
Abstract
This paper proposes that Ca2+ in steel slag (SS) has a weak early hydration activity when SS and granulated blast-furnace slag (GBFS) are used as alkali-activated cementitious materials. Herein, alkaline activators and Ca(OH)2 are used as indicators of Ca2+ reaction in the SS and GBFS system. It is found that the 28-day compressive strength of the cementitious material with 2 wt% Ca(OH)2 addition increased by 25.9% compared with the blank. Hydration products and microstructures were characterized by XRD, SEM/EDS, FTIR and TG/DTA. Results show that the addition of Ca(OH)2 increased the content of active calcium ions in the reaction system, which promotes the reaction and the formation of C–S–H gel, resulting in an enhanced compressive strength ultimately. It can be also concluded that SS is not suitable as a sole precursor for the production of building materials on a large scale due to the weak early activity of Ca2+ in SS.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Steel slag (SS) is an industry waste produced in the process of iron smelting. In China, the amount of SS is about 80 million tons (Wu et al. 1999; Li et al. 2011). The accumulation of SS results in serious environmental problems and social concerns (Huang et al. 2016). In this sense, the utilization of SS needs to be explored urgently (Li et al. 2013). Currently, steel slag is mainly used as road construction materials, building materials, agricultural fertilizers, etc. Most researchers are interested in the utilization of industry waste into construction field (Motz and Jens 2001; Altun and Yilmaz 2002; Shi and Qian 2000). The utilization of SS in construction is an important way to reuse this waste as a high-value product, which will be able to bring huge economic benefits and reduce the cost of building materials. Further researches on the reaction mechanism of steel slag can improve the utilization rate of steel slag and reduce its environmental impacts. SS can be also used as a granular material in road base or subbase, which is another promising approach widely used in Europe (Netinger et al. 2011) for the several reasons: (1) a large amount of SS can be used without constraint conditions, (2) the process for granular use is economically of technically mature, sound and simple, and (3) the attention on long-term stability under unconstrained conditions is less (Ivanka et al. 2016).
However, the use of SS has not been widely investigated as a precursor material for building materials. The most critical problems of the SS utilization as a building material are the volume instability and poor activity (Wang et al. 2010). During the molten iron processing, fluxes consisting of lime can easily enter into the furnace, which contains a certain amount of free lime (f-CaO) in SS and will finally result in volume expansion (Chen et al. 2015; Ahmedzade and Sengoz 2009). The free lime (f-CaO) present in the steel slag can react with water to form Ca(OH)2, resulting in a volume expansion of 100%. This is considered to be the essence of the volume expansion of the steel slag (Wang et al. 2010; Wang 2016).
In order to enlarge the feasibility of SS utilization, many studies were conducted on how to deal with the weak activity of Ca2+ of SS (Wang et al. 2010; Ameri and Behnood 2012; Li et al. 2010). One common methodology is SS aging over a specified period under the normal circumstances. However, the drawback of this aging method is that the procedure will be time-consuming. According to the regulations in Belgium and in the Netherlands, the qualified SS used in unbounded base layers of pavement will take one year for aging process. To reduce the aging time, steam SS aging method is developed in Japan (Ivanka et al. 2016). In this approach, the best condition to accelerate SS aging is that the steel slag is exposed to a pressure of 0.5 MPa for 3 h. Result shows that the volumetric change of SS is reduced dramatically. To further broaden the utilization SS, some researchers have currently applied SS to concrete and cementitious system as a mineral admixture (Collins and Sanjayan 1999; Hu et al. 2006; Peng et al. 2010; Rosales et al. 2017). It is explained that the hydration activity of SS can be improved by raising the fineness of its particles, the curing temperature and the pH value of hydration condition (Han et al. 2015).
