apiaceae, Fabaceae, Asteraceae, Rutaceae, Moraceae, Oleaceae, and Thymelaeaceae families (Maria João et al. 2015; Sharifi-Rad et al. 2021). The coumarins class is further divided into six different categories based on their core scaffold, i.e. simple coumarins, furocoumarins, dihydrofuro-coumarins, pyrano-coumarins (linear and angular), bis-coumarins, and phenyl-coumarins (Maria João et al. 2015). Coumarin is a versatile nucleus owing to its different substitution sites and potent pharmacophore contributing to its broad pharmacological spectrum. Coumarins possess antioxidant (osthole), antiadipogenic (fraxin), anticancer (xanthotoxol), carbonic anhydrase inhibition, antibacterial (agasyllin), antitubercular (bergapten), antifungal (psoralen), antiviral (calanolide A), anti-inflammatory (imperatorin), neuroprotective (esculetin), anticonvulsant (imperatorin), anticoagulant (dicumarol), antidiabetic (fraxidin), and photosensitizing (methoxsalen) activities (Annunziata et al. 2020; Sharifi-Rad et al. 2021). Amidst coumarin’s broad spectrum of pharmacology, its anticancer potential is quite fascinating as many natural coumarins possess anticancer activity—esculetin, osthole, chartreusin, fraxin, imperatorin, umbelliferone, daphnetin, auroptene, psoralen, oxypeucedanin, seselin, etc. (Sharifi-Rad et al. 2021; Önder 2020). Owing to the potency of coumarin scaffold, various synthetic derivatives of coumarins have also been synthesized that are reported to exhibit potent antitumour and anticancer potential (Abolibda et al. 2023; Alshabanah et al. 2022; Gomha et al. 2018). Coumarins act as anticancer agents via different mechanisms—inhibition of mitosis, cell-cycle arrest, angiogenesis, inhibition of telomerase, hsp90, etc. (Önder 2020; Küpeli Akkol et al. 2020). Among various anticancer mechanisms of coumarins, only a few studies have been done on their anti-angiogenic activity (Naderi Alizadeh et al. 2018; Al-Warhi et al. 2020).
Vascular endothelial growth factor receptors (VEGFRs) are a type of receptor tyrosine kinases involved in angiogenesis and bind to Vascular Endothelial Growth Factors (VEGFs) or vasculotropins to produce their angiogenic effect and aid tumour growth and survival (Lugano et al. 2020; Pinedo and Slamon 2000). There are three types of VEGFRs-1, 2, and 3, and among them, VEGFR-2 is expressed on most epithelial cells and, hence, principally involved in angiogenesis during cancer (Hicklin and Ellis 2005). The human VEGFR-2 receptor is a 1356 amino acid protein containing kinase insert domain (KDR). The VEGFR-2 in mature form is a transmembrane protein that is responsible for intracellular signal transduction (Wang et al. 2020). The signalling pathway of VEGFR-2 involves its binding with VEGF-A, which results in dimerization of VEGFR-2. Following this, autophosphorylation of tyrosine residues occurs in the receptor's intracellular region, causing activation of downstream signalling molecules and their subsequent pathways (Nilsson and Heymach 2006). The signalling transduction pathways that get activated are the PLCγ-PKC-MEK-ERK pathway (involved in cell proliferation), TSAd-Src-PI3K-Akt pathway (involved in cell survival), PLCγ-PKC-eNOS-NO and TSAd-Src-PI3K-Akt pathway (involved in cell permeability); SHB-PI3K-Rac, SHB-FAK-paxillin, and NCK-p38-MAPKAPK2/3 pathway (involved in cell migration) (Wang et al. 2020).
Angiogenesis is the primary requisite of tumours for their sustenance, growth, proliferation, and survival; hence, they interfere with the body’s homeostasis and increase the secretion of angiogenic factors. This results in overexpression and increased kinase activity and expression of VEGFR-2 owing to the increase in the secretion of VEGFs (Peng et al. 2017). Thus, the inhibition of VEGFR-2 is considered an attractive approach to target tumour-related angiogenesis and subsequent tumour growth (S et al. 2023). Several FDA-approved VEGFR-2 inhibitors—Sorafenib, Lenvatinib, Sunitinib, Vandetanib, and Axitinib—are already used for different types of cancers. These inhibitors exert their action by blocking the VEGF-mediated activation of VEGFR-2 receptors and, thus, inhibit subsequent signalling (Aziz et al. 2016).
In this work, we conducted a pharmacophore-based virtual screening to identify the potential coumarin leads from the COCONUT database of natural products against vascular endothelial growth factor receptor-2 (VEGFR-2), an angiogenesis-related target for cancer. Based on a literature search, we found that no high-throughput virtual screening studies are performed on a plant-derived extensive database of natural coumarins against the VEGFR-2 protein. So, in this study, we tried to identify natural coumarins as potential inhibitors of the VEGFR-2 receptor by rigorous in-silico studies. Our approach encompassed several key methodologies—feature-based pharmacophore screening of the coumarin database, high-throughput docking studies, and molecular dynamics studies on the identified hits for determining potent angiogenic inhibitors. This study expands the scope of researchers to work on natural coumarins as anticancer agents, as they might have the potential to inhibit VEGFR-2 selectively.
