
Abstract
Alkali-activated materials are low-environmental-impact binders that can be obtained from the alkaline activation of industrial wastes. In this study, converter steel slag as the major raw material and ground granulated blast furnace slag (GBFS) as the modified material were activated by water glass with a modulus of 1.5 and a Na2O dosage of 4%. The hydration process, microstructure and compressive strength of alkali-activated composite materials were investigated. The results show that adding GBFS accelerates the initial dissolution of the particles, leading to higher first exothermic peaks. But adding GBFS decelerates the formation of hydration products, resulting in the delay of the second exothermic peaks. Adding GBFS has no significant effect on the cumulative hydration heat, Ca(OH)2 content and the type of gel in the alkali-activated steel slag systems. However, with increasing GBFS content, the Ca–Si ratio in the gel decreases, and the Al–Si ratio increases. Adding GBFS can refine the pore structure and produces more Si–O–Si bonds in gels, resulting in a significant increase in the compressive strength. The improvement effect of GBFS on the compressive strength is more obvious at a later stage than at an earlier stage.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Alkali-activated materials (AAMs) are called clinker-free cement and are alternative green materials compared to conventional ordinary Portland cement (OPC) [1, 2]. AAMs are generally prepared by various aluminosilicate precursors and alkaline sources without the need for the calcination process. Sodium hydroxide and sodium silicate are common alkaline activators. The alkali activation of precursors can be called “a complex process of structural destruction and polycondensation of hydration products” [2]. Currently, the emission of substantial industrial solid wastes is increasing rapidly due to the fast-growing metallurgical industry. More studies have confirmed that some types of industrial wastes can be converted into potential precursors for AAMs resulting from their high activity under alkali activation [3,4,5,6]. If AAMs are widely used as building materials, they will not only meet the requirements of sustainable development but also conserve natural resources and preserve the environment. Ground granulated blast furnace slag (GBFS), fly ash and metakaolin are the most generated aluminosilicate precursors. It has been reported that AAMs have various good performance, such as lower costs, better durability, higher ability for hazardous wastes solidification, energy conservation and emission reduction [6, 7]. However, it should be noted that there are also disadvantages associated with AAMs, especially in the case where certain waste is used as the sole precursor. For instance, major limiting factors when GBFS is used as the sole precursor include large shrinkage and quick setting, but the low early strength is the greatest challenge when fly ash is used as the sole precursor due to the failure to achieve reasonable curing conditions [8, 9].
The most efficient way to eliminate or mitigate the negative effects of AAMs presented above is to use two or more precursors as the raw materials. Therefore, the modification effect and mechanism of alkali-activated composite materials have received increasing attention. The volumetric stability of alkali-activated GBFS concrete is one of the most important engineering properties that affects the long-term durability and serviceability of such concrete structures [10]. The drying shrinkage of water glass-activated GBFS concrete is 6 times higher than that of OPC concrete cured at ambient temperature [11]. Compared to OPC materials, the higher drying shrinkage of alkali-activated GBFS concrete is ascribed to the increase in the number of mesopores, where the mesopores account for approximately 50 percent of the total pores and the formation of a shrinkage-prone silica gel [12]. Wang et al. investigated the influence of the types and contents of modification additions on the drying shrinkage of alkali-activated GBFS concrete [13]. They found that adding OPC and simultaneously controlling the dosage of the activator could reduce the drying shrinkage. The values for the drying shrinkage of alkali-activated GBFS concrete are similar to those of OPC concrete when adding 9% sodium silicate and 10% OPC [13]. Both Sarathi et al. and Ma et al. found that replacing GBFS with 10% and 20% fly ash could also reduce the drying shrinkage of alkali-activated GBFS concrete cured at room temperature [14, 15]. This reduction was derived from the presence of less interconnected capillary networks in the composite matrix [14, 15]. In addition to reducing drying shrinkage, Sharmin et al. found that the compressive strength of alkali-activated GBFS concrete could be further improved by adding metakaolin, which contributed stronger Si–O–Al and Al–O–Al bonds than the Si–O–Si bond and finally formed a continuous compact microstructure [16]. The alkali-activated fly ash concrete generally requires high content of activator and high-temperature curing to obtain satisfactory mechanical properties [17]. Ali et al. carried out an extensive investigation to understand the influence of GBFS substitution on alkali-activated fly ash materials [18]. They found that adding GBFS to replace fly ash could obtain medium to high strengths without the requirement for a high curing temperature when lower chemical dosages were used, and the setting time decreased due to the addition of GBFS [18]. This was due to the formation of C–A–S–H gel as a hydration product [18]. Therefore, modified alkali-activated composite materials have more distinct advantages than alkali-activated materials with the sole precursor.
