
Abstract
Protein hydrolysates from soft and hard wheat gluten were prepared using kiwifruit protease, followed by post-treatments using ultrasound (US) and atmospheric cold plasma (ACP). Peptides with different molecular weights (Mw) were determined for in vitro physicochemical and biological properties. US and ACP-treated peptides with molecular weight (Mw) of 451–1002 Da and > 12,400 Da constituted by 63–65% and 36–39%, respectively. The content of free SH groups in US and ACP treated peptides increased and decreased, respectively, while the S–S content was not altered. The surface hydrophobicity of hydrolysate increased with treatment intensity and time. The selected fraction with the smallest Mw namely HGH-U25 (< 1 kDa) exhibited the highest inhibitory activity toward α-glucosidase (14.25 ± 0.11%), and α-amylase (37.56 ± 1.12%). SGH-CP60/30 (10 − 100 kDa) showed the strongest cholesterol-lowering activity (47.08 ± 1.71%). High levels of hydrophobic amino acids and proline in S2 (40.91%, 10.88%) and P3 (54.11%, 29.11%) respectively, were more likely related with α-glucosidase (22.17%) and α-amylase inhibition (37.16%) and cholesterol-lowering (55.26%) activity. Thus, new hydrolysates derived from gluten with in vitro cholesterol-lowering activity could act as an anti-diabetic agent.
Graphical abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Bioactive peptides have gained increasing interest as nutraceuticals. In general, a typical amino acid residue has a molecular weight (MW) of around 100 Da, and a complex of two to twenty of these residues (MW < 6 kDa) are considered as bioactive peptides. There are many methods for generating biological peptides, including fermentation, microbial, enzymatic, and chemical hydrolysis. Bioactivities of peptides such as heavy metals binding, anti-diabetic, antioxidant, antimicrobial, and lowering cholesterol activities depend on their specific structural properties (i.e., hydrophobicity, chain length, net charge, and amino acid composition) [1]. To produce the bioactive peptides, the enzymatic process has been widely implemented. Proteases from different sources have been employed [2]. However, plant proteases, especially from kiwifruit, were documented to be able to hydrolyze a wide range of proteins [1, 2].
Ultrasound (US) and atmospheric cold plasma (ACP) treatments, which are non-thermal treatments, are able to expose the buried functional groups in the peptides and increase their biological properties. In US, ultrasonic waves (20–100 kHz) are applied at high power to liquid-filled systems. Acoustic cavitation occurs whenever sound waves propagate through a liquid, thus creating pressure variations, and causing vapor bubbles to grow, implode, and grow again. A cavitation explosion creates high localized shear force, pressure, and temperature, which can dramatically alter the chemical and physical properties of food [3]. During ultrasound treatment, peptide bonds in protein molecules are broken, thus releasing the biologically active peptides [1]. Revero-Pino et al. [4] stated that the increases in biological activity, following ultrasonic treatment were mainly due to changes in surface hydrophobicity, –SH, S–S, and secondary structure of proteins. Atmospheric cold plasma (ACP) is partially ionized and includes reactive substances (reactive nitrogen and oxygen species) [5]. It contains molecules such as oxygen, nitrogen, electrons, and ions that affected food components including proteins. Various reactions can take place when the radicals interact with proteins or peptides. Those include dimerization, oxidization, degradation, sulfoxidation, dehydrogenation, and hydroxylation [6]. ACP has been shown to effectively modify proteins and amino acids or alter biological functionality [5, 6].
In our previous study, actinidin was employed to produce anti-diabetic bio-peptides from soft and hard wheat gluten subunit proteins [1]. The use of non-thermal processing technology as a pretreatment followed by enzymatic hydrolysis has been reported in many studies [3, 4, 7, 8]. Until now, there has been no report about the use of non-thermal processing after enzymatic digestion to enhance bioactivities of peptides produced. It is well known that US and ACP can have important effects on the modification of proteins or peptides. Therefore, the post-enzymatic treatments with ACP and US might enhance the biological properties of peptides derived from various wheat gluten (hard and soft wheat) hydrolysates (WGH) prepared by partially purified kiwifruit enzyme.
The main goal of this study was to assess the impacts of US and ACP post-treatment (power density and time) on the biological properties of the soft and hard WGH. Some of the structural properties such as molecular weight distribution, surface hydrophobicity and free sulfhydryl group content of treated hydrolysates were demonstrated. Fractions with different Mws were evaluated for their inhibition effects toward α-glucosidase and α-amylase activities as well as cholesterol-lowering activity. Amino acid contents of the fractions with the highest inhibitory effects were determined.