Material and method
Database
The COlleCtion of Open Natural ProdUcTs (COCONUT) database was used for performing pharmacophore-based virtual screening (Sorokina et al. 2021). The database consisted of 407270 entries of natural products and was processed in the KNIME analytics platform (Berthold et al. 2008) (https://www.knime.com/) to obtain molecules with only a ‘coumarin’ scaffold. The ‘substructure search’ node was used to accomplish this, where the coumarin scaffold was drawn and executed. Later, the duplicate ligands were filtered out using the ‘duplicate row filter’ node. This resulted in 20,614 molecules with coumarin scaffold that were used for further studies.
e-Pharmacophore generation and pharmacophore-based virtual screening
The crystallographic structure of VEGFR-2 protein complexed with multikinase inhibitor lenvatinib (PDB: 3WZD) was obtained from the protein data bank in PDB format (Berman et al. 2000) (https://www.rcsb.org/). The PDB 3WZD was selected as it was the only entry in protein data bank where lenvatinib was complexed with VEGFR-2. The chosen PDB had a resolution of 1.57 Å and R-value work of 0.181. The structure of VEGFR-2 with co-crystallized ligand and active site is shown in Fig. 1. The interaction of lenvatinib with VEGFR-2 was studied on Discovery Studio 2021. The pharmacophoric features of lenvatinib interacting with receptors such as hydrophobic, hydrogen-bond acceptor, hydrogen-bond donor, and aromatic ring were chosen for pharmacophore model generation.

Crystal structure of VEGFR-2 protein (PDB: 3WZD) in complex with lenvatinib
The pharmacophore-based virtual screening of coumarins using pharmacophoric features of lenvatinib was performed using the Pharmit server (Sunseri and Koes 2016) (https://pharmit.csb.pitt.edu/). In the server, the pharmacophoric search was limited to ‘one conformer per molecule’ and ‘one molecule per hit.’
Homology modelling and model validation
Homology modelling is a highly accurate and successful method for predicting the tertiary structure of query protein using template protein and sequence alignment (Skariyachan and Garka 2018). It is based on the assumption that proteins share structural similarity owing to common evolutionary ancestry, and based on this concept the 3D structure of the target protein could be determined using known homologous protein as a template (Kumar and Sharma 2023). The VEGFR-2 protein structure was viewed in UCSF Chimera 1.14 to check missing residues (Pettersen et al. 2004). The missing residues in the protein were filled using homology modelling performed via SwissModel (https://swissmodel.expasy.org/) using the user template interface (Waterhouse et al. 2018). The FASTA sequence of the protein was obtained from a protein data bank, and the PDB: 3WZD was used as a PDB template to build the homology model.
The quality of the original protein and modelled protein was evaluated and compared to check the accuracy of the homology build protein. The SwissModel server provides QMEAN and the global model quality estimate (GMQE) parameters to assess the quality. Additionally, other freely available web tools, viz. Molprobity and SAVESv6.0 were also used. Molprobity is an easily accessible web server to validate the protein structure (Williams et al. 2018) (http://molprobity.biochem.duke.edu). The protein validation parameters of Molprobity are protein geometry (Ramachandran plot, Cβ deviation, rotamers, MolProbity score) and all-atom contacts (Clashscore). The SAVESv6.0 is a protein structure validation server consisting of different protein validation programs, viz. ERRAT, VERIFY 3D, and PROCHECK (https://saves.mbi.ucla.edu/). ERRAT evaluates improvement and refinement in the modelled protein structure (Skariyachan and Garka 2018). PROCHECK evaluates the quality of modelled protein by Ramachandran plot, bond angle, torsion angles, atomic distances, and surface area (Skariyachan and Garka 2018).
Energy minimization and preparation of protein
The protein energy minimization helps to achieve an energetically favourable structure by bringing the structure close to its native local minima (Jabeen et al. 2019). PDB2PQR server was used to assign the correct protonation state to the protein residues at pH 7.4 (Dolinsky et al. 2004) (https://server.poissonboltzmann.org/pdb2pqr). In this study, we performed the energy minimization of the modelled VEGFR-2 protein using the Pmemd module of Amber 20. The energy minimization parameters were 1000 steps of conjugate gradient, 4000 steps of steepest descent, and 0.02 Å step size. After energy minimization, the protein was further processed in AutoDockTools-1.5.7 by adding polar hydrogens only, merging non-polar hydrogens, adding Gastieger charge, and finally converted into PDBQT format (Tripathi et al. 2022).
Ligand preparation
The ligands obtained after pharmacophore-based virtual screening and PAINS filtering were processed before docking studies. The first step included ligand energy minimization to remove stearic clashes in the ligand and bring them to energetically favourable spatial conformation. It was done via Open Bable 3.1.1 software (O'Boyle et al. 2011). Finally, the minimized ligands were transformed to AutoDock compatible format.
Grid generation and validation
The grid generation is crucial as it sets the docking boundary and specifies binding sites where ligand interaction might occur (Swetha et al. 2022). The interaction of lenvatinib and VEGFR-2 was visualized using Discovery Studio 2021. The interacting residues were used as reference points for building a grid box around the active site (Singh et al. 2022). The grid maps were calculated using Autogrid 4.0 (Morris et al. 2009). The grid size of the minimized protein was set at XYZ points 48 × 62 × 76 with a grid spacing of 0.375 Å, and the grid centre was set at x, y, and z coordinates 1.311, −6.175, and 15.419, respectively. The grid validation was done by redocking lenvatinib with VEGFR-2. It is essential to achieve valid, reproducible docking results (Swetha et al. 2022). The root-mean square deviation (RMSD) between heavy atoms of the co-crystallized lenvatinib and redocked lenvatinib was calculated using the Desmond Maestro package (D.E. Shaw Research, New York, USA).