The alkaline activation reaction products of solid aluminosilicate precursors are discussed, as shown in Fig. 1 (Provis and Bernal 2014). As for the low-calcium systems, the main binding phase has been observed to be amorphous (such as low-calcium fly ash and metakaolin) N–A–S–H (N refers to monovalent cations such as K+ and Na+) (Provis and Bernal 2014; Němeček et al. 2011; Šmilauer et al. 2011). Amorphous N–A–S–H gel has similar chemical properties to natural zeolite (Li et al. 2010; Fernández-Jiménez et al. 2005). K+ and Na+ are embedded into the three-dimensional network frame structure space in the form of a balanced the charge of the phase to form a long disordered three-dimensional network zeolite structure (N–A–S–H) (Rovnaník 2010; Barbosa et al. 2000). An increased Ca/(Al + Si) ratio can promote the formation of C–A–S–H (such as slag, etc.) (Walkley et al. 2016; Nikolić et al. 2014; Kumar et al. 2010). It has been found that during the reaction, when Al forms a tetracoordinated [AlO4]5− tetrahedron, a positive potential compensation is required; then, Ca2+ can compensate for two [AlO4]5− tetrahedrons at the same time, so that the polymer is more tightly connected. It is believed that if sufficient calcium is added to a geopolymeric system, a C–S–H-based cementitious material may form instead (Mijarsh et al. 2015). However, if large amount of Ca2+ participate in the reaction too late, it will be present in the final product in a free state, which is not conducive to the polymerization of aluminosilicate.

Reaction process and products of calcium aluminosilicate precursors with different calcium content under alkaline conditions (Provis and Bernal 2014)
Herein, alkaline activator and Ca(OH)2 are used for the first time as a reaction indicator of Ca2+ in SS–GBFS system in this research, and different amounts of Ca(OH)2 were added into SS–GBFS system to investigate the early hydration of activity of Ca2+ in SS of SS–GBFS system. It provides a theoretical basis on the utilization of SS. XRD, SEM, FTIR and TG–DTA were used in order to investigate the hydration products and microstructure of the test block.
Experimental
Materials and characterizations
GBFS and SS from Huaiyin Iron & steel company (Huaiyin, PR China) were selected as raw materials in this study. The chemical compositions of the feedstocks are tested by X-ray fluorescence (XRF) spectroscopy, as presented in Table 1. The mineralogy of GBFS and SS is presented in Figs. 2 and 3, respectively. The XRD pattern of GBFS shows that GBFS is mostly composed of amorphous phases with minor peaks of crystalline mineral (Singh and Singh 2019). Figure 3 exhibits the XRD of SS and its main mineral components are C2S, C3S, C4AF, calcite and CaO–FeO–MnO–MgO solid solution (RO phase). The phase composition of SS and GBFS was calculated by external standard method, and the results are shown in Table 2.

XRD pattern of GBFS

XRD pattern of SS
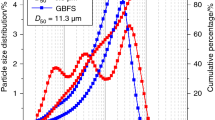
The density of GBFS is 2.62 g/cm3, and the specific surface area is 276 m2/kg determined using the Blaine specific surface area meter. The density and specific surface area of the SS are 3.24 g/cm3 and 411 m2/kg, respectively. The basicity coefficient of the SS is 2.58, and the stability is qualified. Particle size distribution of GBFS and SS is shown in Fig. 4. Water glass (WS) was supplied by Gulf Group Co., Ltd., Qingdao, China, and it contains 64% water with a SiO2/Na2O ratio of 3.3. Sodium hydroxide with purity of around 98% was purchased from Jiangsu Tongsheng Co., Ltd. (Wuxi, China) for regulating the modulus of sodium silicate solution. Calcium hydroxide (analytical grade, > 98%) was purchased from Damao Chemical Reagent Factory, Tianjin, China. Distilled water was used throughout the experiments (Fig. 4).

Particle size distribution of the used GBFS and SS
Samples preparation
The alkali activator (SiO2/Na2O = 1.4) was made by mixing water glass solution (SiO2/Na2O = 3.3) with sodium hydroxide (with 98% purity) in a mass ratio of 6.8. The alkali activator solution was sealed at room temperature for 24 h. Then, GBFS and SS with mass ratio 6/4 were taken into a vortex mixer and stirred for 5 min. Subsequently, alkali activator solution was poured into the vortex mixer. The mixture was initially mixed for 2 min slowly, stopped for 15 s and then stirred quickly for 2 min. After that, the paste was poured into cube specimens of size 20 mm × 20 mm × 20 mm and sealed to prevent any moisture loss. After 24 h, the specimens were demoulded and placed in laboratory conditions at a temperature of 20 ± 2 °C and relative humidity of 90 ± 2% before testing. The specimens were cured for 3, 7 and 28 days before testing. After the specified aging, specimens were put into small bottles containing anhydrous ethanol to stop hydration for 5 h and then placed in a vacuum drying oven (Yang et al. 2018) (Table 3).