The output of steel slag from the steel smelting process accounts for approximately 20% of crude steel production [19]. At present, the utilization rate of steel slag in China is low, and steel slag waste reaches hundreds of millions of tons [20]. This introduces some problems, such as the occupation of land, environmental pollution and the waste of resources [21]. Most steel slag in China is converter slag, which is a material rich in CaO [22]. The presence of C2S and C3S in converter steel slag provides cementitious properties, making it a potential cementitious material [22]. Therefore, alkali-activated steel slag material is one of the most attractive types of AAMs due to its similar composition to OPC. Sodium silicate solution is generally considered to be the most effective activator for converter steel slag [22, 23]. However, previous studies have shown that the compressive strength of alkali-activated steel slag material is only 30–40% of the strength of OPC with the same water–binder ratio of 0.45, even when using water glass with the optimum dosage as an activator [24]. This is because there are less active component contents in converter steel slag than in OPC which leads to less hydration products [24]. More importantly, many inert components in converter steel slag cannot be activated by sodium silicate, which results in a lower bond strength of the matrix [24]. Therefore, it is essential to improve the mechanical strength of alkali-activated steel slag materials before they are used for large-scale applications. The modification of adding other materials, such as GBFS, is the most direct and effective method for improving the strength of alkali-activated steel slag materials.
Although some progress has been made in the alkaline activation of steel slag in the open literature, the modification of the alkali-activated steel slag material is seldom involved. This paper aims to provide more insight into the effect of GBFS on the early hydration process and mechanical properties of the alkali-activated steel slag material. Furthermore, characterization of the microstructure is carried out to better understand the reaction mechanisms.
Experimental program
Raw materials and mix proportion
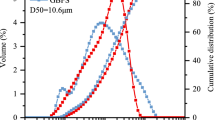
The original converter steel slag and blast furnace slag were broken with a mandible crusher and ground by the ball milling method. As the precursors in this study, the specific surface areas of the steel slag and GBFS were 458 m2 kg−1 and 430 m2 kg−1, respectively. The particle size distributions of the steel slag and GBFS were measured by a laser particle size analyzer (Mastersizer 2000), as shown in Fig. 1. However, the grain composition in the steel slag was obviously poorer than that of the GBFS due to the existence of tough phases. The chemical compositions of the two materials obtained by an X-ray spectrometer are shown in Table 1. According to the alkalinity calculation method from Mason [M = w(CaO)/w(SiO2 + P2O5)], the alkalinity of converter steel slag in this study was 2.25, which was preferable as a cementitious material (M > 1.8) [22]. According to the specification requirement [Mk = w(CaO + MgO + Al2O3)/w(SiO2 + MnO + TiO2) > 1.2] of the Chinese national standard (GB/T 203-2008), the mass fraction of GBFS used in this study was 2.04, which was also suitable as a cementitious material. The mineral phase compositions of the two materials measured by X-ray diffraction analysis are shown in Fig. 2. The main mineral phases are C2S in the steel slag, while amorphous nature in the GBFS is reflected by the presence of a diffuse wide band from the glassy phase. The SiO2 crystals are the main crystalline phase in the GBFS due to the addition of tailings.

Particle size distributions of the precursors

The mineral phase compositions of the precursors
A commercial water glass with a silicate modulus (MS) of 3.12 in liquid form and sodium hydroxide (NaOH, analytical reagent, 99 mass% purity) in the form of pellets were prepared as alkaline activators. ISO standard sand was used as the fine aggregates of the mortar.
A water glass solution (MS = 1.5) was prepared by mixing commercial water glass, NaOH and deionized water. A constant alkali concentration of 4 mass% (Na2O) was used. Three mix proportions of mortars with the same water–binder ratio of 0.45 are presented in Table 2. The sand-binder ratio was 3. Two replacement ratios (10% and 20% by mass) of GBFS were selected.