High-throughput virtual screening (HTVS) and molecular docking study
HTVS is a computational screening methodology complementary to high-throughput screening (HTS). HTVS is extensively used for screening big libraries of compounds to determine the binding affinity of the compounds with target receptor and uses a scoring function to do so. (Subramaniam et al. 2008). Molecular docking is an important part of structure-based drug design that aids in determining ligand–protein interaction and characterizing how ligand acts in the binding pocket of protein. Molecular docking gives valuable information regarding binding affinity, conformation, position, and orientation of ligand in the binding site of the target protein (Meng et al. 2011).
The docking study was performed using AutoDock 4.2.6 and Lamarckian Genetic Algorithm (LGA) in three steps: (1) HTVS, (2) Standard precision (SP) docking, and (3) Extra precision (XP) docking. The coumarins were docked against VEGFR-2, and in each proceeding step, the docking protocol was made stringent to reduce the probability of false positives. The visualization and analysis of docking was performed using Discovery Studio 2021.
In-silico ADMET prediction
The absorption, distribution, metabolism, elimination, and toxicity (ADMET) profile of compounds is an essential aspect in drug discovery as it is the primary determinant of the clinical efficacy of the potential compound. The ADMET determination of compounds at initial stage of the drug discovery process helps in reducing failure at the final stages (Ghosh et al. 2022). The in-silico ADMET estimation of the final hits was done using the freely accessible web tool PreADMET (https://preadmet.webservice.bmdrc.org/) and SwissADME (Daina et al. 2017).
Molecular dynamics (MD) simulation study
Molecular dynamic simulation is an important part of drug discovery as it provides insights into the motion of protein at different time intervals which could not be provided by NMR, XRD, etc. (Durrant and McCammon 2011). When a drug binds to the binding pocket of a target protein it gives rise to conformational changes in protein structure, interferes with its normal function, and exerts agonistic or antagonistic action. Hence, to simulate the body’s environment at the initial developmental stage, MD simulation studies are performed. Thus, molecular docking and molecular dynamics studies are closely intertwined for screening of potential drug-like candidates.
The MD studies of final hits with binding energy less than lenvatinib and cluster size more than lenvatinib were performed via Amber 20. The ligand–protein complex was obtained from Discovery Studio 2021. The complex's parameter and coordinate files were created using tleap and the Amber ff14SB force field. Utilizing TIP3P water molecules in a cubic periodic box with a 10 Å edge length, the refined structures were solvated. Sodium ions were added to the system to balance out its overall charge. Long-range electrostatic interactions were handled using the Particle Mesh Ewald (PME) approach and a non-bonded cut-off of 10 Å (Singh et al. 2022). To refine the system, energy minimization was performed using a combination of steepest descent and conjugate gradient algorithms. The system was heated to 310 K using a Langevin thermostat and the collision frequency was tuned to 5 ps−1 using the NVT ensemble. Once the desired temperature was reached, a 100 ps molecular dynamics (MD) run was carried out in the NPT ensemble at a pressure of one bar, using the Berendsen barostat. Subsequently, a 100 ns production MD simulation was performed once the system reached equilibrium. Using the cpptraj tool of Amber 20, the trajectories acquired from the MD simulations were examined for root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), and radius of gyration (Rg) studies (Tripathi et al. 2022).
Binding free energies
The binding free energy is the difference in free energies of bound and unbound states, where the bound state is the explicit complex state and the unbound state is the one where protein–ligand interaction is negligible (Hata et al. 2021). The calculation of binding free energy was executed via MMPBSA.py package of Amber 20 and comprised of molecular mechanics, Poisson-Boltzmann surface area (MM-PBSA) and molecular mechanics, Generalised Born model for solvent accessibility (MM-GBSA). The energy calculation was done using GB method in the last 50 ns of MD simulation at 10 ns interval. The salt concentration, inner dielectric, and exterior dielectric were set at 0.1 nM, 1, and 80, respectively (Singh et al. 2022).
Results
e-Pharmacophore generation and pharmacophore-based virtual screening
Lenvatinib is a small molecule multiple kinase inhibitor with IC50 value range between 4–100 nm (Hao and Wang 2020). Figure 2 shows the pharmacophoric features of lenvatinib used for generating e-pharmacophore. The pharmacophore-based virtual screening of the COCONUT-coumarin database using this e-pharmacophore resulted in 6301 hits. The RMSD cut off was set at 0.5 Å and the molecules with RMSD less than that were selected for HTVS. This resulted in the final 910 hits.

Pharmacophoric features of lenvatinib selected for virtual screening (A represents Hydrogen-bond acceptor, H represents Hydrophobic, R represents Aromatic Ring)
Homology modelling and model validation
The structure alignment between the template and modelled protein was checked by superimposing them using ‘Protein Structure Alignment’ in Desmond Maestro. The alignment score and RMSD of the modelled VEGFR-2 protein were found to be 0.007 and 0.414 Å, respectively. The sequence alignment of template and modelled proteins have been provided in Supplementary Material (Figure S2). The QMEAN Z-score was 0.89 ± 0.05 and validation results are provided in Supplemental Material (Figure S3).