Analysis technique
Thermometric TAM air was selected to determine the rate of hydration heat liberation and cumulative heat of the mixtures. About 5 g of premixed fresh pastes was poured into a flask. The flasks were then sealed and placed into the calorimeter until the calorimeter was stable. The mixture was mechanically stirred for approximately 1 min, and then the heat evolution was measured during 5 h for the entirety of the hydration process (Hu et al. 2018; Gao et al. 2015c).
20 mm × 20 mm × 20 mm cubes were prepared and tested at the duration of 3 days, 7 days and 28 days, respectively. The compressive strength of each sample was obtained from the average value of six specimens.
The mineral composition of the samples was analyzed by XRD. After being dried for 24 h in a vacuum drying oven, the samples were cracked and ground into fine powders of 20 μm for the analysis of XRD. The X-ray diffraction patterns were recorded on a DX-2700 automated diffractometer using Cu-Kɑ radiation. The tests were run in a 2θ range of 20°–70° at a scanning rate of 5°/min.
Scanning electron microscopy (SEM) (Model Nano SEM450, Hitachi Co., Japan) with energy-dispersive spectroscopy (EDS) and the accelerating voltage of the SEM/EDS was maintained at 15 kV and the working distance was maintained at 6.5 mm, which was used to observe the morphology of the reaction products. The samples were coated with gold to make them conductive before test (Xu et al. 2019).
FTIR analyses were operated on an infrared spectrophotometer (NICOLET, model NEXUS 670) in the range from 4000 to 400 cm−1 and a resolution of 4 cm−1.
The thermogravimetic analysis (TG-DTG) was conducted on STA-449C at a heating rate of 10 °C/min in N2, from 25 to 1000 °C, and the mass loss of the samples was recorded (Abdel-Gawwad et al. 2019).
Results and discussion
Isothermal calorimetry
The calorimetric curve for the slurry is known to have five distinctive periods: dissolution, dormancy, acceleration, deceleration and steady state (Zhu et al. 2018). Figure 5a and b shows the effects of different Ca(OH)2 contents on hydration heat release rate and exothermic curve of the mixture. The hydration process mainly exhibits two main hydration stages, and it is completed within 5 h. The first hydration heat peak which happened during the first 30 min possesses the exothermic peak caused by the wetting and dissolution of SS and GBFS. Meanwhile, the second stage indicates the periods of acceleration and deceleration, which is associated with hydration products (C–(A)–S–H, etc.). It is noteworthy to mention that the main product is C–A–S–H when GBFS is activated under relatively moderate alkaline conditions (Bilim et al 2015; Fernández-Jiménez 2000; Shi et al. 2003). The second exothermic rate peak in Fig. 5a shows that the higher the content of Ca(OH)2 is, the faster the exothermic rate is. And the addition of 2% Ca(OH)2 can make the heat evolution reach a maximum of 14.6 mW/g. This is attributed to that the active Ca2+ is provided by Ca(OH)2, which facilitates the formation of geopolymeric gel, and thus released a lot of heat. It can also be seen from Fig. 5a that exothermic reaction rate was accelerated with an increasing dosage of Ca(OH)2, and it can be explained by the increase in alkali content which led to the accelerated breaking of chemical bonds. Therefore, the heat release rate became faster and the cumulative heat release rate was higher with the increased incorporation of Ca(OH)2.

Curves of hydration exothermic rate and cumulative heat of SS and GBFS with different Ca(OH)2 content, respectively
Compressive strength
The compressive strength tests of 3 days, 7 days and 28 days for the different Ca(OH)2 additions of mortars are shown in Fig. 6. It is found that the compressive strength of the slurry improves with the increase in Ca(OH)2 addition, which exhibits more obviously when at later curing stages. For example, the 28-day compressive strength of the specimens with 2% Ca(OH)2 addition is 25.9% higher than the blank, which is due to the highly reactive Ca2+ provided from Ca(OH)2, and it promoted the hydration reaction. Meanwhile, Ca2+ stimulated the formation of the silicate, polyaluminosilicate network and the hardening of the matrix phase, which promotes the development of strength. Furthermore, Ca(OH)2 can also be used as a nucleating matrix to induce the growth of calcium silicate gels (Yip et al. 2008), which also promotes the development of strength.

