
Abstract
α-Glucosidase is responsible for glucose release of oligosaccharides and disaccharides in the intestine and increase postprandial hyperglycemia. Inhibition of this enzyme is a beneficial therapeutic method for glycemic control in diabetes. This study deals with the design and synthesis of 4,5-diphenylimidazole-N-phenylacetamide derivatives 7a–l and the screen of these compounds for their potential for α-glucosidase inhibition. All the synthesized compounds exhibited superior α-glucosidase inhibition (IC50 = 90.0–598.5 µM) as compared to standard inhibitor acarbose (IC50 = 750.0 µM). In contrast, these compounds were inactive against α-amylase. Among the synthesized compounds, compound 7h was the most potent inhibitor of this library and was a competitive inhibitor into α-glucosidase with Ki value = 86.3 μM. Docking study of the most potent compounds was performed to evaluate the binding interactions of these compounds with the active site of enzyme and to determine of binding energies of ligand–enzyme complexes. The results of this in silico study are in complete agreement with the results obtained from in vitro α-glucosidase inhibition assay. Docking study of the most potent compound demonstrated that it interacted with important residues in the active site of α-glucosidase. In vitro cytotoxic activity of the most potent compounds and in silico druglikeness/ADME/toxicity study of these compounds were evaluated.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Diabetes mellitus is a metabolic disorder that affect people of different ages, from children to the elderly [1]. This disease is classified to two main types: type 1, that is, insulin dependent and due to defects in pancreatic β-cells in insulin secretion and type 2 diabetes, that is, non-insulin dependent and insulin resistance in body cells is usually observed in it [2, 3]. The high prevalence and increasing number of diabetic patients has led to the use of various treatment methods to control blood sugar in these patients. The main treatment for type 1 diabetes is insulin, and the main treatment for type 2 diabetes is oral medications that lower postprandial hyperglycemia [4, 5]. One of the important classes of the latter medications are α-glucosidase inhibitors, such as acarbose, voglibose, and miglitol [6]. These drugs effectively inhibited α-glucosidase that is responsible for breaking down oligosaccharides and disaccharides to monosaccharides and decreased glucose level in blood [7]. Acarbose showed intestinal complications such as flatulence, bloating, cramping, and abdominal pain that several enzymatic studies have suggested that α-amylase inhibition is the cause of these side effects [8]. α-Amylase is an important carbohydrate hydrolyzing enzyme that converts polysaccharides to oligosaccharides and disaccharides. α-Amylase inhibition in addition to helping to lower blood glucose leads to the secretion of undecomposed polysaccharides to long intestine and causes intestinal complications. Mechanism of actions of α-glucosidase and α-amylase is schematically shown in Fig. 1.

Hydrolysis of carbohydrates by α-amylase and α-glucosidase
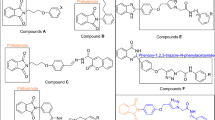
Nitrogen-containing heterocycles have broad range of biological activities and are important blokes for drug design [9]. One of the most important of these heterocycles is imidazole, which is found broadly in the natural and synthetic molecules with biological properties, such as antifungal, anti-inflammatory, antitubercular, anticancer, and antiviral activities [10,11,12,13,14]. 4,5-Diphenylimidazole as a valuable imidazole derivative also exhibited various biological activities [11]. This moiety is found in the several potent α-glucosidase inhibitors, such as compounds A and B (Fig. 2) [15, 16]. Furthermore, 4,5-diphenylimidazole-2-phenoxy derivatives C also exhibited high inhibitory activity against α-glucosidase [17]. On the other hand, N-phenylacetamid moiety is found in the several series of new potent α-glucosidase inhibitors such as compounds D–F (Fig. 2) [18,19,20]. Therefore, herein, novel structures 7 by connecting 4,5-diphenylimidazole-2-phenoxy to N-phenylacetamid were designed, synthesized, and evaluated as new α-glucosidase inhibitors.

Design strategy for 4,5-diphenylimidazole-N-phenylacetamide derivatives 7 as new α-glucosidase inhibitors
Material and methods
General chemistry
Melting points of 4,5-diphenylimidazole-N-phenylacetamide derivatives 7a–l were carried out on a Kofler hot stage apparatus and are uncorrected. Infrared spectra were recorded using KBr disks for solid materials on a Nicolet Magna FTIR 550 instrument. The 1H nuclear magnetic resonance (NMR) and 13C NMR spectra were run on a Bruker FT-500. Compounds were dissolved in dimethyl sulfoxide (DMSO)-d6, and chemical shifts were referenced to tetramethylsilane. Mass spectra were obtained on an Agilent Technology (HP; MS Model: 5973 Network Mass Selective Detector) analysis system. Elemental analyses were performed on an Elementar Analysensysteme GmbH Vario EL CHNS mode instrument.
General procedure for the synthesis of 2-chloro-N-phenylacetamide derivatives (3a–l)
A mixture of aniline derivatives 1a–l (1 mmol) and chloroacetyl chloride 2 (1.2 mmol) in acetone (15 mL) was stirred at room temperature for 30 min. Then the reaction mixture was poured in water and the obtained precipitate was filtered and washed with water to obtain pure 2-chloro-N-phenylacetamide derivatives 3a–l.