The Ramachandran plot obtained from the Molprobity server showed 99.7% residues in the allowed region and only 0.3% in the disallowed region with only one outlier residue, i.e. Asp 260. The Ramachandran plot obtained from PROCHECK indicated no residues in the disallowed region (Fig. 3).

Ramachandran plot of homology model of VEGFR-2 protein obtained from—a Molprobity and b PROCHECK
The Rama Z-score is a global metric for assessing the overall quality of homology-built model and a score between − 2 to 2 is indicative of the normal backbone geometry of the model. The modelled protein had a Rama Z-score value of 1.49 ± 0.49, which was within permissible limits (Sobolev et al. 2020). The Cβ deviation is the difference between the predicted position of the Cβ atom and its ideal expected position. The Cβ deviation greater than 0.25 Å indicates incompatibility in side chain and main chain conformation. No Cβ deviation (> 0.25 Å) was found in the modelled protein (Davis et al. 2007). The Clashscore is number of serious stearic clashes (> 0.4 Å) per 1000 atoms including hydrogen atoms, and for modelled protein it was 1.03 which is considered very well. MolProbity score is the weighted sum of clashes, Ramachandran favoured, and rotamer outliers, and for modelled protein it was 0.80 (Williams et al. 2018).
ERRAT assesses the statistical properties of non-bonded interactions among various atom types within a model protein, comparing them to a database of dependable high-resolution structures (Colovos and Yeates 1993). The overall quality factor of the modelled protein on the ERRAT program was found to be 98.639 and the plot is shown in Supplemental Material (Figure S4). The VERIFY3D assesses how well a 3D atomic model aligns with its corresponding 1D amino acid sequence (Jitendra and Vinay 2011). In this, at least 80% amino acids of the model should score > = 0.1 in 3D/1D profile for model to be considered good. In the modelled VEGFR-2 protein, 85.48% of the residues were found to have averaged 3D/1D score > = 0.1 and the plot is shown in Supplemental Material (Figure S4).
Energy minimization and preparation of protein
The potential energy of the system was minimized from initial − 276,910 kcal/mol to − 420,330 kcal/mol and the graph depicting the same has been provided in the Supplementary Material (Figure S5). Following minimization, the protein was saved into PDBQT format (Singh et al. 2022).
Grid generation and validation
Lenvatinib displayed hydrophobic and hydrogen bond interactions with the active site residues of VEGFR-2, viz. Glu885, Ile888, Leu889, Cys919, Cys1045, Asp1046, Phe1047. The RMSD between the two pose was found to be 0.9280 Å and the poses have been shown in the Supplemental Material (Figure S10).
High-throughput virtual screening (HTVS) and molecular docking study
The coumarin ligands from the COCONUT database were docked against the VEGFR-2 receptor protein (PDB Id: 3WZD). Lenvatinib was employed as an internal reference for selecting the compounds. In HTVS, the ligands having binding energy less than lenvatinib were filtered out for further SP docking. The HTVS was performed on a total 909 ligands and from that, 485 compounds were selected for SP docking. From SP docking, 30 compounds were chosen for XP docking as their binding energy was less than reference standard. Through XP docking, the compounds were tapered to 12 and the docking was analysed and visualized using Discovery Studio. Among 12 compounds, three compounds viz. CNP0056360, CNP0340213, and CNP036628 (Fig. 4) with good binding energy, cluster size (Table 1) and ADMET properties were selected for further molecular dynamic studies. The interaction diagrams of CNP0056360, CNP0340213, and CNP036628 with VEGFR-2 protein along with lenvatinib are shown in Fig. 5. The binding energy, ligand efficiency, and cluster size of the rest of the compounds is provided in Supplemental Material (Table S1).

Structures of final hits and standard

3D and 2D interaction diagram of best hits obtained through HTVS with VEGFR-2 protein (PDB Id. 3WZD). a Lenvatinib, b CNP0056360, c CNP0340213, and d CNP0366287
In-silico ADMET prediction
The in-silico ADMET profiling gives primitive information about the pharmacokinetics of virtually identified leads. The different parameters viz. Blood–brain barrier (BBB) penetration, Aqueous solubility, Human Intestinal Absorption (HIA), human ether-a-go-go-related gene (hERG) inhibition, rodent carcinogenicity, plasma protein binding (PPB%), CYP2D6 inhibition, and Rule of Five (Ro5) violation are important in deciding if the molecule will be successful in drug discovery pipeline. The BBB penetration is a crucial parameter as the CNS active drugs are required to penetrate BBB, while CNS inactive drugs should not cross BBB. The classification used by PreADMET for BBB penetration is BB (Cbrain/Cblood) > 2.0—High CNS absorption, between 2.0 to 0.1—Moderate CNS absorption, and < 0.1—Low CNS absorption (Ma et al. 2005). The PPB controls the amount of drug available in plasma for pharmacological action. According to PreADMET, PPB% > 90% is strong binding, while PPB% < 90% is weak binding. The aqueous solubility of a drug is essential for its absorption and subsequent action. HIA represents the absorption of orally administered drugs from the gastrointestinal system into the bloodstream. The %HIA between 0–20, 20–70, and 70–100 indicates poor, moderate, and high absorption, respectively. The hERG inhibition results in cardiotoxicity by prolongation of the QT interval. Hence, it is important to determine if a lead has high or medium risk for hERG inhibition. CYP2D6 enzymes are responsible for the metabolism of approximately 20% of drugs, and thus, their inhibition will reduce drug metabolism and increase toxicity. Rodent carcinogenicity is determined to extrapolate its carcinogenic effects in humans. The Rule of Five (Ro5) is considered important to predict the drug-likeness of a molecule. The general paradigm is—for a molecule to become drug-like, it must fall within the permissible limits of Ro5 but, in many natural products inspired FDA approved drugs this is not true. The ADMET profile of compounds CNP0056360, CNP0340213, and CNP036628 was comparatively better than rest of the compounds as shown in Table 2. The in-silico ADMET properties of rest of the compounds is provided in Supplemental Material (Table S2).
