
Abstract
One of the therapeutic approaches in the management of type 2 diabetes is delaying the glucose absorption through α-glucosidase enzyme inhibition, which can reduce the occurrence of postprandial hyperglycemia. Based on this thought, a series of novel chloro-substituted 2-(2-methyl-1-phenyl-1H-pyrrol-3-yl)-2-oxo-N-(pyridin-3-yl) acetamide derivatives 5a–i were synthesized and their α-glucosidase inhibitory activities were evaluated. All the synthesized compounds have shown moderate to excellent in vitro α-glucosidase inhibitory activity with IC50 values in the range of 111–673 µM) as compared to acarbose, the standard drug (750 ± 9 µM). Compound 5e (111 ± 12 µM), among the series, was the most potent inhibitor of α-glucosidase in a competitive mode of action based on the kinetic study. The molecular docking study of compounds 5e and 5a revealed that they have a lower free binding energy (− 4.27 kcal/mol and − 3.17 kcal/mol, respectively) than acarbose (− 2.47 kcal/mol), which indicates that the target compound binds more easily to the enzyme than acarbose does. The outcomes from the molecular docking studies supported the results obtained from the in vitro assay. In conclusion, the overall results of our study reveal that the synthesized compounds could be a potential candidate in the search for novel α-glucosidase inhibitors to manage postprandial hyperglycemia incidence.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
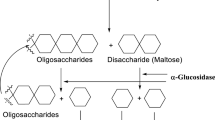
Diabetes mellitus (DM) is a chronic progressive metabolic disease worldwide due to carbohydrate, fat, and protein metabolism that is characterized by hyperglycemia resulting from defects of insulin secretion, insulin action, and/or both (WHO 2016). Autoimmune destruction of the β cells of the islets of Langerhans is the causative factor for type 1 DM, while a combination of genetic predisposition, physical inactivity, and obesity are the causes that contribute to defects in insulin secretion and/or insulin resistance which leads to type 2 DM (WHO 2016; Tundis et al. 2010). DM leads to acute and chronic complications in many parts of the body including diabetic ketoacidosis (DKA), hyperosmolar hyperglycemic state (HHS), neuropathy, nephropathy, retinopathy, ischemic heart disease, myocardial infarction, stroke, and peripheral arterial disease (Powers et al. 2015; Kitabchi et al. 2009; Deshpande et al. 2008). During the digestion of food, α-glucosidase hydrolyzes carbohydrates and produces α-d-glucose, which is absorbed in the blood stream, increases postprandial blood glucose levels, and causes diabetes. Therefore, for the control and prevention of diabetes, α-glucosidase inhibitors are of particular interest, as they can help to reduce the carbohydrate digestion and subsequent monosaccharide absorption (Santos et al. 2018).
Various oral hypoglycemic agents are currently used to lower or maintain the blood glucose level to nearly normal with different therapeutic approaches (Ramprasad and Madhusudhan 2016; Ghani 2015). One of the therapeutic approaches in the management of type 2 diabetes is reducing the postprandial hyperglycemia by slowing down the glucose absorption through enzyme inhibition, α-glucosidase and α-amylase, which hydrolyzes the carbohydrates in the digestive tract (Gollapalli et al. 2018; Sugihara et al. 2014). Acarbose, miglitol, and voglibose are currently used to inhibit the major carbohydrate-hydrolyzing enzymes (Joshi et al. 2015).
On the other hand, long-term high plasma level of glucose causes impairment of the endogenous antioxidant agents such as glutathione, coenzyme-Q, and α-lipoic acid. Decreasing the level of this antioxidant increases the level of reactive oxygen species (ROS) and hydroxyl and nitric oxide (NO) radicals that induces pancreatic β-cell destruction. Furthermore, uncontrolled free radical generation leads to various microvascular and macrovascular complications (Rouzbehan et al. 2017; Ibrahim et al. 2014; Mccue et al. 2005). Consequently, the inhibition of α-glucosidase is the most effective therapeutic approach to prevent the impact of type 2 diabetes through controlling the postprandial glucose levels and reducing postprandial hyperglycemia by delaying the absorption of glucose (Gollapalli et al. 2018; Kim et al. 2008).
The currently available α-glucosidase inhibitors have some side effects such as hypoglycemia at higher doses, liver problems, meteorism, diarrhea, and lactic acidosis (Rouzbehan et al. 2017; Ibrahim et al. 2014). Therefore, the discovery and development of potential α-glucosidase inhibitors can be regarded as a promising topic in recent years.
Pyrrole scaffold is a key structural moiety in numerous synthetic medicinal agents and natural products (Jacobi et al. 2000). Pyrroles and their derivatives are considered as one of the most pharmaceutically significant heterocyclic molecules and extensively used in drug discovery (Abdelrazek & Metwally 2006). They have demonstrated significant biological activities in various areas such as anti-mycobacterial activity (Biava et al. 1999, 2009; Joshi et al. 2008), anti-inflammatory and analgesic activity (Biava et al. 2010), HIV-1 non-nucleoside reverse transcriptase inhibition (Famiglini et al. 2013), antibacterial and antifungal activities (Joshi et al. 2008), acetylcholinesterase Inhibition (Sangnoi et al. 2008), angiotensin AT1 receptor antagonist activities (Wu et al. 2008), and hypoglycemic and hypotriglyceridemic activities (Lohray and Lohray 2005; Mohamed et al. 2014).
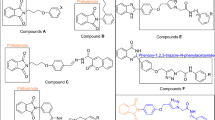
Several pyrrole scaffolds containing compounds have been discovered and marketed to exhibit various therapeutic properties, e.g., drugs tolmetin, atorvastatin, chlorfenapyr, premazepam, pyrvinium, roseophilin, and zomepirac (Domagala et al. 2015). On the other side, there are reports on α-glucosidase inhibitory activity of different heterocyclic compounds (Dhameja and Gupata 2019). For example, a series of 5,6-diphenyl-1,2,4-triazine derivatives (Fig. 1a) showed good activity for inhibition of α-glucosidase enzyme with IC50 ranging from 12 to 72 µM (Wang et al. 2017a). The core of the heterocyclic ring, 1,2,4-triazine, was also used to introduce other α-glucosidase inhibitors (Fig. 1b, c) with different side chain length between the core and third aryl group (Wang et al. 2016a, 2017b). Other heterocyclic rings such as imidazole and pyrrole as central core containing 1,2-diaryl substitution (Fig. 1d, e) have been reported as anti-hyperglycemic agents (Wang et al. 2016b; Goel et al. 2004).

Chemical structures of some compounds with α-glucosidase inhibitory and anti-hyperglycemic activity
With respect to the above background, in the search of new, simple, and easily manageable compounds that could be active against α-glucosidase, herein, we reported the design and synthesis of new 1,5-cis-diaryl pyrrole derivatives. As presented in Fig. 1, our designed structure has been inspired from a and c derivatives. Pyridine ring was used as the third aryl group at the end of the side chain to increase water solubility of the target compounds. After structural characterization, in vitro α-glucosidase inhibitory activity and kinetic study were done for understanding the mode of interaction of the most potent compounds with enzyme-active site, and docking study was also performed.