Test procedures
The hydration process of all the pastes was monitored by hydration heat analysis with an isothermal calorimeter (TAM Air Thermometric, set at 25 °C) for 96 h. To examine the hydration products, microstructure and pore structure, the fresh pastes were cast into plastic centrifuge tubes and cured at 25 °C to certain stages. The Ca(OH)2 content was calculated by thermogravimetric (TG)/derivative thermogravimetric (DTG) analyses. TG/DTG curves were obtained by a thermogravimetric analyzer (NETZSCH STA 449F3) from 30 to 900 °C in a N2 atmosphere. The microstructures of the hardened pastes were observed by scanning electron microscopy (SEM) and energy-dispersive X-ray (EDX) analysis. The pore structures of the hardened pastes were obtained by a mercury intrusion porosimeter (MIP, Autopore II 9220). To examine the compressive strength of the mortar, the fresh mortars were cast into stainless-steel molds with nominal dimensions of 40 × 40 × 160 mm and then covered with plastic foil to prevent water evaporation. The molds were removed after 1 day, and hardened mortars were cured at a standard temperature (20 °C, 99% RH) until reach the testing ages. The compressive strength of the mortar was determined according to Chinese standard GB177-85.
Results and discussion
Hydration heat
The method of isothermal calorimetry was used to test the hydration heat of the composite binders. The exothermic rate curves and the cumulative hydration heat curves for 96 h are shown in Fig. 3a, b, respectively. According to previous research, similar to the hydration process of OPC, the hydration process of alkali-activated steel slag binder can be divided into five periods: the rapid dissolution period with a high exothermic rate, the induction period with a low exothermic rate, the acceleration period, the deceleration period and the stabilization period [22, 25].

Hydration heat evolution curves for all mixtures at 25 °C. a Exothermic rate; b cumulative hydration heat
In general, the rapid dissolution of the particles leads to the first exothermic peak in the curve [26, 27]. The aluminate phase dissolves rapidly due to the high alkalinity of the activators, forming the initially dissolved aluminate units. As shown in Fig. 3a, the first exothermic peaks of samples SB10-M1.5 and SB20-M1.5 almost coincide with each other, and the peak values are higher than that of sample S-M1.5. This indicates that the dissolution rate of GBFS particles is higher than that of steel particles because the amorphous phase in GBFS is more easily depolymerized by an alkaline activator than the crystalline phases in steel slag. Therefore, the addition of GBFS allows alkali-activated steel slag systems to have a higher initial heat release rate. After the first peak, there is a very short induction period with a low exothermic rate in the curve, and the hydration rate decreases obviously with increasing GBFS content. This is because the early dissolution of samples SB10-M1.5 and SB20-M1.5 is too rapid, leading to slower further dissolution due to near saturation points. After a short induction period, sample S-M1.5 first starts the acceleration period of hydration at 1.3 h and reaches the peak rate at 2.52 h, which is 2.19 mW g−1 and lasts for 1.22 h. For sample SB10-M1.5, its acceleration period starts a little later at approximately 1.33 h, reaches the peak rate at 3.31 h, which is 2.12 mW g−1, and lasts for 1.98 h. For sample SB20-M1.5, its acceleration period proceeds from 1.79 to 5.02 h and reaches the peak rate of 1.75 mW g−1, which starts at the latest time compared to the other samples and lasts for 3.23 h. The durations of the deceleration periods for all the mixtures are very long, and the hydration heat release rate levels off after approximately 36 h. The second exothermic peak includes the acceleration period and the deceleration period. Therefore, with the increase in the GBFS content, the occurrence time of the second exothermic peak is prolonged, and the peak exothermic rate decreases. The second exothermic peak is related to the formation of hydration products, including C–A–S–H gels from the hydration of GBFS and steel slag with alkaline activator and Ca(OH)2 resulting from the hydration of steel slag. This indicates that with increasing GBFS content, the hydration rate of the total active components decreases. Previous research shows that the exothermic rate curves of alkali-activated GBFS materials have a long induction period and a large second exothermic peak [28,29,30]. Although the contents of calcium silicate and other active minerals from converter steel slag in the composite binder are reduced, more amorphous components are added with the substitution of GBFS.