Molecular dynamics (MD) simulation study
To check the binding of CNP0056360, CNP0340213, and CNP036628 with VEGFR-2 in a real time simulated environment and study the ligand–protein interaction in detail, the molecular dynamics studies were performed.
The RMSD is the measure of average deviation in the position or coordinates of an atom in a particular frame compared to the reference frame (Ganeshpurkar et al. 2020). Typically, the perfect overlap of structure is inevitable, so it is desirable to obtain minimum deviation to ensure protein stability. One can understand the protein’s structural conformation by monitoring its RMSD throughout the MD simulation. The average RMSD of protein between 1–3 Å is considered acceptable for small globular proteins. The ligand RMSD gives insights into the ligand–protein interactions as it enables assessment of stability about protein and its binding pocket. When the ligand RMSD values significantly exceed that of protein, it indicates that the ligand may have moved away from the binding pocket.
The average RMSD for VEGFR-2 complexed with CNP0056360, CNP0340213, CNP036628, and lenvatinib was found to be 1.0161, 1.7137, 1.6695, and 1.5233 Å, respectively. CNP0056360-VEGFR-2 complex exhibited the lowest RMSD, even smaller than lenvatinib indicating better stability of protein-ligand complex. The RMSD of VEGFR-2 bound to other two ligands were slightly higher than the Lenvatinib-VEGFR-2 complex. The mean ligand RMSD for CNP0056360, CNP0340213, CNP036628, and lenvatinib when bound to VEGFR-2 was found to be 1.4949, 0.9353, 1.6727, and 0.8356 Å, respectively. There was not much variation between ligand and protein RMSD of three ligands and lenvatinib, indicating that ligands didn’t diffuse out of the binding pocket during the simulation run (Fig. 6).

a RMSD of the protein–ligand complexes (Lenvatinib (red), CNP0056360 (green), CNP0340213 (purple), CNP0366287 (blue)). b RMSD of heavy atoms of the ligands (Lenvatinib (red), CNP0056360 (green), CNP0340213 (purple), CNP0366287 (blue))
The RMSF monitoring gives an idea about the flexibility and local changes occurring in the protein during the simulation run. In general, a high fluctuation is observed in the C- and N-terminal compared to other parts. In the RMSF plot, peaks specify protein residues that fluctuate most. The average RMSF values for VEGFR-2 complexed with CNP0056360, CNP0340213, CNP036628, and lenvatinib were 1.105, 1.1258, 1.1194, and 1.128 Å, respectively. The high fluctuations were observed in the tail region which are inevitable. The high fluctuations near residues 840–890, 915–925, and 1035–1050 could be observed in Fig. 7 because they are active site residues and involved in interacting with the ligands. The residues Cys919 and Asp1046 are involved in H-bond interaction, showing high fluctuations in the RMSF plot. Other than active site residues, the rest of the protein was stable throughout the run.

Residue-wise RMSF deviation of complexes (Lenvatinib (red), CNP0056360 (green), CNP0340213 (purple), CNP0366287 (blue))
Hydrogen bonds are crucially involved in the binding of ligands with the protein active site. The H-bond analysis is important as it highly influences absorption and metabolism of drugs. The H-bond is also essential for stabilizing the protein–ligand complex and is indicative of strong and stable binding. Lenvatinib showed a maximum two H-bond interactions present at most of the simulation time. In the case of CNP0056360, the maximum number of H-bond interactions observed was four that kept varying between two and three, and one H-bond was present at most of the run. Except for CNP0056360, no ligand including lenvatinib showed four H-bond interactions. For CNP0340213, three H-bond interactions were observed at the start of the simulation that were reduced to two and subsequently one. In CNP0366287, one H-bond interaction was present for almost whole run, while two and three H-bonds were present only for a brief period of time (Fig. 8).

Number of intermolecular H-bond interactions shown by the ligand during simulation run a CNP0056360 b CNP0340213 c CNP0366287 d Lenvatinib
The radius of gyration (Rg) is done to know how protein atoms are distributed around the axis of protein or its extendedness (Sneha and George Priya Doss 2016). Since, it is a basic measurement of the overall size of a protein chain, Rg analysis is done to get information regarding conformational change in protein structure (Jiang et al. 2019). The average Rg values for CNP0056360-VEGFR-2 complex, CNP0340213-VEGFR-2 complex, CNP0366287-VEGFR-2 complex, and Lenvatinib-VEGFR-2 complex were 20.2873, 20.3322, 20.3348, and 20.4940 Å, respectively (Fig. 9).