Compressive strength of different Ca(OH)2 content
XRD results
Figure 7a, b shows XRD patterns of the slurry hydration products at 3 days, 7 days and 28 days with the blank and 2% Ca(OH)2 addition, respectively. The main mineral compositions are calcite, C3S, C2S, mullite, RO phase, calcium silicide aluminum and sodium calcium silicide (Pacheco-Torgal 2015). In Fig. 7a, the diffraction peak at around 27° of calcium silicide aluminum at 7 days is more obvious than 3 days, indicating that with the progress of reaction, a large amount of calcium silicide aluminum precipitates and there is no enough time to be consumed. However, this peak disappeared at 28-day sample, implying that most of calcium silicide aluminum has been transformed into calc-silicate and silicoaluminate gels, which is in high agreement with the diffraction peaks between 30° and 35°. Calcium silicide aluminum and sodium calcium aluminum are not precipitated in large amounts. At 28 days, it is well observed that the peaks are weakened, indicating that they have been consumed, resulting in the generation of large amount of calc-silicate and silicoaluminate gels. In addition, a small amount of sodium-based zeolite (N–A–S–H) is formed (Singh and Subramaniam 2017). It can also be seen from Fig. 7a that more and more calcite is produced with the prolonged curing age. Many mineral-enhanced phases such as C2S, C3S and mullite are also shown in the figure. For the sample with the addition of 2% Ca(OH)2, the intensity of the peak at around 30° is enhanced obviously, which means that structure of C–S–H gel moves toward more regular structure. The peak at around 27° of calcium silicide aluminum is only present in 3 days and 7 days; while the peak at 30° and 35° has been weak, calcite diffraction peak has been strong all the time. Therefore, the addition of Ca(OH)2 promotes the formation of reaction products in the whole system and the increase of Ca/Si ratio, which is beneficial to the formation of calc-silicate gels.

X-ray diffraction patterns of a samples with the blank and b samples with 2% Ca(OH)2
SEM analysis
Figure 8 shows the microstructures of the cementitious material with the blank (a and b) and Ca(OH)2 of 2% (c and d) hydrated for 3 days and 28 days. It can be seen from Fig. 8a that the flocculent gels were produced by hydration of the cementitious material at 3 days. From the EDS spectrum, the main products are C–A–S–H and C–S–H, which agrees well with XRD results. However, due to the existence of a large number of unhydrated SS and GBFS particles, the structure is relatively loose. Figure 8b shows the microstructure of 28-day hydration product of the blank. It demonstrates a plate-like structure which is obviously denser than hydration products in Fig. 8a. EDS spectrum shows that Na, Al, Si and Ca peaks are much stronger than those at 3 days, indicating that more hydration products including C–A–S–H are formed at 28 days, and the hydration process is more complete. Thus, the more gelatinous hydration products were generated, the denser the microstructure was. Compared with Fig. 8a, a dense plate-like hydration structure is formed in Fig. 8c, indicating that the addition of 2% Ca(OH)2 promoted the reaction and compacted the microstructure. Figure 8d shows the hydration product mixed with 2% Ca(OH)2 at the age of 28 days. The matrix is compact and consolidated as a whole, which is much denser than in Fig. 8b (Wang et al. 2019; Yang et al. 2012; Liu et al. 2019). From the EDS spectrum, it is found that the Ca peak is enhanced while Na and Al peaks become weaker, indicating that after the addition of Ca(OH)2, the extra Ca2+ appears to reduce the formation of sodium-based zeolitic phases, leading to an increase in the C–S–H gel and enhanced strength of the reaction system. The SEM observations agree well with compressive strength results. EDS shows that the hydration products were mainly C–S–H gel in the sample group (c) and (d) with 2% Ca(OH)2, while the hydration products in the sample group (a) and (b) with the blank were mainly C–A–S–H gel. It can be seen that the Ca2+ of the SS does not participate in the reaction in this system, which indicates that the SS does not react as a blending material.