Experimental
All chemical compounds and reagents were purchased from Merck (Darmstadt, Germany). Melting points were determined with a Kofler hot stage apparatus (Reichert, Vienna, Austria) and are uncorrected. 1H NMR and 13C NMR spectra were recorded with Bruker FT-400 and Bruker FT-100 spectrometer (Bruker, Darmstadt, Germany), respectively, using TMS as the internal standard and CDCl3 as a solvent. The chemical shift (δ) values were expressed in parts per million (ppm), the coupling constants (J) in hertz, and spin multiplicities as s (singlet), d (doublet), t (triplet), and m (multiplet). Mass spectrum was performed with an Agilent Technology (HP) mass spectrometer operating at an ionization potential of 70 eV.
Chemistry
The synthetic route for preparation of 5a–I is outlined in schemes 1 and 2.

Synthesis pathway of the target compounds 5a–g

Synthesis pathway of the target compounds 5 h, 5i
General procedure for the preparation of pentane-1, 4-dione derivatives (3a, b)
An appropriate solution of benzaldehyde 1a, b (31.2g, 294 mmol) in DMF (150 mL) was added dropwise over 10 min to vigorously stirred sodium cyanide (1.5g, 30.6 mmol) in DMF (100 mL) at 35 °C in a round-bottom flask under N2 according to the Stetter reaction. The reaction mixture was stirred for 10 min and then methyl vinyl ketone (16.0g, 228 mmol) in DMF (150 mL) was added dropwise within 20 min with continued vigorous stirring at the same temperature leading to 3a, b formation (Peloquin et al. 2012; Biava et al. 2008; Stetter 1976). The obtained compound was concentrated under reduced pressure and purified with column chromatography using ethyl acetate and hexane as an eluent.
General procedure for the preparation of pyrrole derivatives 4a–i
The diketones 3a–c (2.28 mmol) were dissolved in ethanol (3 mL) in a round-bottom flask. A corresponding aniline derivative (2.28 mmol) and a catalytic amount of para-toluene sulfonic acid (pTSA) were added to the mixture and allowed to reflux for 8 h. The reaction progress was inspected by TLC. The reaction becomes cyclized through the process following the Paal–Knorr condensation to yield the desired pyrrole derivatives 4a–i. The obtained crude compounds were concentrated under reduced pressure and purified by column chromatography using hexane and ethyl acetate as an eluent.
General procedure for the synthesis of target compounds 5a–i
To the mixture of appropriate pyrrole derivatives 4a–i (4.82 mmol) in dichloromethane (5 mL) and triethylamine (4.82 mmol), 4.82 mmol of oxalyl chloride was added dropwise at 0 °C. The reaction mixture was stirred for 5 h at room temperature. After 5 h, the reaction mixture was concentrated under reduced pressure to remove the excess oxalyl chloride, and the corresponding α-keto acid chloride was formed as an intermediate at C-3 of the pyrrole nucleus. Then, the residue was dissolved in dichloromethane (5 mL) and para-aminopyridine (6.4 mmol) and triethylamine (1.2 mmol) were added to the mixture in the presence of a catalytic amount of 4-N,N-dimethylaminopyridine to obtain the target compounds. The reaction mixture was refluxed for 12 h at 45 °C. After the 12 h reflux, the reaction progress was inspected by TLC. The mixture was concentrated and the resulting residue was purified with column chromatography using silica gel Merck 60 F254 in hexane/ethyl acetate (3:1 v/v) as eluent. Finally, the purified residue was crystallized to obtain the corresponding target compounds 5a–i from diethyl ether.
2-(1-(2, 4-dichlorophenyl)-2-methyl-5-phenyl-1H-pyrrol-3-yl)-2-oxo-N-(pyridin-3-yl) acetamide (5a)
Yellow solid; yield 45%; m.p. 197–199 °C; 1H-NMR (500 MHz, CDCl3) δ: 2.42 (s, 3H, CH3), 7.33 (s, 1H, Ar), 7.34–7.37 (m, 3H, Ar), 7.38-7.39 (m, 3H, Ar), 7.48 (d, J = 2.0 Hz, 1H, Ar), 8.23-8.25 (m, 1H, pyridine), 8.41 (d, J = 9.0 Hz, 1H, pyridine), 8.48 (d, J = 4.5 Hz, 1H, pyridine), 8.82 (s, 1H, Pyrrole), 9.32 (s, 1H, pyridine), 9.88 (s, 1H, NH). 13C-NMR (125 MHz, CDCl3) δ: 13.29, 112.44, 113.70, 117.02, 127.55, 128.20, 128.26, 128.35, 130.41, 131.17, 133.92, 134.14, 135.23, 135.99, 143.97, 144.10, 150.83, 160.91, 180.79. Mass, m/z (%): 449 (9), 328(40), 290 (47), 83(59), 55(100).
2-(1-(2, 5-dichlorophenyl)-2-methyl-5-phenyl-1H-pyrrol-3-yl)-2-oxo-N-(pyridin-3-yl) acetamide (5b)
Yellow solid; yield 47%; m.p. 107–109 °C; 1H-NMR (500 MHz, CDCl3) δ: 2.43 (s, 3H, CH3), 7.14 (d, J = 2.0 Hz, 1H, Ar), 7.16 (d, J = 2.5 Hz, 1H, Ar), 7.31 (s, 1H, Ar), 7.37–7.39 (m, 3H, Ar), 8.26-8.28 (m, 2H, Ar), 8.48-8.50 (m, 1H, pyridine), 8.51 (d, J = 5.0 Hz, 1H, pyridine), 8.54 (d, J = 2.5 Hz, 1H, pyridine), 8.87 (s, 1H, H- Pyrrole), 9.38 (s, 1H, pyridine), 9.90 (s, 1H, NH). 13C-NMR (125 MHz, CDCl3) δ: 13.23, 120.61, 121.69, 125.20, 127.08, 127.29, 128.00, 128.55, 128.99, 129.60, 129.93, 130.12, 133.14, 133.38, 134.61, 158.23, 160.50, 181.20. Mass, m/z (%): 449 (52), 330 (100), 293 (10), 264 (32), 230 (12), 128 (56), 78 (27), 51(10).
2-(1-(3, 4-dichlorophenyl)-2-methyl-5-phenyl-1H-pyrrol-3-yl)-2-oxo-N-(pyridin-3-yl) acetamide (5c)
Yellow solid; yield 57%; m.p. 213–215 °C; 1H-NMR (500 MHz, CDCl3) δ: 2.51 (s, 3H, CH3), 7.01 (d, J = 7.0 Hz, 1H, Ar), 7.10 (d, J = 7.0 Hz, 1H, Ar), 7.22–7.23(m, 5H, Ar), 7.26-7.33 (m, 1H, pyridine), 7.50 (d, J = 8.5 Hz, 1H, pyridine), 7.63 (s, 1H, Ar), 8.33 (d, J = 7.5 Hz, 1H, pyridine), 8.43 (s, 1H, Pyrrole), 8.79 (s, 1H, pyridine), 9.30 (s, 1H, NH). 13C-NMR (125 MHz, CDCl3) δ: 13.77, 112.79, 113.72, 117.12, 127.45, 127.61, 128.36, 128.40, 129.96, 131.00, 131.06, 133.34, 133.41, 135.00, 136.22, 143.39, 143.96, 150.8, 150.83, 160.97, 180.88. Mass, m/z (%): 449 (10), 328 (100), 264 (50), 163 (7), 128 (12), 78 (8), 51(4).