As shown in Fig. 3b, the cumulative hydration heat of samples S-M1.5, SB10-M1.5 and SB20-M1.5 at 96 h is 147 J g−1, 153 J g−1 and 150 J g−1, respectively. Although the cumulative hydration heat of the alkali-activated composite binder is substantially higher than that of the alkali-activated steel slag binder after 12 h, the hydration heat exhibits little difference at 96 h due to the delay effect of the GBFS. This also indicates that the effect of GBFS on the hydration heat of the whole binder system decreases over time.
Ca(OH)2 content
The Ca(OH)2 content is used to characterize the influence of GBFS on the content of hydration products [22]. Figure 4 shows the TG/DTG curves of the hardened pastes at 1 day, 3 days, 7 days, 28 days and 90 days. There are four common endothermic peaks in the DTG curve. The first peak corresponds to the initial decomposition of the C–A–S–H gel before 350 °C. The second peak represents the dehydration of Ca(OH)2 at approximately 350–420 °C. Another endothermic peak is located at approximately 500–700 °C, corresponding to the later dehydration of the C–A–S–H gel. The last inconspicuous peak is located at approximately 600–800 °C, corresponding to the decarbonization of CaCO3. CaCO3 is formed due to the carbonization reactions of the gel and Ca(OH)2. However, the dehydration temperatures of Ca(OH)2 and CaCO3 in this system are lower than the usual decomposition temperatures, which usually occur at 400–500 °C and 800–900 °C, respectively. This may be because the presence of an alkaline activator improves the decomposition of CaCO3 [31]. The second reason may be due to the presence of poorly crystallized Ca(OH)2 and amorphous carbonates in the alkali-activated steel slag system. The calculation method proposed by Taylor [32] for the dehydration of Ca(OH)2 is described in formula (1):
where \({\text{ML}}_{{{\text{Ca(OH)}}_{ 2} }}\) and \({\text{ML}}_{{{\text{CaCO}}_{ 3} }}\) are the mass loss during the dehydration of Ca(OH)2 and during the decomposition of CaCO3, respectively. The numbers 74, 18 and 44 are the molecular masses of Ca(OH)2, water and CO2, respectively. The Ca(OH)2 contents of hardened pastes are presented in Fig. 5. Ca(OH)2 is derived from the hydration of calcium silicate (C3S and C2S) in converter steel slag. Compared to the Ca(OH)2 content in the OPC binder, there is very low content in the alkali-activated steel slag binder due to less calcium silicate in the latter. The highest Ca(OH)2 content is less than 7%, which appears at the age of 90 days. Previous research has shown that the Ca(OH)2 content in OPC systems is approximately 4 times higher than that in alkali-activated steel slag systems with the same water–binder ratio of 0.45 [24]. The addition of GBFS has little effect on the Ca(OH)2 content. Although adding GBFS to replace steel slag reduces the amount of calcium silicate in the cementing system, it is possible that the hydration reaction of calcium silicate is improved due to the dispersion effect. Meanwhile, the pozzolanic reaction of GBFS can consume Ca(OH)2 to form a C–A–S–H gel. As a result, the Ca(OH)2 content changes little as a result of the two reverse trends.

TG/DTG curves for all the mixtures. a 1 day; b 3 days; c 7 days; d 28 days; e 90 days

The Ca(OH)2 content of the hardened paste
Microstructure analysis
The SEM image clearly reflects the formation of product layers. Figure 6 presents the microstructure of hardened pastes at 90 days under the same magnification of 5000 times. As shown in Fig. 6a, it is difficult for some particles to dissolve in sample S-M1.5, which has an adverse effect on the compactness and adhesion of the matrix. Those particles are mainly γ-C2S and RO phase. However, it is obvious that this phenomenon can be dramatically improved by adding GBFS. With increasing GBFS content, the distinct particles in the observed surface decrease due to the uniform distribution of the gel. Therefore, the addition of GBFS can improve the microstructure of alkali-activated steel slag systems.