Materials and methods
Materials
The soft and hard wheats were kindly supplied by Shahdineh Flour Co., (Iran). The selected kiwifruit (Actinidia deliciosa var. Hayward) was collected from the local farms (Rasht, Iran) (May June 2020). Chemicals, such as C2H5OH, C3OH6, NaOH, K2HPO4, KH2PO4, (NH4)2SO4, Dithiothreitol (DTT), Ethylenediaminetetraacetic acid (EDTA), l–Cysteine, Casein, Trichloroacetic acid (TCA), Tyrosine, Polyacrylamide, 1-aniline-8-naphthalene sulfonate (ANS), trifluoroacetic acid (TFA), PNPG (4-nitrophenyl α-d-glucopyranoside), porcine pancreatic α-amylase (Cat no. A3176), DNS (3,5-Dinitrosalicylic acid), rat intestinal α-glucosidase (Cat no. I1630), color indicators (coomassie brilliant blue G, bromophenol blue) and dialysis membrane (Mw cut-off 14 kDa) were purchased from Merck (Darmstadt, Germany). All chemicals were of analytical grade.
Preparation of hydrolysates
Kiwifruit extract containing protease was used to hydrolyze wheat gluten proteins (pH 6.5, 35 °C). To prepare the partially purified protease from the kiwifruit extract, the method of Mousavi et al. [1] was adopted. Wheat gluten powder (hard gluten and soft gluten) was dispersed in distilled water (5% w/v) in a reaction chamber (100 mL flask bioreactor), heated (10 min, 85 °C), and the pH was adjusted to 6.5 before adding the enzyme (kiwifruit extract 7.3 U/mg protein). The dispersion (5% gluten protein) was hydrolyzed by kiwifruit extract (6 h, 37 °C) using an enzyme to the substrate (1:10) and the pH of the mixture was fixed using 1 N NaOH throughout the hydrolysis process (pH–stat). The enzyme was subsequently inactivated by submerging the reaction chamber in hot water (90 °C, 10 min) and then cooling in an iced bath. After cooling, solutions were centrifuged (8000 g, 15 min, 4 °C) and the supernatant was collected, lyophilized and the obtained powder was stored in an amber glass bottle (4 °C) until further analysis [1]. Based on the raw materials used, hard gluten hydrolysates (HGH) and soft gluten hydrolysates (SGH) were obtained.
Treatment of hydrolysates
The obtained hydrolysates were then subjected to US and ACP.
For US treatment, the lyophilized wheat gluten hydrolysates (5 g) were mixed with distilled water (100 mL) followed by stirring (1 h, 25 °C) and then sonicated at different amplitudes: 30 W (amplitude of 25%), 50 W (amplitude of 50%), and 73 W (amplitude of 75%) for 10 min using the pulse mode (pulse duration of 10 s and off-time 5 s). To maintain the temperature during US treatment, the hydrolysate samples were kept in an iced bath [7]. After US treatment, HGH-U and SGH-U samples were lyophilized before the subsequent analyses.
In another set of experiments, each hydrolysate (5 g) was placed in a polystyrene petri dish (150 mm × 15 mm) and distributed evenly. A dielectric plasma barrier reactor was formed by placing a petri dish between two aluminum plate electrodes (outer diameter = 158 mm) using polypropylene dielectric layers (2 mm thick) and sealed in a tray with O2 inside. Then, the tray was treated directly at 60 and 70 kV for the 30 s and 60 s. After the treatment, a cloud of hydrolysate powder was continuously forming in the petri dish. Treated samples (HGH-CP and SGH-CP) were kept (4 °C, 24 h) before analysis [9].
Determination of molecular weight distribution of treated hydrolysates
The molecular weight distribution was determined by gel permeation chromatography on a TSKgel G2000SWXL column (7.8 mm × 300 mm i.d., 5 µm) with a HPLC system (Tosoh, Tokyo, Japan) [10]. A mixture of water, acetonitrile and trifluoroacetic acid (85:15:0.1, v/v/v) was used as the mobile phase. Then, the molecular weight distribution of the samples was recorded (flow rate: 0.6 m/L; λ = 220 nm) at room temperature. The calibration curve was determined using the following standards: leucine–enkephalin (451 Da), His-Leu-Ser- Thr-Ala-Phe-Ala-Arg-Val (1002 Da), and tochrome-C (12,400 Da).