ESEM micrographs (magnification of 2500 ×) of SS and GBFS system at the age of 3 (left) and 28 days (right): a, b showing 0% Ca(OH)2 at 3 and 28 days; followed by 2% Ca(OH)2 (c, d)
Fourier transform infrared spectroscopy (FT-IR)
Figure 9a, b shows the FTIR spectra of the samples with the blank and 2% Ca(OH)2 at curing age of 3, 7 and 28 days (Angulo-Ramírez et al. 2017). The absorption bands near 3443 cm−1, 3446 cm−1 and 3447 cm−1 in Fig. 9a are the asymmetric stretching vibration absorption bands of O–H in gels. The absorption bands around 1633 cm−1 and 1635 cm−1 are the bending vibration absorption band of O–H in C–(A)–S–H. An increase in the peak value was well observed, indicating that the higher the hydration degree of silicon aluminate ions and silicate ions in the product and more C–(A)–S–H gel is generated. 1420 cm−1 and 1421 cm−1 are the asymmetric stretching vibration peaks of C–O, which are the characteristic peaks of calcite. 872 cm−1, 865 cm−1 and 860 cm−1 are associated with the bending vibration peak of C–O and the asymmetric stretching vibration peak of Al–O. As the aging increases, the peak decreases which is in the direction of Al–O and produced by C–A–S–H gel. The absorption band near 989 cm−1, 987 cm−1 and 986 cm−1 is Si–O–T (T stands for Si or Al) (Pasupathy et al. 2016; Gijbels et al. 2019). The peak value decreases with time, indicating that Al replaces Si in C–S–H to construct C–A–S–H gel. The band at 458 cm−1, 455 cm−1 and 456 cm−1 is related to Si–O bending vibration peak and asymmetric tensile vibration peak Al–O (Shi et al. 2018). The decrease in the peak values indicates that the intensity of Al–O band increases and the content of C–A–S–H increases. In general, the main gel product in the hydration sample is C–A–S–H, and some others including C–S–H and calcite are also generated (Angulo-Ramírez et al. 2017). The infrared absorption band of Fig. 9b is similar to (a), indicating that the main hydration products are mainly C–S–H, C–A–S–H and calcite. However, the bending vibration peaks of Si–O–T and C–O increase with prolonged aging time. The absorption bands near 452 cm−1, 453 cm−1 and 454 cm−1 also show the same trend. The increase in wavenumbers of these band indicates that the degree of silicate polymerization is higher after adding Ca(OH)2 and more C–S–H gel was formed, which agrees well with the result of SEM.

Fourier transform infrared spectra of a samples with the blank and b samples with 2% Ca(OH)2
Thermogravimetric analysis (TGA)
The thermogravimetry results of blank sample and Ca(OH)2 addition of 2% are presented in Fig. 10 (Walkley et al. 2016). The high mass loss before 110 °C is attributed to the release of physically bound water within the paste. The continuous mass loss after 110 °C is assigned to the progressive decomposition of hydrated calcium silicate gels and calcium aluminosilicate gels (Gao et al. 2015b). Notably, a small weight loss can be observed at 650–700 °C, which might be caused by the decomposition of CaCO3 produced by carbonization of Ca(OH)2 (Yang et al. 2019). After 700 °C, all mixtures have a slight and continuous mass loss until 1000 °C. Thermogravimetric analysis shows that the reaction products are mainly amorphous phases with bound water (Gao et al. 2015a ). As can be seen from Fig. 10, the mass loss of the samples 28 days doped with 2% Ca(OH)2 is 20.85%, while the mass loss of the blank sample is 18.77%. The more significant loss of quality after adding Ca(OH)2 indicates that more hydrated calcium silicate gels were generated with the addition of Ca(OH)2, which is consistent with the SEM results. The higher amounts of hydration products formed will definitely lead to a denser microstructure, which is the major reason for the increase in compressive strength (Gao et al. 2015a).

TG/DTG analysis of a samples with the blank and b samples with 2% Ca(OH)2
Conclusions
-
1.
When 2% Ca(OH)2 is added into the cementitious material, the compressive strength of the 28-day cementitious material increased by 25.9% compared with the blank sample.
-
2.
The incorporation of Ca(OH)2 promotes the hydration reaction, accelerates the growth of C–S–H gel, and the resultant microstructure is denser, thereby enhancing the strength.
-
3.
The hydration product of C–A–S–H (from blank) and C–S–H (from 2% Ca(OH)2 addition) confirmed that steel slag is not suitable as a sole precursor for the production of building materials on a large scale due to the weak early activity of Ca2+.