General procedure for the synthesis of 2-(4-formylphenoxy)-N-phenylacetamides (5a–l)
A suspension of 2-chloro-N-phenylacetamide derivatives 3a–l (1 mmol), 4-hydroxybenzaldehyde 4 (1 mmol), KI (1 mmol), and K2CO3 (2 mmol) in acetone (15 mL) was stirred at 80 °C for 4 h. Similar to previous step, the reaction mixture was poured in water and residue was filtered and washed with water to obtain pure 2-(4-formylphenoxy)-N-phenylacetamides 5a–l.
General procedure for the synthesis of 4,5-diphenylimidazole-N-phenylacetamide derivatives (7a–l)
2-(4-Formylphenoxy)-N-phenylacetamides 5a–l (1 mmol), benzil 6 (1 mmol), ammonium acetate (10 mmol), and dimethylformamide (DMF) were stirred at 100 °C for 8 h. Then the reaction mixture was poured into cold water, the precipitate was filtered off, and washed with water to afford 4,5-diphenylimidazole-N-phenylacetamide derivatives 7a–l.
2-(4-(4,5-diphenyl-1H-imidazol-2-yl)phenoxy)-N-phenylacetamide (7a)
White solid; isolated yield: 82%, mp 179–181 °C. Infrared (IR) (KBr, υ): 3289, 3052, 2935, 1637 cm−1. 1H NMR (500 MHz, DMSO-d6) δ 12.54 (s, 1H), 8.03 (d, J = 8.5 Hz, 2H), 7.73 (d, J = 7.6 Hz, 2H), 7.59–7.56 (m, 3H), 7.54–7.52 (m, 4H), 7.45–7.42 (m, 2H), 7.41–7.32 (m, 5H), 7.08 (d, J = 8.6 Hz, 2H), 4.51 (s, 2H, O-CH2). 13C NMR (126 MHz, DMSO-d6) δ 170.27, 164.37, 158.38, 145.97, 139.69, 132.89, 130.98, 130.84, 130.58, 129.53, 129.40, 129.25, 129.14, 129.00, 128.95, 128.73, 127.10, 125.66, 124.15, 115.36, 67.25. MS (70 eV, m/z [M+]) = 445. Anal Calcd for C29H23N3O2, C, 78.18, H, 5.20, N, 9.43. Found C, 78.16, H, 5.27, N, 9.41.
2-(4-(4,5-diphenyl-1H-imidazol-2-yl)phenoxy)-N-(p-tolyl)acetamide (7b)
White solid; isolated yield: 84%, mp 204–206 °C. IR (KBr, υ): 3277, 3069, 2921, 1655 cm−1. 1H NMR (500 MHz, Chloroform-d) δ 12.84 (s, 1H), 7.99 (d, J = 8.7 Hz, 2H), 7.88 (d, J = 7.4 Hz, 4H), 7.78–7.68 (m, 1H), 7.58–7.55 (m, 1H), 7.49–7.44 (m, 4H), 7.44–7.38 (m, 3H), 7.35 (d, J = 8.8 Hz, 2H), 7.30–7.27 (m, 1H), 7.16–7.08 (m, 1H), 5.25 (s, 2H, O-CH2), 2.17 (s, 3H, CH3). 13C NMR (126 MHz, DMSO-d6) δ 170.58, 158.40, 145.97, 138.41, 136.47, 134.01, 132.59, 131.43, 128.86, 128.19, 127.51, 127.14, 125.34, 124.03, 122.20, 120.94, 119.65, 118.15, 115.36, 115.12, 115.12, 65.02, 19.10. MS (70 eV, m/z [M+]) = 459. Anal Calcd for C30H25N3O2, C, 78.41, H, 5.48, N, 9.14. Found C, 78.44, H, 5.41, N, 9.13.
2-(4-(4,5-diphenyl-1H-imidazol-2-yl)phenoxy)-N-(4-ethylphenyl)acetamide (7c)
White solid; isolated yield: 83%, mp 167–169 °C. IR (KBr, υ): 3271, 3073, 2938, 1631 cm−1. 1H NMR (500 MHz, DMSO-d6) δ 12.67 (s, 1H), 8.10–7.95 (m, 3H), 7.58–7.50 (m, 5H), 7.43–7.35 (m, 5H), 7.33–7.24 (m, 3H), 7.11–6.95 (m, 3H), 4.77 (s, 2H, O-CH2), 2.69 (q, J = 7.0 Hz, 2H, CH2), 1.26 (t, J = 7.2 Hz, 3H, CH3). 13C NMR (126 MHz, DMSO-d6) δ 170.59, 158.42, 145.98, 138.95, 136.90, 134.85, 133.82, 132.77, 131.52, 129.98, 128.86, 128.19, 127.52, 127.16, 125.62, 124.00, 121.56, 119.57, 117.80, 115.12, 65.02, 56.52, 19.03. MS (70 eV, m/z [M+]) = 473. Anal Calcd for C31H27N3O2, C, 78.62, H, 5.75, N, 8.87. Found C, 78.58, H, 5.67, N, 8.82.