2-(1-(3-chlorophenyl)-2-methyl-5-phenyl-1H-pyrrol-3-yl)-2-oxo-N-(pyridin-3-yl)acetamide (5d)
Yellow solid; yield 54%; m.p. 164–166 °C; 1H-NMR (500 MHz, CDCl3) δ: 2.50 (s, 3H, CH3), 7.02–7.26 (m, 8H, Ar), 7.38–7.43 (m, 3H, pyridine), 7.59 (s, 1H, Ar), 7.82 (s, 1H, Pyrrole), 8.60 (s, 1H, pyridine), 9.62 (s, 1H, NH). 13C-NMR (125 MHz, CDCl3) δ: 13.23, 112.98, 113.66, 116.54, 125.41, 127.77, 128.81, 129.13, 130.19, 130.40, 134.13, 134.14, 136.32, 143.29, 143.98, 150.84, 161.18, 180.37. Mass, m/z (%): 415 (< 1), 328 (37), 264(10), 149 (15), 129 (18), 97 (40), 71 (60), 55(100).
2-(5-(4-chlorophenyl)-1-(2, 4-dichlorophenyl)-2-methyl-1H-pyrrol-3-yl)-2-oxo-N-(pyridin-3-yl) acetamide (5e)
Yellow solid; yield 87%; m.p. 98–100 °C; 1H-NMR (500 MHz, CDCl3) δ: 2.42 (s, 3H, CH3), 7.33–7.34 (m, 2H, Ar), 7.37 (s, 1H, Ar), 7.38 (d, J = 4.5 Hz, 2H, Ar), 7.48 (d, J = 2.0 Hz, 2H, Ar), 8.25 (d, J = 9.0 Hz, 1H, pyridine), 8.39–8.41 (m, 1H, pyridine), 8.48 (d, J = 4.5 Hz, 1H, pyridine), 8.82 (s, 1H, Pyrrole), 9.24 (s, 1H, pyridine), 9.85 (s, 1H, NH). 13C-NMR (125 MHz, CDCl3) δ: 13.23, 112.87, 117.23, 123.73, 126.75, 128.25, 128.37, 128.47, 128.60, 129.12, 129.34, 129.72, 130.25, 130.36, 130.52, 131.46, 133.48, 133.70, 133.82, 133.87, 134.10, 136.18, 141.44, 144.10, 145.85, 160.70, 181.20. Mass, m/z (%): 483 (11), 362 (100), 324(5), 149 (50), 55(75).
2-(5-(4-chlorophenyl)-1-(2, 5-dichlorophenyl)-2-methyl-1H-pyrrol-3-yl)-2-oxo-N-(pyridin-3-yl) acetamide (5f)
Yellow solid; yield 84%; m.p. 101–103 °C; 1H-NMR (500 MHz, CDCl3) δ: 2.43 (s, 3H, CH3), 7.06 (d, J = 8.5 Hz, 1H, Ar), 7.20 (d, J = 8.0 Hz, 2H, Ar), 7.23 (d, J = 1.5 Hz, 1H, Ar), 7.42 (d, J = 8.5 Hz, 2H, Ar), 7.46–7.48 (m, 2H, pyridine), 7.61 (s, 1H, Ar), 8.47 (d, J = 4.5 Hz, 1H, pyridine), 8.56 (s, 1H, Pyrrole), 9.11 (s, 1H, pyridine), 9.72 (s, 1H, NH). 13C-NMR (125 MHz, CDCl3) δ: 13.23, 112.91, 117.25, 123.72, 126.76, 128.24, 128.49, 128.61, 129.05, 129.28, 129.63, 130.38, 130.84, 131.36, 131.48, 133.48, 133.74, 133.88, 134.83, 136.05, 141.46, 145.85, 160.69, 181.25. Mass, m/z (%): 483 (8), 362 (100), 264(4), 163 (9), 126 (38), 78(10).
2-(5-(4-chlorophenyl)-1-(3, 4-dichlorophenyl)-2-methyl-1H-pyrrol-3-yl)-2-oxo-N-(pyridin-3-yl) acetamide (5 g)
Yellow solid; yield 76%; m.p. 176–178 °C; 1H-NMR (500 MHz, CDCl3) δ: 2.49 (s, 3H, CH3), 6.98 (d, J = 2.5 Hz, 1H, Ar), 7.00 (d, J = 2.5 Hz, 1H, Ar), 7.02 (d, J = 8.5 Hz, 2H, Ar), 7.21 (d, J = 8.5 Hz, 2H, Ar), 7.36–7.38 (m, 1H, pyridine), 7.52 (d, J = 8.5 Hz, 1H, pyridine), 7.61 (s, 1H, Ar), 8.35 (d, J = 8.0 Hz, 1H, pyridine), 8.43 (s, 1H, Pyrrole), 8.84 (s, 1H, pyridine), 9.39 (s, 1H, NH). 13C-NMR (125 MHz, CDCl3) δ: 13.77, 113.17, 113.76, 113.95, 117.19, 119.36, 121.83, 127.58, 128.71, 129.51, 129.94, 130.83, 131.28, 133.48, 133.67, 133.75, 136.29, 143.94, 150.87, 160.88, 180.85. Mass, m/z (%): 483 (11), 362 (100), 327 (5), 298(7), 264 (7), 163 (10), 78 (12), 51(9).
2-(1-(2, 4-dichlorophenyl)-2, 5-dimethyl-1H-pyrrol-3-yl)-2-oxo-N-(pyridin-3-yl) acetamide (5 h)
Yellow solid; yield 44%; m.p. 140–142 °C; 1H-NMR (500 MHz, CDCl3) δ: 1.95 (s, 3H, CH3), 2.32 (s, 3H, CH3), 7.21–7.24 (m, 1H, pyridine), 7.25 (d, J = 1 Hz, 1H, Ar), 7.36 (d, J = 4.5 Hz, 1H, Ar), 7.44 (d, J = 8.5 Hz, 1H, pyridine), 7.62 (s, 1H, Ar), 8.33 (d, J = 7.0 Hz, 1H, pyridine), 8.41 (s, 1H, Pyrrole), 8.83 (s, 1H, pyridine), 9.36 (s, 1H, NH). 13C-NMR (125 MHz, CDCl3) δ: 12.15, 13.22, 110.46, 116.35, 121.81, 123.67, 127.15, 128.42, 129.68, 130.72, 131.76, 133.40, 134.12, 136.18, 142.26, 146.57, 157.09, 161.19, 181.03. Mass, m/z (%): 387 (31), 266 (100), 231 (5), 202(37), 167 (20), 78 (18), 51(14).