References
Abdel-Gawwad HA, Mohammed MS, Alomayri T (2019) Single and dual effects of magnesia and alumina nano-particles on strength and drying shrinkage of alkali activated slag. Constr Build Mater. https://doi.org/10.1016/j.conbuildmat.2019.116827
Ahmedzade P, Sengoz B (2009) Evaluation of steel slag coarse aggregate in hot mix asphalt concrete. J Hazard Mater 165:300–305. https://doi.org/10.1016/j.jhazmat.2008.09.105
Altun IA, Yilmaz I (2002) Study on steel furnace slags with high MgO as additive in Portland cement. Cem Concr Res 32:1247–1249. https://doi.org/10.1016/S0008-8846(02)00763-9
Ameri M, Behnood A (2012) Laboratory studies to investigate the properties of CIR mixes containing steel slag as a substitute for virgin aggregates. Constr Build Mater 26:475–480. https://doi.org/10.1016/j.conbuildmat.2011.06.047
Angulo-Ramirez DE, de Gutierrez RM, Puertas F (2017) Alkali-activated Portland blast-furnace slag cement: mechanical properties and hydration. Constr Build Mater 140:119–128. https://doi.org/10.1016/j.conbuildmat.2017.02.092
Barbosa VFF, MacKenzie KJD, Thaumaturgo C (2000) Synthesis and characterisation of materials based on inorganic polymers of alumina and silica: sodium polysialate polymers. Int J Inorg Mater 2:309–317. https://doi.org/10.1016/S1466-6049(00)00041-6
Bilim C, Karahan O, Atis CD, Ilkentapar S (2015) Effects of chemical admixtures and curing conditions on some properties of alkali-activated cementless slag mixtures. KSCE J Civ Eng 19:733–741. https://doi.org/10.1007/s12205-015-0629-0
Chen ZW, Wu SP, Wen J, Zhao ML, Yi MW, Wan JM (2015) Utilization of gneiss coarse aggregate and steel slag fine aggregate in asphalt mixture. Constr Build Mater 93:911–918. https://doi.org/10.1016/j.conbuildmat.2015.05.070
Collins FG, Sanjayan JG (1999) Workability and mechanical properties of alkali activated slag concrete. Cem Concr Res 29:455–458. https://doi.org/10.1016/S0008-8846(98)00236-1
Fernández-Jiménez AM (2000) Cementos de Escorias Activadas Alcalinamente: Influencia de Las Variables y Modelizacion Del Proceso. Les Sources de l'histoire de France - Des origines aux guerres d'Italie 5:A140. https://doi.org/10.1108/TR-07-2014-0039
Fernández-Jiménez A, Palomo A, Criado M (2005) Microstructure development of alkali-activated fly ash cement: a descriptive model. Cem Concr Compos 35:1204–1209. https://doi.org/10.1016/j.cemconres.2004.08.021
Gao X, Yu QL, Brouwers HJH (2015a) Characterization of alkali activated slag-fly ash blends containing nano-silica. Constr Build Mater 98:397–406. https://doi.org/10.1016/j.conbuildmat.2015.08.086
Gao X, Yu QL, Brouwers HJH (2015b) Properties of alkali activated slag-fly ash blends with limestone addition. Cem Concr Compos 59:119–128. https://doi.org/10.1016/j.cemconcomp.2015.01.007
Gao X, Yu QL, Brouwers HJH (2015c) Reaction kinetics, gel character and strength of ambient temperature cured alkali activated slag-fly ash blends. Constr Build Mater 80:105–115. https://doi.org/10.1016/j.conbuildmat.2015.01.065
Gijbels K, Lacobescu RI, Pontikes Y, Schreurs S, Schroeyers W (2019) Alkali-activated binders based on ground granulated blast furnace slag and phosphogypsum. Constr Build Mater 215:371–380. https://doi.org/10.1016/j.conbuildmat.2019.04.194
Han FH, Zhang ZQ, Wang DM, Yan PY (2015) Hydration heat evolution and kinetics of blended cement containing steel slag at different temperatures. Thermochim Acta 605:43–51. https://doi.org/10.1016/j.tca.2015.02.018
Hu SG, He YJ, Lu LN, Ding QJ (2006) Effect of fine steel slag powder on the early hydration process of Portland cement. J Wuhan Univ Technol Mater Sci Ed 21:147–149
Hu YY, Li WF, Ma SH, Wang QQ, Zou HR, Shen XD (2018) The composition and performance of alite–ye'elimite clinker produced at 1300 degrees. Cem Concr Res 107:41–48. https://doi.org/10.1016/j.cemconres.2018.02.009