2-(4-(4,5-diphenyl-1H-imidazol-2-yl)phenoxy)-N-(4-methoxyphenyl)acetamide (7d)
White solid; isolated yield: 82%, mp 193–195 °C. IR (KBr, υ): 3279, 3071, 2920, 1644 cm−1. 1H NMR (500 MHz, DMSO-d6) δ 12.56 (s, 1H), 8.02 (d, J = 8.4 Hz, 2H), 7.62–7.17 (m, 15H), 7.04 (d, J = 8.4 Hz, 2H), 4.76 (s, 2H, O-CH2), 3.74 (s, 3H, O-CH3). 13C NMR (126 MHz, DMSO-d6) δ 170.58, 158.39, 145.97, 140.02, 138.08, 136.53, 135.14, 133.49, 132.05, 130.71, 128.86, 128.04, 127.12, 125.64, 124.05, 122.18, 120.99, 119.31, 117.93, 115.11, 65.03, 55.67. MS (70 eV, m/z [M+]) = 475. Anal Calcd for C30H25N3O3, C, 75.77, H, 5.30, N, 8.84. Found C, 75.83, H, 5.28, N, 8.83.
N-(4-chlorophenyl)-2-(4-(4,5-diphenyl-1H-imidazol-2-yl)phenoxy)acetamide (7e)
White solid; isolated yield: 86%, mp 239–241 °C. IR (KBr, υ): 3330, 2905, 1742, 1647 cm−1. 1H NMR (500 MHz, DMSO-d6) δ 13.58 – 12.21 (m, 1H), 8.04 (d, J = 8.9 Hz, 2H), 7.65–7.35 (m, 13H), 7.31 (t, J = 7.3 Hz, 2H), 7.06 (d, J = 8.9 Hz, 2H), 4.77 (s, 2H, O-CH2). 13C NMR (126 MHz, DMSO-d6) δ 170.54, 158.57, 150.64, 145.87, 138.73, 133.33, 132.69, 130.80, 128.90, 128.25, 127.67, 127.32, 125.72, 124.60, 123.52, 122.03, 120.97, 118.84, 115.39, 115.16, 115.16, 65.01. MS (70 eV, m/z [M+]) = 479. Anal Calcd for C29H22ClN3O2, C, 72.57, H, 4.62, N, 8.75. Found C, 72.52, H, 4.61, N, 8.69.
N-(2,6-dichlorophenyl)-2-(4-(4,5-diphenyl-1H-imidazol-2-yl)phenoxy)acetamide (7f)
White solid; isolated yield: 84%, mp 199–201 °C. IR (KBr, υ): 3336, 2953, 1649 cm−1. 1H NMR (500 MHz, DMSO-d6) δ 12.58 (s, 1H), 8.06 (d, J = 8.4 Hz, 2H), 7.64–7.47 (m, 8H), 7.46–7.32 (m, 6H), 7.17 (d, J = 8.9 Hz, 2H), 4.85 (s, 2H, O-CH2). 13C NMR (126 MHz, DMSO-d6) δ 167.62, 157.75, 147.99, 145.83, 140.30, 138.80, 136.48, 135.52, 133.87, 132.96, 132.31, 130.44, 128.85, 128.24, 127.24, 125.91, 125.44, 124.72, 124.22, 115.50, 67.61. MS (70 eV, m/z [M+]) = 514. Anal Calcd for C29H21Cl2N3O2, C, 67.71, H, 4.11, N, 8.17. Found C, 67.76, H, 4.12, N, 8.15.
N-(2-bromophenyl)-2-(4-(4,5-diphenyl-1H-imidazol-2-yl)phenoxy)acetamide (7g)
White solid; isolated yield: 85%, mp 236–238 °C. IR (KBr, υ): 3326, 2987, 1648 cm−1. 1H NMR (500 MHz, DMSO-d6) δ 12.59 (s, 1H), 8.10 (d, J = 7.8 Hz, 2H), 7.89 (d, J = 7.5 Hz, 1H), 7.71 (d, J = 7.6 Hz, 1H), 7.60–7.50 (m, 4H), 7.49–7.27 (m, 7H), 7.25–7.03 (m, 4H), 4.87 (s, 2H, O-CH2). 13C NMR (126 MHz, DMSO-d6) δ 167.12, 157.99, 145.91, 140.26, 137.61, 135.86, 134.33, 133.14, 131.59, 130.38, 128.77, 128.43, 127.79, 127.47, 127.23, 125.87, 124.60, 123.43, 121.85, 120.32, 117.44, 115.52, 115.52, 67.56. MS (70 eV, m/z [M+]) = 523. Anal Calcd for C29H22BrN3O2, C, 66.42, H, 4.23, N, 8.01. Found C, 66.44, H, 4.21, N, 8.08.
N-(3-bromophenyl)-2-(4-(4,5-diphenyl-1H-imidazol-2-yl)phenoxy)acetamide (7h)
White solid; isolated yield: 86%, mp 214–216 °C. IR (KBr, υ): 3273, 3072, 2937, 1643 cm−1. 1H NMR (500 MHz, DMSO-d6) δ 12.53 (s, 1H), 8.02 (d, J = 7.8 Hz, 2H), 7.62 – 7.48 (m, 6H), 7.47 – 7.17 (m, 9H), 7.04 (d, J = 7.8 Hz, 2H), 4.75 (s, 2H, O-CH2). 13C NMR (126 MHz, DMSO-d6) δ 170.57, 158.39, 145.97, 138.95, 136.93, 135.92, 134.52, 133.23, 131.34, 128.81, 127.11, 124.05, 122.73, 121.52, 120.80, 119.71, 118.63, 117.69, 116.60, 115.11, 115.11, 113.36, 111.63, 65.03. MS (70 eV, m/z [M+]) = 523. Anal Calcd for C29H22BrN3O2, C, 66.42, H, 4.23, N, 8.01. Found C, 66.49, H, 4.22, N, 8.06.