2-(1-(3, 4-dichlorophenyl)-2, 5-dimethyl-1H-pyrrol-3-yl)-2-oxo-N-(pyridin-3-yl) acetamide (5i)
Yellow solid; yield 36%; m.p. 150–152 °C; 1H-NMR (500 MHz, CDCl3) δ: 2.02 (s, 3H, CH3), 2.39 (s, 3H, CH3), 7.10 (d, J = 8.5 Hz, 1H, Ar), 7.21–7.27 (m, 1H, pyridine), 7.27 (s, 1H, Ar), 7.37 (d, J = 7.5 Hz, 1H, Ar), 7.64 (d, J = 8.5 Hz, 1H, pyridine), 8.34 (d, J = 8.0 Hz, 1H, pyridine), 8.42 (s, 1H, Pyrrole), 8.82 (s, 1H, pyridine), 9.36 (s, 1H, NH). 13C-NMR (125 MHz, CDCl3) δ: 12.65, 13.71, 110.63, 113.66, 116.15, 127.29, 129.86, 129.94, 131.40, 133.79, 133.85, 136.15, 142.22, 144.01, 150.82, 161.14, 180.48. Mass, m/z (%): 387 (5), 266 (100), 231 (4), 202(7), 167 (5), 78 (8), 51(4).
Biological evaluation
In vitro α-glucosidase inhibition activity
The α-glucosidase inhibitory activity of the synthesized compounds was evaluated using p-nitrophenyl‐α‐d‐glucopyranoside (pNPG) as a substrate based on a previously reported method (Nikookar et al. 2018). The α-glucosidase enzyme (EC3.2.1.20, Saccharomyces cerevisiae, 20 U/mg) and substrate (pNPG) were purchased from Sigma-Aldrich. The desired concentrations of enzyme were prepared in potassium phosphate buffer (pH 6.8, 50 mM), and the target compounds 5a–i were dissolved in DMSO (10% final concentration). A volume of 20 μL of α‐glucosidase solution (0.5 U/mL), different concentrations of the target compounds 5a–i (20 μL), and 135 μL of potassium phosphate buffer were added to the 96-well plate and incubated at 37 °C for 10 min. Then, 25 µL of 4 mM p‐nitrophenyl‐α‐d‐glucopyranoside solution in 0.05 M phosphate buffer (pH = 6.8) was added to each well and the reaction mixture was allowed to incubate for 20 min at 37 °C. Finally, the reaction was terminated by adding 50 µL of 0.2 M sodium carbonate solution and then the change in absorbance was measured at 405 nm with a spectrophotometer (Gen5, Power wave xs2, BioTek, America). DMSO and acarbose were used as the control and standard inhibitor, respectively. Three independent assays were performed in each concentration. The mean of three experiments was used to calculate IC50. The α‐glucosidase inhibitory activity was expressed as the percentage of enzyme inhibition for each synthesized compound and calculated by the following formula (Rouzbehan et al. 2017; Hati et al. 2015):
The concentration of the target compounds required to inhibit 50% of the enzyme activity (IC50) values was obtained from non-linear regression curve using the logit method.
Kinetic study
The inhibition mode of the most potent compound 5e as identified with its lowest IC50 was investigated against α-glucosidase activity. The enzyme solution (1 U/mL, 20 µL) was incubated with different concentrations (0, 70, 95, and 111 µM) of compound 5e (20 µL) at 30 °C for 15 min. The reaction was initiated by adding different concentrations of p-nitrophenyl α-d glucopyranoside (1–10 mM) as substrate, and change in absorbance was measured in the absence and presence of compound 5e for 20 min at 405 nm by using a spectrophotometer (Gen5, Power wave xs2, BioTek, USA) (Nikookar et al. 2018). A Lineweaver–Burk plot was produced to identify the mode of inhibition and the Michaelis–Menten constant (Km) value was determined from the plot between the reciprocal of the substrate concentration (1/[S]) and the reciprocal of enzyme rate (1/V) over various inhibitor concentrations. The value of inhibition constant (Ki) was made by secondary plots of the inhibitor concentration [I] versus Km.
Molecular docking studies
Molecular docking studies were performed to comprehend the possible interactions of the most potent compounds with the amino acid residues on the active site of the target enzyme. AutoDock version 4.2.6, AutoDock Tools (ADT) version 1.5.6 and Discovery studio v16.1.0.15350 were used to conduct the docking studies. The most potent compounds from the series were generated as a ligand with their 3D structure, 5e and 5a and the reference drug, acarbose, using ChemDraw Ultra 12.0.2 version of Cambridge University. To perform the docking studies, we retrieved the three-dimensional structure of isomaltase from Saccharomyces cerevisiae (PDB ID: 3A4A; Resolution 1.6 Å) from the Brookhaven protein database (www.rcsb.org/pdb/) as there is no report yet about the three-dimensional structure of α-glucosidase from Saccharomyces cerevisiae. Then, the protein structure was prepared by removing the water molecules and original inhibitors. Subsequently, the AutoDock format of protein and potent compounds was prepared and then the attained structure of the enzyme was used as an input for the AutoGrid program. AutoGrid was used to prepare the grid map using a grid box. The dimension of the grid size was set to 60 × 60 × 60 with a grid spacing of 0.375 Å. The center of grid box was designated in coordinates x = 21.272, y = −0.751, and z = 18.633. Fifty runs of the AUTODOCK search by the Lamarckian genetic algorithm were performed for each docked system. To analyze the interactions between the target enzyme and the inhibitor, the best pose of each ligand was designated and the results were visualized with Discovery Studio Visualizer v16.1.0.15350 (Accelrys, San Diego, USA).
Results and discussion
Chemistry
The general procedure for synthesis of the final target compounds 5a–i is described in Schemes 1 and 2. Briefly, pentane-1,4-dione derivatives 3a, b were obtained by reacting appropriate benzaldehydes 1a, b with methyl vinyl ketone 2 in the presence of sodium cyanide as a catalyst in dimethylformamide (DMF) at 35 °C following Stetter reaction and other previously reported methods (Peloquin et al. 2012; Biava et al. 2008; Stetter 1976). The corresponding diketones 3a–c (3c was purchased) were dissolved in ethanol (EtOH) and refluxed for 8 h with aniline derivatives (3-chloroaniline, 2,4-dichloroaniline, 2,5-dichloroaniline, and 3,4-dichloroaniline) in the presence of a catalytic amount of p-toluenesulfonic acid (PTSA). Following the Paal–Knorr condensation, the reaction mixture was cyclized to yield the corresponding 1 N-phenylpyrrole derivatives 4a–i (Biava et al. 2011; Stetter 1976). The target compounds 5a–i were achieved with a two-step reaction. Primarily, the 1 N-phenylpyrrole derivatives 4a–i dissolved in dichloromethane (DCM) and oxalyl chloride and triethylamine (Et3N) were added dropwise to the reaction mixture in an ice bath. The corresponding α-keto acid chloride was formed through regioselective acylation as the side chain of 4a–i at C3 of the pyrrole ring reacted with the oxalyl chloride. Then, after removing the unreacted oxalyl chloride with a rotary evaporator, the residue was dissolved in DCM and reacted with 3-aminopyridine in the presence of Et3N and a catalytic amount of 4-N,N-dimethylaminopyridine at 45 °C for 12 h. After 12 h, the reaction progress was examined by thin layer chromatography and UV light. Finally, the excess solvents were removed under reduced pressure and the target compounds 5a–i were purified with column chromatography using hexane/ethyl acetate (3:1, v/v) as eluent and recrystallized from diethyl ether. Altogether, nine new target compounds (5a–i) were synthesized in good to moderate yields. The chemical structure of the final compounds was confirmed by 1HNMR, 13CNMR, and mass spectroscopy.