Huang XY, Wang ZJ, Liu Y, Hu WT, Ni W (2016) On the use of blast furnace slag and steel slag in the preparation of green artificial reef concrete. Constr Build Mater 112:241–246. https://doi.org/10.1016/j.conbuildmat.2016.02.088
Ivanka NG, Ivana B, Aleksandra F, Bansode SS (2016) Characteristics and uses of steel slag in building construction. Woodhead Publishing, Cambridge
Kumar S, Kumar R, Mehrotra SP (2010) Influence of granulated blast furnace slag on the reaction, structure and properties of fly ash based geopolymer. J Mater Sci 45:607–615. https://doi.org/10.1007/s10853-009-3934-5
Li C, Sun HH, Li LT (2010) A review: the comparison between alkali-activated slag (Si plus Ca) and metakaolin (Si plus Al) cements. Cem Concr Res 40:1341–1349. https://doi.org/10.1016/j.cemconres.2010.03.020
Li JX, Yu QJ, Wei JX, Zhang TS (2011) Structural characteristics and hydration kinetics of modified steel slag. Cem Concr Res 41:324–329. https://doi.org/10.1016/j.cemconres.2010.11.018
Li ZB, Zhao SY, Zhao XG, He TS (2013) Cementitious property modification of basic oxygen furnace steel slag. Constr Build Mater 48:575–579. https://doi.org/10.1016/j.conbuildmat.2013.07.068
Liu Z, Zhang DW, Li L, Wang JX, Shao NN, Wang DM (2019) Microstructure and phase evolution of alkali-activated steel slag during early age. Constr Build Mater 204:158–165. https://doi.org/10.1016/j.conbuildmat.2019.01.213
Mijarsh MJA, Johari MAM, Ahmad ZA (2015) Compressive strength of treated palm oil fuel ash based geopolymer mortar containing calcium hydroxide, aluminum hydroxide and silica fume as mineral additives. Cem Concr Compos 60:65–81. https://doi.org/10.1016/j.cemconcomp.2015.02.007
Motz H, Jens G (2001) Products of steel slags an opportunity to save natural resources. Waste Manag 21:285–293. https://doi.org/10.1016/S0956-053X(00)00102-1
Němeček J, Šmilauer V, Kopecký L (2011) Nanoindentation characteristics of alkali-activated aluminosilicate materials. Cem Concr Compos 33:163–170. https://doi.org/10.1016/j.cemconcomp.2010.10.005
Netinger I, Bjegović D, Vrhovac G (2011) Utilisation of steel slag as an aggregate in concrete. Mater Struct 44:1565–1575. https://doi.org/10.1617/s11527-011-9719-8
Nikolić I, Karanović L, Častvan IJ, Radmilović V, Mentus S, Radmilović V (2014) Improved compressive strength of alkali activated slag upon heating. Mater Lett 133:251–254. https://doi.org/10.1016/j.matlet.2014.07.021
Pacheco-Torgal F (2015) Introduction to handbook of alkali-activated cements, mortars and concretes. In: Pacheco-Torgal F, Labrincha J, Leonelli C, Palomo A, Chindaprasit P (eds) Handbook of alkali-activated cements, mortars and concretes. Woodhead Publishing, Cambridge
Pasupathy K, Berndt M, Castel A, Sanjayan J, Pathmanathan R (2016) Carbonation of a blended slag-fly ash geopolymer concrete in field conditions after 8 years. Constr Build Mater 125:661–669. https://doi.org/10.1016/j.conbuildmat.2016.08.078
Peng YZ, Hu SG, Ding QJ (2010) Preparation of reactive powder concrete using fly ash and steel slag powder. J Wuhan Univ Technol 25:349–354. https://doi.org/10.1007/s11595-010-2349-0
Provis JL, Bernal SA (2014) Geopolymers and related alkali-activated materials. Annu Rev Mater Res 44:299–327. https://doi.org/10.1146/annurev-matsci-070813-113515
Rosales J, Cabrera M, Agrela F (2017) Effect of stainless steel slag waste as a replacement for cement in mortars. mechanical and statistical study. Constr Build Mater 142:444–458. https://doi.org/10.1016/j.conbuildmat.2017.03.082
Rovnaník P (2010) Effect of curing temperature on the development of hard structure of metakaolin-based geopolymer. Constr Build Mater 24:1176–1183. https://doi.org/10.1016/j.conbuildmat.2009.12.023
Shi CJ, Pavel VK, Della R (2003) Alkali-Activated Cements and Concretes. Taylor & Francis, Boca Raton
Shi CJ, Qian J (2000) High performance cementing materials from industrial slags. Resour Conserv Recycl 29:195–207. https://doi.org/10.1016/S0921-3449(99)00060-9