N-(4-bromophenyl)-2-(4-(4,5-diphenyl-1H-imidazol-2-yl)phenoxy)acetamide (7i)
White solid; isolated yield: 86%, mp 224–226 °C. IR (KBr, υ): 3325, 2954, 1649 cm−1. 1H NMR (500 MHz, DMSO-d6) δ 13.01 – 11.95 (m, 1H), 8.03 (d, J = 8.5 Hz, 2H), 7.60 – 7.23 (m, 15H), 7.05 (d, J = 8.5 Hz, 2H), 4.76 (s, 2H, O-CH2). 13C NMR (126 MHz, DMSO-d6) δ 170.58, 158.41, 145.98, 136.09, 134.81, 133.08, 132.27, 131.13, 130.27, 128.86, 128.18, 127.51, 127.16, 125.77, 125.09, 124.00, 122.28, 121.28, 119.55, 115.12, 65.02. MS (70 eV, m/z [M+]) = 523. Anal Calcd for C29H22BrN3O2, C, 66.42, H, 4.23, N, 8.01. Found C, 66.40, H, 4.25, N, 8.01.
2-(4-(4,5-diphenyl-1H-imidazol-2-yl)phenoxy)-N-(2-methyl-3-nitrophenyl)acetamide (7j)
White solid; isolated yield: 81%, mp 195–197 °C. IR (KBr, υ): 3288, 3098, 2963, 1655 cm−1. 1H NMR (500 MHz, DMSO-d6) δ 12.58 (s, 1H), 8.03 (d, J = 8.3 Hz, 2H), 7.61–7.45 (m, 6H), 7.45–7.21 (m, 9H), 7.05 (d, J = 8.3 Hz, 2H), 4.76 (s, 2H, O-CH2), 2.07 (s, 3H, CH3). 13C NMR (126 MHz, DMSO-d6) δ 170.58, 158.40, 145.98, 136.68, 134.99, 133.13, 131.55, 130.59, 129.76, 128.86, 128.16, 127.51, 127.14, 126.12, 125.08, 124.03, 122.45, 120.83, 118.76, 117.90, 115.12, 65.02, 19.03. MS (70 eV, m/z [M+]) = 504. Anal Calcd for C30H24N4O4, C, 71.42, H, 4.79, N, 11.10. Found C, 71.44, H, 4.76, N, 11.16.
N-(3-chloro-2-methylphenyl)-2-(4-(4,5-diphenyl-1H-imidazol-2-yl)phenoxy)acetamide (7k)
White solid; isolated yield: 83%, mp 166–168 °C. IR (KBr, υ): 3283, 3086, 2963, 1650 cm−1. 1H NMR (500 MHz, DMSO-d6) δ 12.43 (s, 1H), 8.23 (d, J = 8.3 Hz, 1H), 8.14 (d, J = 8.3 Hz, 1H), 8.09 (d, J = 8.9 Hz, 2H), 7.80 (t, J = 7.8 Hz, 1H), 7.57–7.52 (m, 4H), 7.42–7.29 (m, 7H), 7.19 (d, J = 8.9 Hz, 2H), 4.86 (s, 2H, O-CH2), 2.05 (s, 3H, CH3). 13C NMR (126 MHz, DMSO-d6) δ 195.29, 180.73, 167.92, 156.48, 144.75, 143.98, 142.84, 141.38, 140.41, 139.08, 138.34, 136.95, 136.02, 134.06, 132.71, 131.35, 130.07, 129.99, 127.18, 125.37, 123.63, 122.65, 120.98, 64.04, 18.85. MS (70 eV, m/z [M+]) = 493. Anal Calcd for C30H24ClN3O2, C, 72.94, H, 4.90, N, 8.51. Found C, 72.91, H, 4.88, N, 8.50.
N-(2-chloro-3-fluorophenyl)-2-(4-(4,5-diphenyl-1H-imidazol-2-yl)phenoxy)acetamide (7l)
White solid; isolated yield: 87%, mp 177–19 °C. IR (KBr, υ): 3287, 3080, 2966, 1651 cm−1. 1H NMR (500 MHz, DMSO-d6) δ 12.56 (s, 1H), 8.07 (d, J = 8.5 Hz, 2H), 7.80 (t, J = 7.5 Hz, 1H), 7.55 (s, 5H), 7.47–7.39 (m, 3H), 7.38–7.30 (m, 3H), 7.27 – 7.20 (m, 2H), 7.14 (d, J = 8.5 Hz, 2H), 4.89 (s, 2H, O-CH2). 13C NMR (126 MHz, DMSO-d6) δ 167.52, 164.28, 158.32, 149.55, 145.94, 137.31, 135.81, 130.39, 129.41, 129.32, 128.97, 128.81, 128.70, 128.23, 128.13, 127.81, 127.55, 127.41, 127.32, 127.17, 126.86, 126.01, 125.46, 124.36, 120.87, 120.50, 120.37, 115.36, 67.39. MS (70 eV, m/z [M+]) = 497. Anal Calcd for C29H21ClFN3O2, C, 69.95, H, 4.25, N, 8.44. Found C, 69.91, H, 4.24, N, 8.49.