In vitro α-glucosidase inhibition activity
α-Glucosidase activities are associated with two small intestinal membrane-bound enzymes: maltase-glucoamylase (MGAM) and sucrase-isomaltase (SI). As they are involved in the breakdown of dietary sugars and starches, MGAM and SI are attractive targets for inhibition, as a means of controlling blood glucose levels in individuals with type 2 diabetes (Santos et al. 2018; Flores-Bocanegra et al. 2015; Palcic et al. 1999). Therefore, the discovery and development of potential α-glucosidase inhibitors can be regarded as a promising topic in recent years.
The activity of the target compounds 5a–i was assessed in vitro against yeast α-glucosidase (EC 3.2.1.20) inhibition. The usage of this source of enzyme has been reported for inhibitory properties of α-glucosidase in numerous papers (Guerreiro et al. 2013; Nikookar et al. 2018; Wang et al., 2016a, Wang et al., 2016b; Wang et al. 2017a; Wang et al. 2017b). Chiba and Shimomura proposed a slightly different classification of α-glucosidases that is based on recognition of homologous sites of the enzymes’ amino acid sequences. According to the mentioned classification, E. coli and other bacterial species constitute family I of α-glucosidases, and α-glucosidases from mammalian tissues, plants, and fungi constitute family II. Three-dimensional structures have also been obtained for the enzymes from A. oryzae, pig pancreas, A. niger, barley, and Pseudomonas stutzeri. All these structures have similar topological features. In all these enzymes, the active site is a slit formed at the C-terminus of the β-sheet of the (β/α) eight barrel (Chiba and Shimomura 1978; Krasikov et al. 2001; Ghasemi and Mehraban 2015). However, some structural features appear to have been conserved between α-glucosidase from lower and higher eukaryotes. Alignment of the plant and yeast glucosidase with their mammalian counterparts (Homo sapiens Swiss-Prot Q13724, Mus musculus Swiss-Prot Q9Z2W5, and Rattus rattus Swiss-Prot O88941) and the enzyme from C. elegans (Swiss-Prot Q19426) shows Ala and His residues at the binding site of α-glucosidases (Faridmoayer and Scaman 2005). In Saccharomyces cerevisiae, CWH41 encodes processing α-glucosidase (Cwh41p) (Romero et al. 1997) which is a member of family 63 of the glycoside hydrolases (Cantarel et al. 2009). Cwh41p is a type II membrane glycoprotein with a proposed domain orientation comparable to its mammalian counterpart (Shailubhai et al. 1991; Kalz-Fuller et al.1995; Jiang et al. 1996). Sequence comparison between enzyme orthologs (i.e., human, Caenorhabditis elegans and S. cerevisiae) shows higher identity (34–49%) at the end of the C-terminus domain where the putative catalytic domain is located (Romero et al. 1997).
Furthermore, the similar results have been reported for assessment of α-glucosidase inhibitory activity using two different kinds of enzymes (α-glucosidase EC 3.2.1.20; Sigma, and rat intestinal α-glucosidase source). For kaempferol, a natural product separated from herb, IC50 were 61 µM and 59 µM, respectively. The percentage of enzyme inhibition at concentrations of 0.83 µM was reported as 68% and 55%, respectively (Kang et al. 2011).
Compound 5e was bound to active site on the enzyme and competed with the substrate for binding to the active site, so the type of inhibition was competitive inhibition. The results were compared with acarbose, the marketed α-glucosidase inhibitor, as a standard drug. The activity of acarbose has been reported in a range of 600–800 µM related to the source of the enzyme. In our experiments, 750 ± 9 µM was found for IC50 of acarbose in triplicated tests. The in vitro α-glucosidase inhibitory activity was carried out based on the previously reported method (Nikookar et al. 2018). The results were described as the concentration of the target compounds required to inhibit 50% of the α-glucosidase activity (IC50 values) (Sun et al. 2017; Biava et al. 2011) and are summarized in Table 1. Accordingly, a careful remark of the bioactivity data revealed that all the target compounds 5a–i exhibited better inhibitory activity than the standard drug, acarbose. Optimization of the α-glucosidase inhibitory activity of 2-(2-methyl-1-phenyl-1H-pyrrol-3-yl)-2-oxo-N-(priding-3-yl) acetamide derivatives 5a–i was performed using chloride substituents on a different position of the phenyl rings. The differences in the pattern of inhibition of the synthesized compounds are mainly due to the position of the substituents on the phenyl moiety.
The most potent compounds were 5e, 5a, and 5c with IC50 values 111 ± 12, 196 ± 10, and 197 ± 8 µM, respectively, while the IC50 value of acarbose was 750 ± 8 µM (Table 1). The other target compounds showed a moderate inhibitory activity around 1.11- to 2.72-fold more than acarbose with the IC50 values in the range of 276 ± 10 to 673 ± 12 µM. The greater inhibitory activity of compound 5e over compound 5a might be due to the introduction of an electron-withdrawing group through its strong inductive effect as a para substituent on the aromatic ring attached to the pyrrole moiety, as both compounds contain 2, 4-dichloro substituents on the 1N-phenyl ring. The presence of a para-chloro substituent on the 5-phenyl ring of 5e improves its binding interaction with the amino acid residue in the active site of the enzyme through hydrophobic interaction as the experimental findings were confirmed by molecular docking studies. However, when the position of 2,4-dichloro substituents of compound 5e on the 1N-phenyl ring was shifted to 2,5-dichloro and 3,4-dichloro positions on the same moiety as in the case of compound 5f (IC50 = 573 ± 12 µM) and compound 5 g (IC50 = 639 ± 13 µM), respectively, the inhibitory activity noticeably diminished by more than fivefold as compared to the most active compound 5e. Even though the inhibitory activity of these compounds is reduced in comparison with the most potent one, they have still a better inhibitory activity than the standard drug, acarbose (IC50 = 750 ± 9 µM). This result indicates that the orientation of the substituents on the phenyl ring has a great impact on the α-glucosidase inhibitory activity.
Similarly, when the position of 2,4-dichloro substituents of compound 5a (IC50 = 196 ± 10 µM) on the 1N-phenyl ring was shifted to 2,5-dichloro as in the case of compound 5b (IC50 = 663 ± 11 µM), the inhibitory activity decreased by more than threefold, whereas there was no difference when it was shifted to position 3,4-dichloro as in compound 5c (IC50 = 197 ± 8 µM). On the other hand, the replacement of substituted 5-phenyl moiety of the pyrrole ring with methyl moiety reduces the inhibitory activity greatly as in the case of compound 5 h (IC50 = 494 ± 10 µM) and 5i (IC50 = 673 ± 12 µM); however, the inhibition potency was still better than that of acarbose. This indicated that the nature of substituents at position 5 of the pyrrole ring exhibited a significant effect on the inhibition potency of the α-glucosidase enzyme.