Determination of the contents of free sulfhydryl (SH) group and disulfide bond (SS) contents
The sulfhydryl (–SH) and disulfide bonds (S–S) contents of treated hydrolysates were measured using the method of Mahdavian and Koochaki [11]. The sample solutions were diluted (5 times) by Tris-Gly buffer (0.086 M Tris, 0.09 M glycine, 4 mM EDTA, pH 8.0). Thereafter, 20 µL of DTNB (4 mg/mL) was added to 2 mL of sample solution and mixed well After incubation for 25 min at 25 °C, the absorbance was read (λ = 412 nm, Agilent-Carry 60, Santa Clara, CA, USA). Equation (1) was used to calculate the –SH group content.
The mixing ratio of samples (200 µL), β-mercaptoethanol, and Tris-Gly buffer was 5:1:10 (v/v/v). Following the addition of the TCA (10 mL, 12%, w/v), the mixture was centrifuged (15 min, 5000 g). The precipitate was collected and mixed with DTNB (0.15 mL, 4 mg/mL) at 25 °C for 25 min in a water-bath and the absorbance was read (λ = 412 nm). Equation (2) was used to calculate the S–S content.
Surface hydrophobicity index (SHI) determination
A hydrophobic fluorescence probe, ANS, was used to determine the surface hydrophobicity index (SHI) of treated hydrolysates. The sample was serially diluted [0.002–0.01% (w/v)] in phosphate buffer (0.1 mol/L, pH 7.0, 25 °C). Then sample solution (4 mL) was mixed with ANS (20 μL, 8 mM in 0.1 M phosphate buffer, pH 7) followed by vortexing (10 S). A spectrofluorometer (Cary Eclipse, Varian Inc., Palo Alto, CA, USA) was used to measure the fluorescence intensity of each sample at its excitation (295 nm) and emission (475 nm), after storage in dark (15 min). A surface hydrophobicity index (SHI) was calculated from the slope of relative fluorescent intensity (RFI) vs. protein concentrations. RFI was defined as (F − F0)/F0, where F represented the fluorescence of ANS conjugate protein and F0 represented the fluorescence of ANS solution without protein [11].
Preparation of ultrafiltration fractions
The treated hydrolysate having the highest bioactivities were subjected to an ultrafiltration with different molecular weight (MW) cut-offs (100, 10, and 1 kDa). Fractions with varying MW distributions > 100, 10–100, 1–10, and < 1 kDa were recovered separately, lyophilized, and their biological activities were measured.
In vitro bioactivity assay
The treated hydrolysates and the obtained fractions obtained from the ultra-filtration and RP-HPLC separation were tested for the in vitro anti-diabetic activity (α-glucosidase and α-amylase enzymatic inhibitory activities) and in vitro inhibition activity of micellar solubility of cholesterol.
The α-glucosidase inhibitory activity of samples was assayed based on the modified method of Revero-Pino et al. [4] The lyophilized treated (US and ACP) samples (100 µL, 30 mg/mL PBS (pH 6.8, 0.1 M) were incubated (37 °C, 10 min) with the α-glucosidase solution. Then, pNPG (100 μL, 3 mM) was added to the mixture and incubation was performed (37 °C, 30 min). After the addition of the Na2CO3 (0.1 M, 750 μL), the reaction was terminated and the absorbance was recorded (λ = 405 nm) every 2 min. The inhibitory activity (%) was calculated using the Eq. (3).
where Ac and As represent the slope of curve for absorbance of control and sample, respectively.
The in vitro α-amylase inhibitory effect was assayed according to the method of Revero-Pino et al. [4] The freeze-dried sample (100 μL, 30 mg/mL) was incubated (37 °C, 10 min) with enzyme solution (100 μL, 3.75 U/mL) and then the starch solution (100 μL, 0.5% w/v) was added to the mixture. To stop the reaction, 200 µL of DNS solution was added. Subsequently, the test tubes were placed in boiling water (5 min) to inactive the enzyme. After cooling in the iced bath, 500 μL of distilled water was added to every tube and the absorbance (λ = 540 nm) was read.
The inhibitory activity (%) was calculated using the Eq. (4).
where Ac and As represent the absorbance of control and sample, respectively.