Microstructure of hardened pastes at 90 days. a Sample S-M1.5; b sample SB10-M1.5; c sample SB20-M1.5
EDX spectroscopy is used to analyze the chemical compositions of the amorphous gels at 90 days, and at least 20 different points are selected to obtain the atomic percentages of the various elements. Representative EDX spectra are shown in Fig. 7. The major elements in the gels are Ca, Si and O, and there are small amounts of Al, Mg and Fe. The amorphous products of the three samples are C–A–S–H gel, indicating that adding GBFS has no effect on the types of gels. The atomic percentage of each element in the gel is calculated based on the statistical results. Then, the average Ca–Si and Al–Si ratios are also calculated and presented in Fig. 8. The Ca–Si ratios in the gels of samples S-M1.5, SB10-M1.5 and SB20-M1.5 are 1.393, 1.235 and 1.164, respectively. The Al–Si ratios of the gel are 0.137, 0.192 and 0.261, respectively. Obviously, with the increase in GBFS content, the Ca–Si ratio significantly decreases, while the Al–Si ratio increases. Although the alkaline environment of the three mixtures is identical, the chemical compositions of the raw materials are obviously different. The results in Table 1 show that the contents of CaO, Al2O3 and SiO2 in converter steel slag are 41.02%, 5.85% and 16.93%, respectively. The contents of the three components above in the GBFS are 40.05%, 15.71% and 31.01%, respectively. Although the CaO contents in the two raw materials are similar, the Al2O3 content in GBFS is approximately 3 times than in the steel slag, and the SiO2 content is approximately 2 times that in the steel slag. With the addition of GBFS, the CaO content in the composite binder is basically unchanged, while the contents of Al2O3 and SiO2 obviously increase.

EDX spectra in Fig. 6. a–c Scanning positions 1–3

Ca–Si ratio and Al–Si ratio in C–A–S–H gels
Pore structure
The pore structure of the matrix is an important index for the development of mechanical strength and durability. MIP analysis was used to obtain the early (3 days) and late (90 days) pore structures of the hardened pastes. At 3 days, the total porosities of samples S-M1.5, SB10-M1.5 and SB20-M1.5 are 37.45%, 38.44% and 40.20%, respectively. With the increase in the GBFS content, the total porosities change little or even increase slightly at 3 days. At 90 days, the total porosities of the three samples are 33.74%, 31.82% and 23.51%, respectively. With the increase in the GBFS content, the total porosity decreases obviously at 90 days. Adding GBFS has a more significant impact on the late pore structure than the early pore structure. The total porosity is even reduced by 10% at 90 days in the case where 20% steel slag is replaced.
The pores in cement-based materials can be classified as follows: harmless pores (< 20 nm), little harmful pores (20–50 nm), harmful pores (50–200 nm) and much harmful pores (> 200 nm) [22, 25, 33]. It is generally believed that micropores less than 50 nm (harmless pores and little harmful pores) have no adverse effect on the matrix. The cumulative pore volumes of the hardened pastes at 3 days and 90 days are shown in Fig. 9. At 3 days, the total micropore volumes only slightly increase with increasing GBFS content. However, the changing trend shows the opposite pattern for pores larger than 50 nm in diameter. The total macropore (harmful pores and much harmful pores) volumes decrease with increasing GBFS content. At 90 days, the total micropore volumes obviously decrease with increasing GBFS content. The total macropore volumes also decrease, among which sample SB20-M1.5 contains almost no harmful pores and only a very small number of much harmful pores. Therefore, adding GBFS can obviously reduce the number of macropores in hardened pastes, which further indicates that adding GBFS can refine the pore structure. The distribution of the hydration products is more uniform due to the addition of GBFS, and more hydration products fill the pores to improve the pore structure, which is consistent with the microstructure observed by SEM.

Pore volume distributions of the hardened paste. a 3 days; b 90 days
Compressive strength
Figure 10 shows the compressive strengths of the mortar of the three samples at different ages when the modulus of sodium silicate solution is 1.5. Overall, the compressive strength of the mortar increases with increasing GBFS content at all ages. On the one hand, the change in the compressive strength is directly related to the pore structure distributions. From the analysis results of the pore structure, it can be seen that adding GBFS can refine the pore structure, and this phenomenon is more obvious at a later stage than at an earlier stage. On the other hand, the improved strength is also attributed to changes in gel structure with the increase in GBFS content, the Ca–Si ratio significantly decreases. It indicates that more formation of stronger Si–O–Si bonds exists in C–A–S–H gel, thus resulting in strength gain [34, 35].