As shown in Fig. 1, a heterocyclic core containing di-aryl rings (a, b, c) or tri-aryl rings (d, e) have been reported as either α-glucosidase inhibitor or anti-hyperglycemic agents. For a–d derivatives (Fig. 1), a 5–70 μM range of IC50 was reported that shows the triazine or imidazole served as a suitable core for design of α-glucosidase inhibitor with high potency. For pyrrole derivatives (e, Fig. 1), the study involved an in vivo test (goel et al. 2004) without any data related to α-glucosidase inhibitory activity. On the other hand, there are several reports that have shown α-glucosidase inhibitory activity of pyrrolidine derivatives. Between a large numbers of the related structure (Kasturi et al. 2017, Damsud et al. 2013, and Guerreiro et al. 2013), only two compounds were active, namely 3-hydroxypyrrolidine-2,5-diyl)dimethanol, a polyhydroxylated pyrrolidine, and 6-hydroxy-5-(hydroxymethyl)tetrahydro-1H-pyrrolizin-3(2H)-one (an oxapyrrolizidine molecule). On the other hand, Damsud et al. 2013 isolated three new phenylpropanoyl amides: chaplupyrrolidones a and b, which contain a 5-oxygenated-∆3-2-pyrrolidone moiety, and deacetylsarmentamide b (a dihydroxy pyrrolidine derivative). The chaplupyrrolidones a and b displayed a weak α-glucosidase inhibitory activity with IC50 values of 7820 and 430 µM, respectively, while deacetylsarmentamide b was inactive as compared to acarbose (Damsud et al. 2013). Five-membered iminocyclitol α-glucosidase inhibitors were reported (Guerreiro et al. 2013) that indicates the reported compounds were weaker than acarbose with IC50 ranging from 10.9 to 38 mM. Some studies showed that the pyrrolidine derivatives exhibited either moderate α-glucosidase inhibitory activity or were totally inactive as compared to acarbose.
However, our compounds showed less activity than triazole or imidazole derivatives, and the pyrrole scaffold illustrated remarkable better α-glucosidase inhibitory effects when compared with the activity of pyrrolidine derivatives.
Kinetic study
The mechanism of enzyme inhibition of the most potent compound 5e on α-glucosidase was evaluated using a kinetic study. The inhibition mode and value of inhibition constant (Ki) were determined by Lineweaver–Burk plots and secondary re-plot of Lineweaver–Burk plots, respectively (Nikookar et al. 2018).
In the presence of different concentrations of compound 5e, the Lineweaver–Burk plot showed that the Michaelis–Menten constant (Km) gradually increased while Vmax remained unaffected (Fig. 2a), which indicates that the type of inhibition is competitive. The same mechanism for enzyme inhibition of acarbose was observed and the results are presented in Fig. 3. Again, the gradual increase of Km and unchanged Vmax indicates competitive inhibition for acarbose. This result suggests that compound 5e competes with the substrate for binding to the active site of the enzyme. Moreover, the plot of Km versus various concentrations of compound 5e presented an estimate of the value of inhibition constant Ki as 106 µM (Fig. 2b).

Kinetics of α-glucosidase inhibition by compound 5e. a The Lineweaver–Burk plot in the absence and presence of different concentrations of compound 5e (µM); b the secondary plot between Km and different concentrations of compound 5e

Kinetics of α-gluosidase inhibition by acarbose. The Lineweaver–Burk plot in the absence and presence of different concentrations of acarbose
Molecular docking
Molecular docking study was carried out on the possible interaction of the target compounds with the amino acid residues in the active site of the modeled α-glucosidase. The most active compounds 5e and 5a and the reference drug, acarbose were docked using AutoDock Tools version 1.5.6. To date, there has been no report regarding the crystal structure of α-glucosidase from Saccharomyces cerevisiae. It is obvious that molecular models attained from crystallography are used widely as a tool for enlightening molecular information of life progressions. All methods that might be obtained from a model have their own weakness and strength; however, the majority of crystal structures are formed identical to the aqueous structure. Furthermore, it yields high atomic resolution irrespective of the molecular weight of the sample and is also more suitable for membrane proteins, water-soluble proteins, and macromolecular complexes. It is also an influential tool, if handled properly, to convey a consistent structural data of biological macromolecules and regulate the structure and position of the active center, which assists in comprehending in what way the protein recognizes and binds to the ligand fragments (Rhodes 2006). But, when the crystal structure of a specified molecule is not manageable, homology modeling can be an outstanding replacement to upkeep drug discovery determinations (Levoin et al. 2011). Hence, we used the three-dimensional structure of isomaltase from Saccharomyces cerevisiae (PDB ID: 3A4A; Resolution 1.6 Å) to prepare the protein for docking, which has a 71% identity and 84% similarity to the α-glucosidase from Saccharomyces cerevisiae (Leong et al. 2018; Sun et al. 2017). The binding mode of acarbose, as shown in Fig. 4, demonstrates that it interacts with the active site of isomaltase via ten hydrogen bonds, two carbon–hydrogen bonds, and one hydrophobic interaction with the residues Asp69, Phe159, Gln182, Asp215, Asp242, Glu277, Asp307, Arg315, Gln353, Glu411, and Arg442.

The predicted binding mode of acarbose in the active site pocket
Compound 5e, the most active one from the target compounds, recognized hydrogen bond interaction with the active site residue His280 through the carbonyl oxygen of the amide unit of the acetamide chain (Fig. 5). π– π T-shaped interaction was formed with Tyr158 through the 5-phenyl ring. The pyridine ring established π–anion interactions with Asp307 residue, while the 5-phenyl and 1N-phenyl rings of compound 5e formed the same interaction with the Asp352 residue. Moreover, the para-chloro substituents of 5-phenyl and 1N-phenyl rings of compound 5e created π–alkyl interactions with the residues Phe178 and Phe303, respectively (Fig. 5a). Other hydrophobic interactions were also detected through the 5-phenyl ring with the residue Val216, 2-methyl substituent of pyrrole ring with the residue Phe303, pyridine ring with the residue Arg315, and the ortho-chloro substituent of the 1N-phenyl ring with the residue Arg442 (Table 2).

The predicted mode of binding of compounds 5e and 5a in the active site pocket. a The interactions between compound 5e and the active site of the modeled α-glucosidase b The interactions between compound 5a and the active site of the modeled α-glucosidase
Compound 5a, the second most active of the series, interacted with the active site residue Arg442 via hydrogen bond with the carbonyl oxygen of the acetamide chain. π–π stacked interaction was formed with the residues Phe303 and Phe178 through the 5-phenyl and pyridine rings, respectively. Hydrophobic interactions were also observed with the residues Tyr158, Val216, His280, Phe303, and Arg315 (Fig. 5b). The hydrophobic interactions of residues Tyr158, His280, and Phe303 were observed via 2-chloro substituent of the 1N-phenyl ring, while residues Arg315 and Val216 formed the hydrophobic interaction through 1N-phenyl and pyridine rings, respectively. Moreover, residues Arg315 and Glu411 made π–cation and π–anion interactions with the 5-phenyl and pyrrole rings, respectively.
On top of their interactions, the binding energies of compounds 5e, 5a, and acarbose, as observed from the best docking conformations, showed that both compounds 5e and 5a have a lower free binding energy (− 4.27 kcal/mol and − 3.17 kcal/mol, respectively) than acarbose (2.47 kcal/mol). Therefore, the results emphasize that the target compounds bind more easily to the target enzyme (α-glucosidase) than the reference drug, acarbose. In general, the outcomes from the molecular docking studies supported the results obtained from the in vitro assay.