According to Ashraf et al., [8] cholesterol micelles were prepared. The mixture of cholesterol (2 mM), NaCl (132 mM), sodium taurocholate (10 mM), oleic acid (1 mM), and sodium phosphate buffer (15 mM) was sonicated (400 W, 20 kHz, 20 min) to prepare an emulsion (pH 7.4). After adding 100 µL of the sample (2 mg/mL) to the emulsion (100 µL), the solution was incubated (37 °C, 24 h) and then centrifuged (8000×g, 30 min). Total cholesterol (TC) kits were used to measure cholesterol levels in the supernatants. The total cholesterol was measured by a total cholesterol kit (TC) from Biorex-fars (Iran). The mean values for each set of three measurements were reported. Using the Eq. (5), the percent inhibition of micelle cholesterol uptake was computed.
where Co represents the cholesterol concentration of the original micelles and Cs is the cholesterol concentration after the peptide fraction was added.
Peptide separation with RP-HPLC
Lyophilized fractions from ultrafiltration process with high inhibitory (anti-diabetic & preventing micelle cholesterol uptake) activity were re-suspended in HPLC-grade water (10% w/v). The separation was performed after the sample (60 µL) was injected into the semi-preparative C18 column (Agilent ZORBAX SB-C18, 5 µm, 4.6 mm × 250 mm) of the RP-HPLC system (Agilent Technologies, Santa Clara, CA, USA) equipped with a fraction collector. Eluents A (0.1% (v/v) TFA in ultrapure water) and eluent B [0.1% TFA in acetonitrile (ACN)] were used for a gradient elution as follows: 1–5 min, 20% B; 5–35 min, 20–45% B; 35–45 min, 45–90% B then back to 20% B for 5 min and flow rate (1 mL/min). Absorbance at 280 nm was monitored. Five fractions (S1, S2, P1, P2, and P3) were detected. The collected peptide fractions were freeze-dried and subjected to bioactivity tests [12].
Amino acid analysis
The RP-HPLC fractions with the highest biological activity were collected, and the amino acid compositions of these fractions were determined. After samples (2 mg) were hydrolyzed in HCl (4 mL, 6 N, 110 °C and 24 h) and derivatized with diethyl ethoxy methylene emalonate, the amino acid composition of the hydrolysate fractions was measured using an amino acid analyzer equipped with a Hitachi ion exchange resin column (60 mm × 4.6 mm id, 1 µm). Twenty different amino acids in the samples were examined, and the results were expressed as g/100 g using d, l-a-aminobutyric acid as an internal standard [8, 12].
Statistical analysis
All experiments were carried out in triplication; except the amino acid composition of each sample, which was analyzed in duplicate. Comparison of sample means was carried out via one-way analysis of variance (ANOVA) and Duncan’s multiple range test under the significance level of p < 0.05. The less significant differences (LSD) of post hoc tests were presented as the mean ± standard deviations (SD). Excel software was used to plot the bar graphs and Normal distribution was tested using SPSS statistics 18 software (IBM, New York, USA).
Results and discussion
Molecular weight distribution
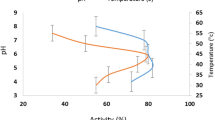
The molecular weight distribution of the all hydrolysates was detected by GPC (Fig. 1). The molecular weight of the un-treated hydrolysates mainly ranged from 451 to 12,400 Da (78–79%), indicating that the actinidin enzyme in kiwifruit extract effectively hydrolyzed the gluten and high-molecular-weight protein (≥ 10 kDa). The most frequent molecular weight distribution of US-treated samples was mainly (63–65%) in the range of 451–1002 Da with minor distribution at Mw > 12,400 (2%). ACP-treated hydrolysates contained the large quantity (> 98%) of peptides with Mw > 451 and the peptides larger than 12,400 was found (36–39%). The application of both US and ACP treatments did not have the marked impact on hydrolysis improvement. However cavitation explosion in ultrasound treatments altered the physical properties of hydrolysate and created high amount of peptides ranging from 451 to 1002 kDa. In ACP-treated hydrolysates various reactions such as dimerization and oxidization led to the formation of peptides with Mw larger than 12,400 kDa. Non-thermal technologies such as US and ACP altered the structure of hydrolysates, mainly via unfolding. Di- and tripeptides could be absorbed more efficiently with greater efficiency than free amino acids and bigger peptide molecules [7]. These peptides were postulated to possess antioxidative, antihypertensive, and anti-diabetic properties [13]. Jayawardana et al. [10] also demonstrated that the highest antioxidant activity and α-amylase inhibitory activity was found in gluten hydrolysate fraction with Mw less than 3 kDa.