Compressive strengths of the mortar
It is worth noting that the change in the 1-day compressive strength with increasing GBFS content is not obvious. This is because the hydration degree is very low at 1 day, and the addition of GBFS is not sufficient to substantially improve the hydration degree, which can be reflected by the Ca(OH)2 content in Fig. 5. Adding GBFS can obviously improve the compressive strength of mortar after 3 days. At 90 days, the maximum compressive strength is 47 MPa (sample SB20-M1.5), while the compressive strength of sample S-M1.5 is 33 MPa. Therefore, adding 20% GBFS increases the 90-day compressive strength by 10%. Thus, it can be seen that the improvement effect of GBFS on the compressive strength is very obvious.
Conclusions
In this paper, the influence of GBFS on the early hydration and microstructure of alkali-activated steel slag binders is investigated from multiple perspectives. The following conclusions can be drawn according to this study:
-
1.
Adding GBFS accelerates the initial dissolution of the particles, leading to higher first exothermic peaks. However, the second exothermic peaks are delayed with the addition of GBFS. Adding GBFS has no significant effect on the cumulative hydration heat.
-
2.
Adding GBFS has little effect on the Ca(OH)2 content and does not change the type of gel. However, with increasing GBFS content, the Ca–Si ratio significantly decreases, and the Al–Si ratio increases.
-
3.
Adding GBFS can refine the pore structure and produces more Si–O–Si bonds in C–A–S–H gel, resulting in a higher compressive strength. The improvement effect of the GBFS on the compressive strength is more obvious at a later stage than at an earlier stage.
References
Wang D, Wang Q, Fang Z. Influence of alkali activators on the early hydration of cement-based binders under steam curing condition. J Therm Anal Calorim. 2017;130(1):1–16.
Bernal SA, Provis JL, Walkley B, Nicolas SR, Deventer JSJ. Gel nanostructure in alkali-activated binders based on slag and fly ash, and effects of accelerated carbonation. Cem Concr Res. 2013;53:127–44.
Wang D, Wang Q, Huang Z. New insights into the early reaction of NaOH-activated slag in the presence of CaSO4. Compos B Eng. 2020;198:108207.
Wang Q, Yan P, Feng J. A discussion on improving hydration activity of steel slag by altering its mineral compositions. J Hazard Mater. 2011;186(2–3):1070–5.
Li LG, Wang YM, Tan YP, Kwan AKH. Filler technology of adding granite dust to reduce cement content and increase strength of mortar. Powder Technol. 2019;342:388–96.
Wang D, Wang Q, Xue J. Reuse of hazardous electrolytic manganese residue: detailed leaching characterization and novel application as a cementitious material. Resour Conserv Recycl. 2020;154:1–11.
Liu J, Guo R. Hydration properties of alkali-activated quick cooled copper slag and slow cooled copper slag. J Therm Anal Calorim. 2020;139:3383–94.
Yliniemi J, Walkley B, Provis JL, Kinnunen P, Illikainen M. Influence of activator type on reaction kinetics, setting time, and compressive strength of alkali-activated mineral wools. J Therm Anal Calorim. 2020. https://doi.org/10.1007/s10973-020-09651-6.
Chi M. Effects of modulus ratio and dosage of alkali-activated solution on the properties and micro-structural characteristics of alkali-activated fly ash mortars. Constr Build Mater. 2015;99:128–36.
Mastali M, Kinnunen P, Dalvand A, Firouz RM, Illikainen M. Drying shrinkage in alkali-activated binders-a critical review. Constr Build Mater. 2018;190:533–50.
Lee NK, Jang JG, Lee HK. Shrinkage characteristics of alkali-activated fly ash/slag paste and mortar at early ages. Cem Concr Compos. 2014;53:239–48.
Cao J, Chung DDL. Microstructural effect of the shrinkage of cement-based materials during hydration, as indicated by electrical resistivity measurement. Cem Concr Res. 2004;34:1893–7.
Wang F, Sun R, Cui Y. Study on modification of the high-strength slag cement material. Cem Concr Res. 2005;35:1344–8.
Sarathi PD, Nath P, Sarker PK. Drying shrinkage of slag blended fly ash geopolymer concrete cured at room temperature. Procedia Eng. 2015;125:594–600.
Ma Y, Ye G. The shrinkage of alkali activated fly ash. Cem Concr Res. 2015;68:75–82.