Pharmacology
In vitro α-glucosidase inhibition assay
α-Glucosidase inhibition assay was evaluated on yeast α-glucosidase (Saccharomyces cerevisiae, EC3.2.1.20, 20 U/mg) according to the previous reports [21,22,23,24]. First, the various concentrations of compounds 7a–l (20 μl), α-glucosidase solution (20 μl), and potassium phosphate buffer (135 μl) were added to a 96-well plate and incubated at 37 °C for 10 min. Then p-nitrophenyl glucopyranoside (substrate, 25 μl, 4 mM) was added to the latter plate and allowed to incubate at 37 °C for 20 min. Finally, the absorbance changes were measured at 405 nm by using spectrophotometer (Gen5, Power wave xs2, BioTek, America). Solvent (DMSO with final concentration of 10%) was used as negative control and acarbose was used as positive control. IC50 values of title compounds 7a–l were calculated from nonlinear regression curve using the Logit method.
In vitro α-amylase inhibition assay
The inhibitory activity of the newly synthesized compounds 7a–l against α-amylase was obtained according to used method in our previous work [23].
Kinetic study
For kinetic study, the most potent compound 7h was selected and 20 μl of it in four different concentrations 0, 30, 60, and 90 μM was incubated with 20 μl of α-glucosidase solution (1 U/ml) for 15 min at 30 °C. After that, the enzymatic reaction was started by adding 20 μl of p-nitrophenyl glucopyranoside as substrate in concentrations 1–4 mM and the absorbance changes was determined for 20 min at 405 nm by using spectrophotometer (Gen5, Power wave xs2, BioTek, America).
Molecular modeling study
Modeled α-glucosidase was constructed according to the described method by Imran et al. [25, 26]. At first, a search was performed by using SWISS-MODEL to find an appropriate protein in the protein data bank (PDB) with a high sequence similarity with S. cerevisiae α-glucosidase (used form of enzyme in enzymatic assay). As a result, isomaltase from S. cerevisiae (PDB code 3A4A) was selected with 72% identical and shares 85% similarity with the S. cerevisiae α-glucosidase. Then this enzyme was subjected through sequence alignment and homology model using automated homology modeling pipeline SWISS-MODEL (managed by Swiss Institute of Bioinformatics) and the quality of the obtained modeled enzyme (in pdb format) was verified using PROCHECK [27]. Pdb format of the modeled enzyme was converted to pdbqt coordinate by Auto dock Tools. Three-dimensional structures of the most potent compounds and standard inhibitor acarbose were built by MarvineSketch 5.8.3, 2012, ChemAxon (http://www.chemaxon.com) and were saved as pdb format and converted to pdbqt coordinate using Auto dock Tools. These pdbqts were used as an input file for the AUTOGRID program. In the latter program, for each atom type in the ligand, maps were calculated with 0.375 Å spacing between grid points and center of grid box was placed at x = 12.5825, y = 7.8955, and z = 12.519 Å with dimensions 40 × 40 × 40 Å. Flexible ligand dockings were applied for the selected ligands. Each docked system was carried out by 50 runs of AUTODOCK search by the Lamarckian genetic algorithm. The best docking pose in term of binding energy for each ligand was selected for study of the interaction mode between α-glucosidase and ligand.
In vitro cytotoxicity assay
In vitro cytotoxic activity of the most potent compounds 7h, 7a, and 7d was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay according to literature [28]. For this propose, normal and cancer human cell lines were used.
In silico druglikeness/ADME/toxicity study
In silico druglikeness/ADME/toxicity properties of standard drug acarbose and the most potent compounds 7h, 7a, and 7d were predicted by using the preADMET online server (http://preadmet.bmdrc.org/) [29].
Results and discussion
Chemistry
The synthetic routes for the synthesis of 4,5-diphenylimidazole-N-phenylacetamide derivatives 7a–l has been depicted in Scheme 1. This synthesis process was started from the reaction between aniline derivatives 1a–l and chloroacetyl chloride 2 in acetone at room temperature for 30 min to give 2-chloro-N-phenylacetamide derivatives 3a–l. The latter compounds reacted with 4-hydroxybenzaldehyde 4 in the presence of KI and K2CO3 in acetone at 80 °C for produce 2-(4-formylphenoxy)-N-phenylacetamides 5a–l. In the final step, 2-(4-formylphenoxy)-N-phenylacetamides 5a–l and benzil 6 in the presence of ammonium acetate in DMF at 100 °C converted to desired 4,5-diphenylimidazole-N-phenylacetamide derivatives 7a–l.