The radius of gyration of complexes (Lenvatinib (red), CNP0056360 (green), CNP0340213 (purple), CNP0366287 (blue))
Binding free energies
The prediction of binding energy between ligand and protein is achieved through free energy calculations, which encompass a set of techniques. The binding-free energy calculations provide a semi-qualitative assessment of binding affinity of a ligand with active site of protein. These methods analyse the atomic-level interactions that contribute to the affinity of the ligand for the protein (Ganeshpurkar et al. 2020). Within the framework of MM-GBSA and MM-PBSA calculations, the calculation involves the assessment of both molecular mechanics (MM) and solvation energies. The solvation energy encompasses two components: a polar component [evaluated using an implicit solvent model (PB or GB)] and a non-polar component [determine the solvent-accessible surface area (SASA)] (Singh et al. 2022).
Tables 3 and 4 display the non-bonded interaction energies (van der Waals energy—ΔEvdw, electrostatic energy—ΔEele, polar part of solvation free energy—ΔGPB, non-polar part of solvation free energy—ΔGNonpolar, and total binding free energy—ΔGMMPBSA) for all the complexes analysed using the MM-GBSA and MM-PBSA methods, respectively. Both approaches demonstrated low overall binding energies, indicating a high level of stability in the complexes. The gas phase energies, specifically ΔEvdw and ΔEele, substantially contribute to the overall stability, suggesting that confirming the ligand relative to the receptor is the primary factor in complex stability. Among the identified compounds, CNP0056360 and CNP0340213 exhibited stronger binding free energy than lenvatinib in the GB model. In the PB model, CNP0056360 and CNP0340213 exhibited stronger binding free energy than lenvatinib.
Discussion
This research investigates potential coumarin lead compounds from the COCONUT database as VEGFR-2 inhibitors, a crucial target in cancer treatment. The study employed a multi-stage virtual screening approach (HTVS, SP, XP docking) to identify promising coumarin leads from a large database. Three compounds—CNP0056360, CNP0340213, and CNP036628—emerged with favourable binding energies and ADMET properties. In-silico ADMET profiling provided preliminary insights into the pharmacokinetics of these leads. CNP0056360, CNP0340213, and CNP036628 displayed good ADMET profiling. The study further evaluated the stability and binding dynamics of the leads using molecular dynamics simulations. All four complexes exhibited good stability throughout the simulation run, as indicated by RMSD and RMSF analyses. CNP0056360 formed the most stable complex with VEGFR-2, showing lower RMSD values than even lenvatinib. H-bond analysis revealed varying degrees of interaction between the ligands and VEGFR-2. CNP0056360 formed the most extensive and consistent H-bond interactions, potentially contributing to its higher stability. Rg analysis suggested minimal conformational changes in the protein structure upon ligand binding, further supporting the stability of the complexes. MM-GBSA and MM-PBSA methods predicted low overall binding energies for all complexes, indicating stable and strong ligand-receptor interactions. Gas phase energies (ΔEvdw and ΔEele) significantly contributed to the binding stability, emphasizing the importance of ligand conformation relative to the receptor. Notably, CNP0056360 and CNP0340213 demonstrated stronger binding free energies than lenvatinib in both GB and PB solvation models, suggesting potentially superior binding affinities. Overall, these leads displayed favourable binding profiles, good stability in complex with VEGFR-2, and potentially superior binding affinities compared to lenvatinib.
Conclusion
In this study, the COCONUT database was subjected to rigorous pharmacophore-based in-silico investigation to identify coumarin leads as VEGFR-2 inhibitors using lenvatinib as a standard for comparison. The specific cut-off for binding energy and cluster size resulted in three hits—CNP0056360, CNP0340213, and CNP0366287 and these ligands possessed acceptable ADMET properties in in-silico ADMET estimation. Molecular dynamics simulations of all three hits demonstrated favourable stability when bound to the VEGFR-2 in a simulated biological environment. The binding free energy calculations also revealed the superior binding free energies of CNP0056360 and CNP0340213 compared to lenvatinib in both solvation models. The obtained results demonstrate the effectiveness and potential of identified leads as inhibitors of VEGFR-2, a crucial target in cancer treatment. These coumarin leads have shown successful inhibition of VEGFR-2 receptors in in-silico studies. While, further in-vitro and in-vivo studies are necessary to validate these findings, the current results establish a strong foundation for developing novel coumarin-based VEGFR-2 inhibitors for cancer therapy.
Future perspective
The study identified three promising natural coumarin leads (CNP0056360, CNP0340213, and CNP0366287) from the COCONUT database. As natural coumarins are relatively unexplored as VEGFR-2 inhibitors, the study highlights their potential for new anticancer therapeutic strategies. While, the in-silico results are encouraging, further chemical modifications on the identified leads that might optimize their binding affinity and pharmacokinetics could be explored, leading to even more potent VEGFR-2 inhibitors. As VEGFR-2 is involved in various cancers, investigating the lead compounds against specific cancer types with VEGFR-2 overexpression could give more precise insights into their role as anti-angiogenic agents. Overall, this research lays a strong foundation for further developing coumarin-based VEGFR-2 inhibitors. With successful experimental validation and continued research, these leads have the potential to translate into novel and effective cancer treatment options.