Sharmin A, Alengaram UJ, Jumaat MZ, Yusuf MO, Kabir SMA, Bashar II. Influence of source materials and the role of oxide composition on the performance of ternary blended sustainable geopolymer mortar. Constr Build Mater. 2017;144:608–23.
Maryam H, Aleksandra R. Shrinkage and strength development of alkali-activated fly ash-slag binary cements. Constr Build Mater. 2017;150:808–16.
Ali R, Vinai R, Soutsos M, Sha W. Effects of slag substitution on physical and mechanical properties of fly ash-based alkali activated binders (AABs). Cem Concr Res. 2019;122:118–35.
Lee KJ, Yoo SY, Koo JS, Cho BS, Lee HH. Experimental study on the characteristics of rapid chilled converter slag by watering. Int J Concr Struct Mater. 2011;5:133–7.
Shi M, Wang Q, Zhou Z. Comparison of the properties between high-volume fly ash concrete and high-volume steel slag concrete under temperature matching curing condition. Constr Build Mater. 2015;98:649–55.
Han F, Zhang Z. Properties of 5-year-old concrete containing steel slag powder. Powder Technol. 2018;334:27–35.
Sun J, Chen Z. Effect of silicate modulus of water glass on the hydration of alkali-activated converter steel slag. J Therm Anal Calorim. 2019;138:47–56.
Liu Z, Zhang D, Li L, Wang D. Microstructure and phase evolution of alkali-activated steel slag during early age. Constr Build Mater. 2019;204:158–65.
Sun J, Zhang Z, Zhuang S, He W. Hydration properties and microstructure characteristics of alkali-activated steel slag. Constr Build Mater. 2020;241:1–9.
Sun J, Wang Z, Chen Z. Hydration mechanism of composite binders containing blast furnace ferronickel slag at different curing temperatures. J Therm Anal Calorim. 2018;131(3):2291–301.
Pacewska B, Iwona W. Usage of supplementary cementitious materials: advantages and limitations: part I. C–S–H, C–A–S–H and other products formed in different binding mixtures. J Therm Anal Calorim. 2020. https://doi.org/10.1007/s10973-020-09907-1.
Zhang L, Li G, Ye W, Chang Y, Liu Q, Song Z. Effects of ordinary Portland cement on the early properties and hydration of calcium sulfoaluminate cement. Constr Build Mater. 2018;186:1144–53.
Yusuf M, Johari M, Ahmad Z. Impacts of silica modulus on the early strength of alkaline activated ground slag/ultrafine palm oil fuel ash based concrete. Mater Struct. 2015;48(3):733–41.
You N, Li B, Cao R, Shi J, Chen C, Zhang Y. The influence of steel slag and ferronickel slag on the properties of alkali-activated slag mortar. Constr Build Mater. 2019;227:1–10.
Langaro E, Costa M, Buth IS, Angulski C, Pereira F, Matoski A. Use of slag (GBFS) generated in charcoal blast furnace as raw material in alkali-activated cement. J Therm Anal Calorim. 2020. https://doi.org/10.1007/s10973-020-09632-9.
Salman M, Pontikes Y, Snellings R. Cementitious binders from activated stainless steel refining slag and the effect of alkali solutions. J Hazard Mater. 2015;286:211–9.
Taylor H. Cement Chemistry. 1st ed. London: Academic Press Limited; 1990.
Nazari A, Riahi S. Microstructural, thermal, physical and mechanical behavior of the self-compacting concrete containing SiO2 nanoparticles. Mater Sci Eng A. 2010;527(29):7663–72.
Leong HY, Ong DEL, Sanjayan JG. The effect of different Na2O and K2O ratios of alkali activator on compressive strength of fly ash based-geopolymer. Constr Build Mater. 2016;106:500–11.
Ryu GS, Lee YB, Koh KT. The mechanical properties of fly ash-based geopolymer concrete with alkaline activators. Constr Build Mater. 2013;47(5):409–18.
Acknowledgements
Authors would like to acknowledge the support of National Natural Science Foundation of China (51778334) and Tsinghua University Initiative Scientific Research Program.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhou, Y., Sun, J. & Liao, Y. Influence of ground granulated blast furnace slag on the early hydration and microstructure of alkali-activated converter steel slag binder. J Therm Anal Calorim 147, 243–252 (2022). https://doi.org/10.1007/s10973-020-10220-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-020-10220-0