Conclusion
The reported derivatives exhibited α-glucosidase inhibition activity better than the reference compound (acarbose). Compounds 5e, 5a, and 5c showed potent activity among the series. Compound 5e bearing a 2,4-dichloro substituent on 1N-phenyl ring and para-chloro substituent of the 5-phenyl ring of the pyrrole scaffold exhibited the most potent α-glucosidase inhibitory activity with an IC50 value of 111 ± 11 μM. The outcomes from the molecular docking studies of compounds 5e and 5a supported the results obtained from the in vitro assay. Moreover, these compounds have a lower free binding energy than the reference drug, acarbose, which indicates that they bind easily to the target enzyme than acarbose. This might be the best clue for the good inhibitory activity of the compounds. In conclusion, the synthesized compounds of this class of heterocyclic motifs have shown promising results for further development of potent, selective, and efficacious inhibitors against the α-glucosidase enzyme that could be a potential candidate for the treatment of diabetes.
References
Abdelrazek FM, Metwally NH (2006) Synthesis of some new N-substituted pyrroles, pyrrolo[1,2-a]quinazoline, and diaza-as-indacene derivatives. Synth Commun 36:83–839
Biava M, Fioravantia R, Porrettaa GC, Deiddab D, Maullub C, Pompei R (1999) New pyrrole derivatives as antimycobacterial agents analogs of BM212. Bioorg Med Chem Lett 9:2983–2988
Biava M, Porretta GC, Poce G, De Logu A, Saddi M, Meleddu R et al (2008) 1,5-Diphenylpyrrole derivatives as antimycobacterial agents. Probing the influence on antimycobacterial activity of lipophilic substituents at the phenyl rings. J Med Chem 51:3644–3648
Biava M, Porretta GC, Poce G, De Logu A, Meleddu R, De Rossi E et al (2009) 1,5-Diaryl-2-ethyl pyrrole derivatives as antimycobacterial agents: design, synthesis, and microbiological evaluation. Eur J Med Chem 44:4734–4738
Biava M, Porretta GC, Poce G, Battilocchio C, Manetti F, Botta M et al (2010) Novel ester and acid derivatives of the 1,5-diarylpyrrole scaffold as anti-inflammatory and analgesic agents. synthesis and in vitro and in vivo biological evaluation. J Med Chem 53:723–733
Biava M, Porretta GC, Poce G, Battilocchio C, Alfonso S, Rovini M et al (2011) Novel analgesic/anti-inflammatory agents: diarylpyrrole acetic esters endowed with nitric oxide releasing properties. J Med Chem 54:7759–7771
Chiba S, Shimomura T (1978) Diversity of substrate specificity of α-glucosidase. J Jpn Soc Starch Sci 25:105–112
Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B (2009) The carbohydrate-active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res 54:7759–7771
Damsud T, Adisakwattana S, Phuwapraisirisan P (2013) Three new phenylpropanoyl amides from the leaves of Piper sarmentosum and their α-glucosidase inhibitory activities. Phytochem Lett 6:350–354
Deshpande AD, Harris-Hayes M, Schootman M (2008) Epidemiology of diabetes and diabetes-related complications. Phys Ther 88:1254–1264
Dhameja M, Gupata P (2019) Synthetic heterocyclic candidate as promising α-glucosidase inhibitors: an overview. Eur J Med Chem 176:343–377
Domagala A, Jarosz T, Lapkowski M (2015) Living on pyrrolic foundations-advances in natural and artificial bioactive pyrrole derivatives. Eur J Med Chem 100:176–187
Famiglini V, Coluccia A, Brancale A, Pelliccia S, La Regina G, Silvestri R (2013) Arylsulfone-based HIV-1 non-nucleoside reverse transcriptase inhibitors. Future Med Chem 5:2141–2156
Faridmoayer A, Scaman CH (2005) Binding residues and catalytic domain of soluble Saccharomyces cerevisiae processing alpha-glucosidase I. Glycobiolog 15:1341–1348
Flores-Bocanegra L, Pérez-Vásquez A, Torres-Piedra M, Bye R, Linares E, Mata R (2015) α-Glucosidase inhibitors from Vauquelinia corymbosa. Molecules 20:15330–15342
Ghani U (2015) Re-exploring promising α-glucosidase inhibitors for potential development into oral anti-diabetic drugs: finding needle in the haystack. Eur J Med Chem 103:133–162
Ghasemi Y, Mehraban MH (2015) A computational comparative study of α-Glucosidase enzyme divergence. J Appl Bioinform Comput Biol 4:2–5
Goel A, Agarwal N, Singh FV, Sharon A, Tiwari P, Dixit M et al (2004) Antihyperglycemic activity of 2-methyl-3,4,5-triaryl-1H-pyrroles in SLM and STZ models. Bioorg Med Chem Lett 14:1089–1092
Gollapalli M, Taha M, Ullah H, Nawaz M, AlMuqarrabun LM, Rahim F et al (2018) Synthesis of Bis-indolylmethane sulfonohydrazides derivatives as potent α-Glucosidase inhibitors. Bioorg Chem 80:112–120
Guerreiro LR, Carreiro EP, Fernandes L, Cardote TA, Moreira R, Caldeira AT et al (2013) Five-membered iminocyclitol α-glucosidase inhibitors: synthetic, biological screening and in silico studies. Bioorg Med Chem 21:1911–1917
Hati S, Madurkar SM, Bathula C, Thulluri C, Agarwal R, Siddiqui FA et al (2015) Design, synthesis and biological evaluation of small molecules as potent glucosidase inhibitors. Eur J Med Chem 100:188–196
Ibrahim MA, Koorbanally NA, Islam MS (2014) Antioxidative activity and inhibition of key enzymes linked to type-2 diabetes (α-glucosidase and α-amylase) by Khaya senegalensis. Acta Pharmaceutica 64:311–324
Jacobi PA, Coutts LD, Guo J, Hauck SI, Leung SH (2000) New strategies for the synthesis of biologically important tetrapyrroles. The “B, C + D + A” approach to linear tetrapyrroles. J Org Chem 65:205–213
Jiang B, Sheraton J, Ranv VFJ, Dijkgraaf GJP, Klis FM, Bussey H (1996) CWH41 encodes a novel endoplasmic reticulum membrane N-glycoprotein involved in P-l,6-glucan assembly. J Bacteriol 178:1162–1171
Joshi SD, Vagdevi HM, Vaidya VP, Gadaginamath GS (2008) Synthesis of new 4-pyrrol-1-yl benzoic acid hydrazide analogs and some derived oxadiazole, triazole and pyrrole ring systems: a novel class of potential antibacterial and antitubercular agents. Eur J Med Chem 43:1989–1996
Joshi SR, Standl E, Tong N, Shah P, Kalra S, Rathod R (2015) Therapeutic potential of α-glucosidase inhibitors in type 2 diabetes mellitus: an evidence-based review. Expert Opin Pharmacother 16:1959–1981
Kalz-Fuller B, Bieberich E, Bause E (1995) Cloning and expression of glucosidase I from human hippocampus. Eur J Biochem 231:344–351