Shi ZG, Shi CJ, Wan S, Li N, Zhang ZH (2018) Effect of alkali dosage and silicate modulus on carbonation of alkali-activated slag mortars. Cem Concr Res 113:55–64. https://doi.org/10.1016/j.cemconres.2018.07.005
Singh J, Singh SP (2019) Development of alkali-activated cementitious material using copper slag. Constr Build Mater 211:73–79. https://doi.org/10.1016/j.conbuildmat.2019.03.233
Singh GVPB, Subramaniam KVL (2017) Evaluation of sodium content and sodium hydroxide molarity on compressive strength of alkali activated low-calcium fly ash. Cem Concr Compos 81:122–132. https://doi.org/10.1016/j.cemconcomp.2017.05.001
Šmilauer V, Hlaváček P, Škvára F, Šulc R, Kopecký L, Němeček J (2011) Micromechanical multiscale model for alkali activation of fly ash and metakaolin. J Mater Sci 46:6545–6555. https://doi.org/10.1007/s10853-011-5601-x
Walkley B, San Nicolas R, Sani MA, Rees GJ, Hanna JV, van Deventer JSJ, Provis JL (2016) Phase evolution of C-(N)-A-S-H/N-A-S-H gel blends investigated via alkali-activation of synthetic calcium aluminosilicate precursors. Cem Concr Res 89:120–135. https://doi.org/10.1016/j.cemconres.2016.08.010
Wang GC (2016) The utilization of slag in civil infrastructure construction. Woodhead Publishing, Cambridge
Wang G, Wang YH, Gao ZL (2010) Use of steel slag as a granular material: volume expansion prediction and usability criteria. J Hazard Mater 184:555–560. https://doi.org/10.1016/j.jhazmat.2010.08.071
Wang JB, Du P, Zhou ZH, Xu DY, Xie N, Cheng X (2019) Effect of nano-silica on hydration, microstructure of alkali-activated slag. Constr Build Mater 220:110–118. https://doi.org/10.1016/j.conbuildmat.2019.05.158
Wu XQ, Zhu H, Hou XK, Li HS (1999) Study on steel slag and fly ash composite Portland cement. Cem Concr Res 29:1103–1106. https://doi.org/10.1016/S0008-8846(98)00244-0
Xu WY, Yang ST, Xu CJ, Sun H (2019) Study on fracture properties of alkali-activated slag seawater coral aggregate concrete. Constr Build Mater 223:91–105. https://doi.org/10.1016/j.conbuildmat.2019.06.191
Yang KH, Cho AR, Song JK, Nam SH (2012) Hydration products and strength development of calcium hydroxide-based alkali-activated slag mortars. Constr Build Mater 29:410–419. https://doi.org/10.1016/j.conbuildmat.2011.10.063
Yang JB, Li DX, Fang Y (2018) Effect of synthetic CaO–Al2O3–SiO2–H2O on the early-stage performance of alkali-activated slag. Constr Build Mater 167:65–72. https://doi.org/10.1016/j.conbuildmat.2018.01.148
Yang J, Huang JX, Su Y, He XY, Tan HB, Yang W, Strnadel B (2019) Eco-friendly treatment of lowcalcium coal fly ash for high pozzolanic reactivity: a step towards waste utilization in sustainable building material. J Clean Prod 238:117962. https://doi.org/10.1016/j.jclepro.2019.117962
Yip C, Lukey GC, Provis JL, van Deventer JS (2008) Effect of calcium silicate sources on geopolymerisation. Cem Concr Res 38:554–564. https://doi.org/10.1016/j.cemconres.2007.11.001
Zhu XH, Tang DS, Yang K, Zhang ZL, Li Q, Pan Q, Yang CH (2018) Effect of Ca(OH)2 on shrinkage characteristics and microstructures of alkali-activated slag concrete. Constr Build Mater 175:467–482. https://doi.org/10.1016/j.conbuildmat.2018.04.180
Acknowledgements
The authors are grateful for the financial support of the National Key Research and Development Program of China (2019YFC1907103) and the National Natural Science Foundation of China (51672237).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declared that they have no conflicts of interest to this work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wang, M., Qian, B., Jiang, J. et al. The reaction between Ca2+ from steel slag and granulated blast-furnace slag system: a unique perspective. Chem. Pap. 74, 4401–4410 (2020). https://doi.org/10.1007/s11696-020-01248-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11696-020-01248-5