Synthetic routes to the target compounds 7a–l: (a) Acetone, r.t .30 min; (b) KI, K2CO3, Acetone, 80 °C, 4 h; (c) NH4OAC, DMF, 100 °C, 8 h
α-Glucosidase and α-amylase inhibitory activities
In vitro anti-α-glucosidase activity of the synthesized 4,5-diphenylimidazole-N-phenylacetamide derivatives 7a–l was evaluated and the obtained results were compared with acarbose as a standard α-glucosidase inhibitor (IC50 = 750.0 ± 9.6 µM). As can be seen in Table 1, the IC50 values of the studied compounds demonstrated that all the synthesized compounds 7a–l were more potent than acarbose (IC50s ≤ 598.5 ± 8.0 µM). In the synthesis of these compounds, aniline and various aniline derivatives with electron-donating groups such as methyl, ethyl, and methoxy and electron-withdrawing groups such as chloro, bromo, nitro, and fluoro were used in order to optimize α-glucosidase inhibition. The most potent compounds were 3-bromo and un-substituted compounds 7h and 7a with IC50 values = 90.0 ± 2.5 and 103.6 ± 4.0 μM, respectively. Furthermore, 4-methoxy derivative 7d, 2,6-dichloro derivative 7f and 2-bromo derivative 7g exhibited good anti-α-glucosidase activity (IC50 values ≤179.8 ± 4.4 µM).
The α-glucosidase inhibitory activity of 4,5-diphenylimidazole-N-phenylacetamide derivatives 7a–l demonstrated that 3-bromo derivative 7h showed the most potent activity, while 4-bromo derivative 7i was less active compound among the synthesized compounds. Moreover, movement of bromo substituent of 3-position to 2-position, as in compound 7g, led to a moderate decrease in the inhibitory activity.
The second potent compound among the synthesized compounds was un-substituted compound 7a. Introduction of 4-methyl, 4-ethyl, or 4-chloro on N-phenylacetamide ring dramatically deteriorated anti-α-glucosidase as observed in compounds 7b, 7c, and 7e while introduction of 4-methoxy substituent on the latter ring, as in compound 7d, slightly decreased inhibitory activity in comparison to un-substituted compound 7a.
Among the two-substituted compounds 7f and 7j–l, 2,6-dichloro derivative 7f exhibited the best inhibitory activity.
α-Amylase inhibition assay of the synthesized compounds 7a–l was also carried out and obtained data revealed that all the title compounds (IC50 values >500 μM) in comparison to acarbose (IC50 = 108 ± 0.71 μM) were inactive.
Kinetic study against α-glucosidase
Kinetic study of the most potent compound 7h was carried out in order to evaluate mechanism of α-glucosidase inhibition of the new compounds. As was shown in Fig. 3a, with increase of concentration of the compound 7h, Vmax remained unchanged while Km increased. Therefore, compound 7h was a competitive inhibitor against α-glucosidase. The value of Ki for compound 7h was 86.3 ± 0.11 μM (Fig. 3b).

a The inhibition type and b Ki value determination of the most potent compound 7h
α-Glucosidase molecular modeling study
In order to clarify interaction modes of the newly synthesized compounds in the active site of α-glucosidase, molecular modeling was conducted on the modeled form of this enzyme using Auto Dock Tools (version 1.5.6) [21]. For docking purposes, acarbose as standard inhibitor and most potent compounds 7h and 7a were selected and docked in the α-glucosidase active site. The superimposed structure of acarbose and the most potent compound 7h in the active site of target enzyme is shown in Fig. 4a. Analysis of the best pose of acarbose revealed that this inhibitor with binding energy of −4.04 kcal/mol, interacted with the α-glucosidase active site residues Gln322, Thr301, Glu304, Ser308, Arg312, Thr307, and Asn241 via hydrogen bonds (Fig. 4b). Furthermore, acarbose formed a hydrophobic interaction with His279, several non-classical hydrogen bonds with Arg312, Glu304, Val305, and His239, and two unfavorable interactions with Arg312 and Thr307.

(a) Superimposition structure of acarbose (cyan) and the most potent compound 7h (blue); (b) interaction mode of acarbose in the active site of α-glucosidase
The most potent compound 7h established a hydrogen bond with Thr307 via NH unit of imidazole ring and a π-anion interaction with Glu304 via phenoxy group (Fig. 5a). Imidazole ring and phenoxy group also interacted with residue Pro309 through hydrophobic interactions. This amino acid also established a non-classical hydrogen bond with compound 7h. Phenyl rings attached to imidazole ring interacted with Ala326 and Val316 via hydrophobic interactions. Furthermore, N-3-bromophenylacetamid moiety of compound 7h established the following interactions: two hydrophobic interactions with residues Phe158 and Arg312 through 3-bromo substituent and phenyl ring, respectively, and a non-classical hydrogen bond with residue Phe311 through carbonyl unit.

(a) The interaction modes of the most potent compounds 7h and (b) 7a in the active site of α-glucosidase
The comparison of interaction modes of the most potent compound 7h containing N-3-bromophenylacetamid moiety with the second potent compound 7a with N-phenylacetamid moiety demonstrated that compound 7h established an additional hydrophobic interaction with active site residue Phe158 via 3-bromo substituent (Fig. 5). It should also be noted that compound 7a created an additional hydrophobic interaction with Pro309 via 5-phenyl ring in comparison to compound 7h (Fig. 5b). Other established interactions are the same in the both compounds 7h and 7a. Details of interaction modes of compounds 7h and 7a are listed in Table 2.
The calculation of binding energies of acarbose (−4.04 kcal/mol) and compounds 7h (−11.32 kcal/mol) and 7a (−10.65 kcal/mol) demonstrated that our newly synthesized compounds bind to α-glucosidase more easily than acarbose. These findings are in agreement with the obtained date of in vitro enzyme inhibition assay (Table 1, compounds 7h and 7a vs. acarbose).