Data availability
The files associated with the research could be freely assessed on “https://zenodo.org/records/10646050”.
References
Abolibda TZ, Fathalla M, Farag B, Zaki MEA, Gomha SM (2023) Synthesis and molecular docking of some novel 3-Thiazolyl-coumarins as inhibitors of VEGFR-2 Kinase. Molecules [Online], 28
Alshabanah LA, Al-Mutabagani LA, Gomha SM, Ahmed HA (2022) Three-component synthesis of some new coumarin derivatives as anticancer agents. Front Chem 9:762248
Al-Warhi T, Sabt A, Elkaeed EB, Eldehna WM (2020) Recent advancements of coumarin-based anticancer agents: an up-to-date review. Bioorg Chem 103:104163
Annunziata F, Pinna C, Dallavalle S, Tamborini L, Pinto A (2020) An overview of coumarin as a versatile and readily accessible scaffold with broad-ranging biological activities. Int J Mol Sci [Online] 21
Aziz MA, Serya RAT, Lasheen DS, Abdel-Aziz AK, Esmat A, Mansour AM, Singab ANB, Abouzid KAM (2016) Discovery of potent VEGFR-2 inhibitors based on furopyrimidine and thienopyrimidne scaffolds as cancer targeting agents. Sci Rep 6:24460
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The protein data bank. Nucl Acids Res 28:235–242
Berthold MR, Cebron N, Dill F, Gabriel TR, Kötter T, Meinl T, Ohl P, Sieb C, Thiel K, Wiswedel B (2008) KNIME: the Konstanz information miner. In: Data analysis, machine learning and applications. Springer, Berlin pp 319–326
Colovos C, Yeates TO (1993) Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci 2:1511–1519
Daina A, Michielin O, Zoete V (2017) SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 7:42717
Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB III, Snoeyink J, Richardson JS, Richardson DC (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucl Acids Res 35:W375–W383
Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA (2004) PDB2PQR: an automated pipeline for the setup of Poisson–Boltzmann electrostatics calculations. Nucl Acids Res 32:W665–W667
Durrant JD, McCammon JA (2011) Molecular dynamics simulations and drug discovery. BMC Biol 9:71
Ganeshpurkar A, Singh R, Gore PG, Kumar D, Gutti G, Kumar A, Singh SK (2020) Structure-based screening and molecular dynamics simulation studies for the identification of potential acetylcholinesterase inhibitors. Mol Simul 46:169–185
Ghosh P, Singh R, Ganeshpurkar A, Swetha R, Kumar D, Singh SK, Kumar A (2022) Identification of potential death-associated protein kinase-1 (DAPK1) inhibitors by an integrated ligand-based and structure-based computational drug design approach. J Biomol Struct Dyn 1–13
Gomha SM, Abdel-Aziz HM, El-Reedy AAM (2018) Facile synthesis of Pyrazolo[3,4-c]pyrazoles bearing coumarine ring as anticancer agents. J Heterocycl Chem 55:1960–1965
Hao Z, Wang P (2020) Lenvatinib in management of solid tumors. Oncologist 25:e302–e310
Hata H, Phuoc Tran D, MarzoukSobeh M, Kitao A (2021) Binding free energy of protein/ligand complexes calculated using dissociation parallel cascade selection molecular dynamics and Markov state model. Biophys Physicobiol 18:305–316
Hicklin DJ, Ellis LM (2005) Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol 23:1011–1027
Jabeen A, Mohamedali A, Ranganathan S (2019) Protocol for Protein structure modelling. Encyclopedia of bioinformatics and computational biology. Academic Press, Oxford
Jiang Z, You L, Dou W, Sun T, Xu P (2019) Effects of an electric field on the conformational transition of the protein: a molecular dynamics simulation study. Polymers [Online] 11
Jitendra S, Vinay R (2011) Structure based drug designing of a novel antiflaviviral inhibitor for nonstructural 3 protein. Bioinformation 6:57–60
Kumar R, Sharma A (2023) Chapter 15—Computational strategies and tools for protein tertiary structure prediction. Basic biotechniques for bioprocess and bioentrepreneurship. Academic Press, Cambridge
Küpeli Akkol E, Genç Y, Karpuz B, Sobarzo-Sánchez E, Capasso R (2020) Coumarins and coumarin-related compounds in pharmacotherapy of cancer. Cancers [Online] 12
Lugano R, Ramachandran M, Dimberg A (2020) Tumor angiogenesis: causes, consequences, challenges and opportunities. Cell Mol Life Sci 77:1745–1770
Ma X-L, Chen C, Yang J (2005) Predictive model of blood-brain barrier penetration of organic compounds. Acta Pharmacol Sin 26:500–512
Maria João M, Lourdes S, Eugenio U, Orlando AA, Enrique M, Estela Guardado Y (2015) Coumarins—an important class of phytochemicals. Phytochemicals. IntechOpen
Meng XY, Zhang HX, Mezei M, Cui M (2011) Molecular docking: a powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des 7:146–157
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 30:2785–2791
NaderiAlizadeh M, Rashidi M, Muhammadnejad A, MoeiniZanjani T, Ziai SA (2018) Antitumor effects of umbelliprenin in a mouse model of colorectal cancer. Iran J Pharm Res 17:976–985
Nilsson M, Heymach JV (2006) Vascular endothelial growth factor (VEGF) pathway. J Thorac Oncol 1:768–770