Kang WY, Yl Song, Zhang L (2011) α-Glucosidase inhibitory and antioxidant properties and antidiabetic activity of Hypericum ascyron L. Med Chem Res 20:809–816
Kasturi S, Surarapu S, Uppalanchi S, Anireddy JS, Dwivedi S, Anantaraju HS, Perumal Y, Sigalapalli DK, Babu BN, Ethiraj KS (2017) Synthesis and α-glucosidase inhibition activity of dihydroxy pyrrolidines. Bioorg Med Chem Lett 27:2818–2823
Kim KY, Nam KA, Kurihara H, Kim SM (2008) Potent α-glucosidase inhibitors purified from the red alga Grateloupia elliptica. Phytochem 69:2820–2825
Kitabchi AE, Umpierrez GE, Miles JM, Fisher JN (2009) Hyperglycemic crises in adult patients with diabetes. Diabetes Care 32:1335–1343
Krasikov VV, Karelov DV, Firsov LM (2001) α-Glucosidases. Biochem (Moscow) 6:267–281
Leong SW, Abas F, Lam KW, Yusoff K (2018) In vitro and in silico evaluations of diarylpentanoid series as α-glucosidase inhibitor. Bioorg Med Chem Lett 28:302–309
Levoin N, Calmels T, Krief S, Danvy D, Berrebi-Bertrand I, Lecomte JM et al (2011) Homology model versus x-ray structure in receptor-based drug design: a retrospective analysis with the dopamine D3 receptor. ACS Med Chem lett 2:293–297
Lohray BB, Lohray V (2005) Novel pyrrole-containing hypoglycemic and hypotriglyceridemic compounds. Pure Appl Chem 77:179–184
Mccue P, Kwon Y-I, Shetty K (2005) Anti-amylase, anti-glucosidase and anti-angiotensin I converting enzyme potential of selected foods. J Food Biochem 29:278–294
Mohamed MS, Ali SA, Abdelaziz DHA, Fathallah SS (2014) Synthesis and evaluation of novel pyrroles and pyrrolopyrimidines as anti-hyperglycemic agents. Biomed Res Int 2014:249780
Nikookar H, Mohammadi-Khanaposhtani M, Imanparast S, Faramarzi MA, Ranjbar PR, Mahdavi M, Larijani B (2018) Design, synthesis and in vitro α-glucosidase inhibition of novel dihydropyrano[3,2-c]quinoline derivatives as potential anti-diabetic agents. Bioorg Chem 77:280–286
Palcic MM, Scaman CH, Otter A, Szpacenko A, Romaniouk A, Li YX, Vijay IK (1999) Processing alpha-glucosidase I is an inverting glycosidase. Glycoconj J 16:351–355
Peloquin AJ, Stone RL, Avila SE, Rudico ER, Horn CB, Gardner KA et al (2012) Synthesis of 1,3-Diphenyl-6-alkyl/aryl-substituted fulvene chromophores: observation of π–π Interactions in a 6-Pyrene-Substituted 1,3-Diphenylfulvene. J Org Chem 77:6371–6376
Powers AC (2015) Diabetes mellitus: diagnossis, classification and pathophysiology. Harrison’s Principles of Internal Medicine.19th edn. The McGraw-Hill Companies, pp 2275–2304
Ramprasad R, Madhusudhan S (2016) In vitro–amylase and–glucosidase inhibitory activities of ethanolic extract of Lactuca runcinata DC. Der Pharmacia Lett 8:231–236
Rhodes G (2006) Crystallography made crystal clear. Elsevier, Burlington, USA
Romero PA, Dijkgraaf GJ, Shahinian S, Herscovics A, Bussey H (1997) The yeast CWH41 gene encodes glucosidase I. Glycobiology 7:997–1004
Rouzbehan S, Moein S, Homaei A, Moein MR (2017) Kinetics of α-glucosidase inhibition by different fractions of three species of Labiatae extracts: a new diabetes treatment model. Pharm Biol 55:1483–1488
Sangnoi Y, Sakulkeo O, Yuenyongsawad S, Kanjana-opas A, Ingkaninan K, Plubrukarn A et al (2008) Acetylcholinesterase-inhibiting activity of pyrrole derivatives from a novel marine gliding bacterium, Rapidithrix thailandica. Mar Drugs 6:578–586
Santos C, Marisa Freitas M, Fernandes E (2018) A comprehensive review on xanthone derivatives as α-glucosidase inhibitors. Eur J Med Chem 157:1460–1749
Shailubhai K, Pukazhenthi BS, Saxena ES, Varma GM, Vijay IK (1991) Glucosidase I, a transmembrane endoplasmic reticular glycoprotein with a luminal catalytic domain. J Biol Chem 266:16587–16593
Stetter H (1976) Catalyzed addition of aldehydes to activated double-bonds-a new synthetic approach. Angew Chem 15:639–647
Sugihara H, Nagao M, Harada T, Nakajima Y, Tanimura-Inagaki K, Okajima F et al (2014) Comparison of three α-glucosidase inhibitors for glycemic control and bodyweight reduction in Japanese patients with obese type 2 diabetes. J Diabetes Investig 5:206–212
Sun H, Ding W, Song X, Wang D, Chen M, Wang K et al (2017) Synthesis of 6-hydroxyaurone analogues and evaluation of their α-glucosidase inhibitory and glucose consumption-promoting activity: development of highly active 5,6-disubstituted derivatives. Bioorg Med Chem Lett 27:3226–3230
Tundis R, Loizzo MR, Menichini F (2010) Natural products as -amylase and -glucosidase inhibitors and their hypoglycaemic potential in the treatment of diabetes: an update. Mini Rev Med Chem 10:315–331
Wang G, Wang J, He D, Li X, Li J, Peng Z (2016a) Synthesis and biological evaluation of novel 1,2,4-triazine derivatives bearing carbazole moiety as potent α-glucosidase inhibitors. Bioorg Med Chem Lett 26:2806–2809
Wang G, Peng Z, Wang J, Li J, Li X (2016b) Synthesis and biological evaluation of novel 2,4,5-triarylimidazole– 1,2,3-triazole derivatives via click chemistry as α-glucosidase inhibitors. Bioorg Med Chem Lett 26:5719–5723
Wang G, Li X, Wang J, Xie Z, Li L, Chen M, Chen S, Peng Y (2017a) Synthesis, molecular docking and α-glucosidase inhibition of 2-((5,6-diphenyl-1,2,4-triazin-3-yl)thio)-N-arylacetamides. Bioorg. Med Chem Lett 27:1115–1118
Wang G, Peng Z, Wang J, Li X, Li J (2017b) Synthesis, in vitro evaluation and molecular docking studies of novel triazine-triazole derivatives as potential α-glucosidase inhibitors. Eur J Med Chem 125:423–429
World Health Organization (2016) Global report on diabetes. World Health Organization. https://apps.who.int/iris/handle/10665/204871/. Accessed 24 Feb 2018
Wu J, Wang Q, Guo J, Hu Z, Yin Z, Xu J, Wu X (2008) Characterization of angiotensin II antagonism displayed by Ib, a novel nonpeptide angiotensin AT1 receptor antagonist. Eur J Pharmacol 589:220–224
Acknowledgements
This research was supported by a grant from Tehran University of Medical Sciences (GN: 39750).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Tafesse, T.B., Moghadam, E.S., Bule, M.H. et al. Synthesis and biological evaluation of 2-(2-methyl-1H-pyrrol-3-yl)-2-oxo-N-(pyridine-3-yl) acetamide derivatives: in vitro α-glucosidase inhibition, and kinetic and molecular docking study. Chem. Pap. 74, 1583–1596 (2020). https://doi.org/10.1007/s11696-019-00999-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11696-019-00999-0