In vitro cytotoxicity assay
In vitro cytotoxicity of compounds 7h, 7a, and 7d was evaluated against normal cells HDF and cancer cells MCF-7 by MTT assay [28]. As can be seen in Table 3, the selected compounds were non-cytotoxic at 200 µM against studied cells in comparison to standard cytotoxic agent etoposide.
In silico druglikeness, ADME, and toxicity studies
Druglikeness/ADME/T properties of acarbose as standard drug and new compounds 7h, 7a, and 7d as the most potent inhibitors were predicted by the online software PreADMET [29]. The obtained results are listed in Table 4. As can be seen in this table, compounds 7a and 7d are drug-like in terms of Rule of Five and MDDR-like rule while compound 7h and acarbose only follow MDDR-like rule. Our new compounds 7h, 7a, and 7d have Caco2 cell permeability and human oral absorption higher of acarbose. The predicted values of blood–brain barrier permeability for any of the studied compounds are not in the acceptable range. Predicting the toxicity of the all selected compounds showed that all the studied compounds were mutagenic (Ames test). Moreover, in silico toxicity assay also predicted that new compounds 7h, 7a, and 7d are high risk in terms of cardiotoxicity while cardiotoxicity of acarbose is ambiguous. Finally, all the test compounds have carcinogenic effect on rat while acarbose has carcinogenic effect on mouse.
Conclusion
In conclusion, we have designed and synthesized a series of new 4,5-diphenylimidazole-N-phenylacetamide derivatives 7a–l and studied α-glucosidase and α-amylase inhibitory activities of these compounds. All the synthesized compounds were more potent than standard inhibitor, and among them, compounds 7h, 7a, 7d, 7f, and 7g exhibited excellent anti-α-glucosidase activity with IC50 ≤ 179.8 ± 4.4 µM. On the other hand, these compounds were inactive against α-amylase in comparison to acarbose. Compound 7h (IC50 = 90.0 ± 2.5 µM) with N-3-bromophenylacetamid moiety was found to be the most active compound against α-glucosidase and was a competitive inhibitor. Furthermore, molecular modeling study was performed to investigate the interaction modes and binding energies of the most active analogs in the α-glucosidase active site. In vitro cytotoxicity assay of the most potent compound revealed that these compounds are non-cytotoxic in comparison to standard cytotoxic agent etoposide. Furthermore, in silico druglikeness/ADME/toxicity properties of the most active compounds were predicted by a reliable online software.
References
Zimmet P, Alberti KG, Kaufman F, Tajima N, Silink M, Arslanian S, et al. The metabolic syndrome in children and adolescents–an IDF consensus report. Pediatr Diabetes. 2007;8:299–306.
Ozougwu JC, Obimba KC, Belonwu CD, Unakalamba CB. The pathogenesis and pathophysiology of type 1 and type 2 diabetes mellitus. J Physiol Pharm. 2013;4:46–57.
Fronzo DE. The triumvirate: SS-cell, muscle, liver-a collusion responsible for NIDDM. Diabetes. 1988;37:667–87.
Chamberlain JJ, Kalyani RR, Leal S, Rhinehart AS, Shubrook JH, Skolnik N, et al. Treatment of type 1 diabetes: synopsis of the 2017 American Diabetes Association Standards of Medical Care in Diabetes. Ann Intern Med. 2017;167:493–8.
Cervera A, Wajcberg E, Sriwijitkamol A, Fernandez M, Zuo P, Triplitt C, et al. Mechanism of action of exenatide to reduce postprandial hyperglycemia in type 2 diabetes. Am J Physiol Endocrinol Metab. 2008;294:E846–52.
Derosa G, Maffioli P. α-Glucosidase inhibitors and their use in clinical practice. Arch Med Sci AMS. 2012;8:899.
Leroux-Stewart J, Rabasa-Lhoret R, Chiasson JL. α-Glucosidase inhibitors. Ther Diabetes Mellit Relat Disord. 2014;2:416.
Dong Y, Zhang B, Sun W, Xing Y. Intervention of prediabetes by flavonoids from Oroxylum indicum. In: Watson RR, Preedy VR, editors. Bioactive food as dietary interventions for diabetes. London: Academic Press, p. 559–575 2019.
Asif M. A mini review: biological significances of nitrogen hetero atom containing heterocyclic compounds. Int J Bioorg Chem. 2017;2:146–52.
Hossain M, Nanda AK. A review on heterocyclic: synthesis and their application in medicinal chemistry of imidazole moiety. Science. 2018;6:83–94.
Shalini K, Sharma PK, Kumar N. Imidazole and its biological activities: a review. Der Chem Sin. 2010;1:36–47.
Bhatnagar A, Sharma PK, Kumar N. A review on “Imidazoles”: their chemistry and pharmacological potentials. Int J PharmTech Res. 2011;3:268–82.
Gupta V, Kant V. A review on biological activity of imidazole and thiazole moieties and their derivatives. Sci Int. 2013;1:253–60.
Rani N, Sharma A, Kumar Gupta G, Singh R. Imidazoles as potential antifungal agents: a review. Mini Rev Med Chem. 2013;13:1626–55.