O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR (2011) Open babel: an open chemical toolbox. J Cheminformatics 3:33
Önder A (2020) Chapter 3—Anticancer activity of natural coumarins for biological targets. In: Atta Ur R (ed) Studies in natural products chemistry. Elsevier, Amsterdam
Peng F-W, Liu D-K, Zhang Q-W, Xu Y-G, Shi L (2017) VEGFR-2 inhibitors and the therapeutic applications thereof: a patent review (2012–2016). Expert Opin Ther Pat 27:987–1004
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera—A visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612
Pinedo HM, Slamon DJ (2000) INTRODUCTION: Translational Research: The Role of VEGF in Tumor Angiogenesis. Oncologist 5:1–2
S V, Kajal K, Mondal S, Wahan SK, Das Kurmi B, Das Gupta G, Patel P (2023) Novel VEGFR-2 kinase inhibitors as anticancer agents: a review focusing on SAR and molecular docking studies (2016–2021). Chem Biodivers 20:e202200847
Sharifi-Rad J, Cruz-Martins N, López-Jornet P, Lopez EP-F, Harun N, Yeskaliyeva B, Beyatli A, Sytar O, Shaheen S, Sharopov F, Taheri Y, Docea AO, Calina D, Cho WC (2021) Natural coumarins: exploring the pharmacological complexity and underlying molecular mechanisms. Oxid Med Cell Longev 2021:6492346
Singh R, Pokle AV, Ghosh P, Ganeshpurkar A, Swetha R, Singh SK, Kumar A (2022) Pharmacophore-based virtual screening, molecular docking and molecular dynamics simulations study for the identification of LIM kinase-1 inhibitors. J Biomol Struct Dyn 41:1–15
Skariyachan S, Garka S (2018) Chapter 1—Exploring the binding potential of carbon nanotubes and fullerene towards major drug targets of multidrug resistant bacterial pathogens and their utility as novel therapeutic agents. In: Grumezescu AM (ed) Fullerens graphenes and nanotubes. William Andrew Publishing
Sneha P, George Priya Doss C (2016) Chapter seven—Molecular dynamics: new frontier in personalized medicine. In: Donev R (ed) Advances in protein chemistry and structural biology. Academic Press
Sobolev OV, Afonine PV, Moriarty NW, Hekkelman ML, Joosten RP, Perrakis A, Adams PD (2020) A global Ramachandran score identifies protein structures with unlikely stereochemistry. Structure 28:1249-1258.e2
Sorokina M, Merseburger P, Rajan K, Yirik MA, Steinbeck C (2021) COCONUT online: collection of open natural products database. J Cheminformatics 13:1–13
Subramaniam S, Mehrotra M, Gupta D (2008) Virtual high throughput screening (vHTS)—a perspective. Bioinformation 3:14–17
Sunseri J, Koes DR (2016) Pharmit: interactive exploration of chemical space. Nucl Acids Res 44:W442–W448
Swetha R, Sharma A, Singh R, Ganeshpurkar A, Kumar D, Kumar A, Singh SK (2022) Combined ligand-based and structure-based design of PDE 9A inhibitors against Alzheimer’s disease. Mol Divers 26:2877–2892
Tripathi N, Goel B, Bhardwaj N, Sahu B, Kumar H, Jain SK (2022) Virtual screening and molecular simulation study of natural products database for lead identification of novel coronavirus main protease inhibitors. J Biomol Struct Dyn 40:3655–3667
Wang X, Bove AM, Simone G, Ma B (2020) Molecular Bases of VEGFR-2-Mediated Physiological Function and Pathological Role. Front Cell Dev Biol 8:599281
Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, De Beer TAP, Rempfer C, Bordoli L, Lepore R, Schwede T (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucl Acids Res 46:W296–W303
Williams CJ, Headd JJ, Moriarty NW, Prisant MG, Videau LL, Deis LN, Verma V, Keedy DA, Hintze BJ, Chen VB, Jain S, Lewis SM, Arendall WB III, Snoeyink J, Adams PD, Lovell SC, Richardson JS, Richardson DC (2018) MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci 27:293–315
Acknowledgements
The authors extend their gratitude toward Professor David A. Case, Department of Chemistry & Chemical Biology, Rutgers University, New Jersey, USA, for granting a licence for Amber 20. NT and NB are also thankful to IIT (BHU) and Ministry of Education (MoE), New Delhi for providing teaching assistantship.
Funding
This work was partially supported by the SERB-CRG Grant (Grant No. CRG/2022/001637).
Author information
Authors and Affiliations
Contributions
Nancy Tripathi: Conceived, designed, and performed the experiments; Analyzed the data; Writing original draft. Nivedita Bhardwaj: Prepared figures and tables; Paper formatting. Bikarma Singh: Conceived and designed the experiments. Shreyans K. Jain: Conceived and designed the experiments; Supervision; Final paper review and formatting.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that the research was conducted without any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Tripathi, N., Bhardwaj, N., Singh, B. et al. In-silico identification of Coumarin-based natural compounds as potential VEGFR-2 inhibitors. Chem. Pap. 78, 4347–4360 (2024). https://doi.org/10.1007/s11696-024-03395-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11696-024-03395-5