Yar M, Bajda M, Shahzad S, Ullah N, Gilani MA, Ashraf M, et al. Organocatalyzed solvent free an efficient novel synthesis of 2, 4, 5-trisubstituted imidazoles for α-glucosidase inhibition to treat diabetes. Bioorg Chem. 2015;58:65–71.
Asgari MS, Mohammadi-Khanaposhtani M, Sharafi Z, Faramarzi MA, Rastegar H, Esfahani EN, et al. Design and synthesis of 4,5‑diphenyl‑imidazol‑1,2,3‑triazole hybrids as new anti‑diabetic agents: in vitro α‑glucosidase inhibition, kinetic and docking studies. Mol Divers. 2020. https://doi.org/10.1007/s11030-020-10072-8
Saeedi M, Mohammadi-Khanaposhtani M, Asgari MS, Eghbalnejad N, Imanparast S, Faramarzi MA, et al. Design, synthesis, in vitro, and in silico studies of novel diarylimidazole-1,2,3-triazole hybrids as potent α-glucosidase inhibitors. Bioorg Med Chem. 2019;27:115148.
Mohammadi-Khanaposhtani M, Rezaei S, Khalifeh R, Imanparast S, Faramarzi MA, Bahadorikhalili S, et al. Design, synthesis, docking study, α-glucosidase inhibition, and cytotoxic activities of acridine linked to thioacetamides as novel agents in treatment of type 2 diabetes. Bioorg Chem. 2018;80:288–95.
Ye GJ, Lan T, Huang ZX, Cheng XN, Cai CY, Ding SM, et al. Design and synthesis of novel xanthone-triazole derivatives as potential antidiabetic agents: α-Glucosidase inhibition and glucose uptake promotion. Eur J Med Chem. 2019;177:362–73.
Asemanipoor N, Mohammadi-Khanaposhtani M, Moradi S, Vahidi M, Asadi M, Faramarzi MA, et al. Synthesis and biological evaluation of new benzimidazole-1, 2, 3-triazole hybrids as potential α-glucosidase inhibitors. Bioorg Chem. 2020;95:103482.
Adib M, Peytam F, Rahmanian-Jazi M, Mahernia S, Bijanzadeh HR, Jahani M, et al. New 6-amino-pyrido[2, 3-d] pyrimidine-2, 4-diones as novel agents to treat type 2 diabetes: a simple and efficient synthesis, α-glucosidase inhibition, molecular modeling and kinetic study. Eur J Med Chem. 2018;155:353–63.
Sepehri N, Asemanipoor N, Mousavianfard SA, Hoseini S, Faramarzi MA, Adib M, et al. New acridine-9-carboxamide linked to 1, 2, 3-triazole-N-phenylacetamide derivatives as potent α-glucosidase inhibitors: design, synthesis, in vitro, and in silico biological evaluations. Med Chem Res. 2020;29:1836–45.
Asgari MS, Mohammadi-Khanaposhtani M, Kiani M, Ranjbar PR, Zabihi E, Pourbagher R, et al. Biscoumarin-1, 2, 3-triazole hybrids as novel anti-diabetic agents: design, synthesis, in vitro α-glucosidase inhibition, kinetic, and docking studies. Bioorg Chem. 2019;92:103206.
Sherafati M, Mohammadi-Khanaposhtani M, Moradi S, Asgari MS, Najafabadipour N, Faramarzi MA, et al. Design, synthesis and biological evaluation of novel phthalimide-Schiff base-coumarin hybrids as potent α-glucosidase inhibitors. Chem Pap. 2020;74:4379–88.
Imran S, Taha M, Ismail NH, Kashif SM, Rahim F, Jamil W, et al. Synthesis of novel flavone hydrazones: in-vitro evaluation of α-glucosidase inhibition, QSAR analysis and docking studies. Eur J Med Chem. 2015;105:156–70.
Imran S, Taha M, Ismail NH, Kashif SM, Rahim F, Jamil W, et al. Synthesis, in vitro and docking studies of new flavone ethers as α‐glucosidase inhibitors. Chem Biol Drug Des. 2016;87:361–73.
Kiefer F, Arnold K, Künzli M, Bordoli L, Schwede T. The SWISS-MODEL Repository and associated resources. Nucleic Acids Res. 2009;37:D387–92.
Abolhasani MH, Safavi M, Goodarzi MT, Kassaee SM, Azin M. Identification and anti-cancer activity in 2D and 3D cell culture evaluation of an Iranian isolated marine microalgae Picochlorum sp. RCC486. DARU J Pharm Sci. 2018;26:105–16.
Bioinformatics and Molecular Design Research Center. Pre-ADMET program. Bioinformatics and Molecular Design Research Center, Seoul, South Korea. 2014. http://preadmet.bmdrc.org
Funding
This work was financially supported by Babol University of Medical Sciences (the grant number: 724132438). The ethics code for this study is IR.MUBABOL.HRI.REC.1398.181.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Mohammadi-Khanaposhtani, M., Nikraftar, A., Asgari, M.S. et al. Synthesis, in vitro and in silico enzymatic inhibition assays, and toxicity evaluations of new 4,5-diphenylimidazole-N-phenylacetamide derivatives as potent α-glucosidase inhibitors. Med Chem Res 30, 1273–1283 (2021). https://doi.org/10.1007/s00044-021-02734-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-021-02734-5