GPC chromatogram (left) and molecular weight distribution (right) of hydrolysates. Values above peaks indicate molecular weight
Surface hydrophobicity
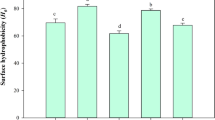
Amphiphilic fluorescent dyes such as ANS can be used to detect hydrophobic patches on protein surface. With US and ACP treatments, hydrophobic groups can be exposed, impacting the stability, structure, and function of the protein [11]. It was observed that (Fig. 2A) the hydrophobicity of the hydrolysates was significantly affected by US and ACP post-treatments. This was more likely correlated with increased biological activity of the treated hydrolysates. The hydrophobicity of the hydrolysates post-treated with ACP (SGH-CP60/30) and US (SGH-U25) reached the maximum value (2122.5 and 1041 a.u), which had 227% and 61% increases, compared to that of the control (649.2 a.u), respectively. In general, ACP upsurged surface hydrophobicity of both SGH and HGH, in comparison with the controls and those subjected to ultrasound post-treatment. The highest hydrophobicity was found in SGH subjected to APC at 60 kVpp for 30 s (p < 0.05). Radicals in plasma plausibly increased surface property of peptides, in the way which hydrophobic domain was more exposed to aqueous phase. Furthermore, the hydrophobicity of the peptide increased as the ultrasonic amplitude of 25% was used in both SGH and HGH. Jin et al. [14] reported that the ultrasound action may cause amino acids that are buried inside native proteins to become exposed, thereby increasing the number of hydrophobic sites. Nevertheless, surface hydrophobicity of both hydrolysates decreased as the amplitude was higher than 25% (p < 0.05). Chandrapala et al. [15] also found that during prolonged ultrasound treatments, surface hydrophobicity could be reduced. These results indicated that both post-treatments could facilitate the accessibility of hydrophobic amino acid residues to ANS binding.

Surface hydrophobicity (A) and Free SH and SS content (B) of treated hydrolysates
Sulfhydryl (SH) group and disulfide bond (SS) contents
The total free SH group (T-SH) and SS contents of wheat gluten hydrolysate treated with US (25–75% amplitude) and ACP (60–70 kV and 30–60 s) are represented in Fig. 2B. Free SH group content in SGH was higher than that of HGH (p < 0.05). As a result of sonication, free SH group content increased significantly (p < 0.05), suggesting the possible exposure of internal SH groups in peptides derived from LMW and HMW glutenin subunits. According to Saleem and Ahmad, [16] the increased SH content were observed after ultrasound pretreatment. The amount of free SH groups were highest in the treated hydrolysate. With ACP post-treatment, the amount of SH content decreased dramatically in all samples (p < 0.05), especially for 70kV treated samples. SH radicals produced by plasma free radicals, specifically OH· could combine with free –SH groups on peptide surfaces [17]. Radicals were very readily active in intramolecular or intermolecular –SS– crosslinking. Furthermore, these SH radicals are capable of further interacting with oxygen molecules to produce thiol peroxyl radicals (SOO·), which would deoxygenate the thiol groups found in LMW and HMW of glutenin to form SS bonds [9]. Plasma discharge produces a large amount of ozone, which oxidizes sulfur-containing amino acids, such as cysteine, and thus decreased the number of free SH groups via oxidizing thiol groups to disulfide bonds [5]. OH radicals exhibited a five-fold higher rate of reaction than O3 radicals [6]. Thus, it was speculated that OH radicals more likely played a more important role in decreasing free—SH groups after ACP post-treatment of both hydrolysates. These findings were also consistent with previous works [18]. No difference (p > 0.05) could be seen in the content of S–S bonds between SGH and HGH. The increases in –SH bonds were attained in the SGH post-treated with ultrasound of 25 and 75%. For the APC post-treatment, the increases were found in SGH subjected to APC at 60 and 70 kV for 30 s (p < 0.05). The exposed free SH groups might undergo oxidation to form S–S bonds to high extent.
Anti-diabetic (α-glucosidase and α-amylase inhibitory activities) of hydrolysates as affected by post-treatments
HGH-U25 (30 mg/mL) showed the highest α-glucosidase inhibitory activity, (9.71 ± 0.23% inhibition). As the amplitude intensity increased to 25%, the inhibitory activity of the sample increased. The activity was then decreased when amplitude of 75% was applied. After sonication of hydrolysates, hidden amino acid residues and side chains with inhibitory activity (which might be concealed within folded peptides) were more exposed, leading to an increase in inhibitory activity. High amplitude of ultrasound post-treatment could increase aggregation of peptides, which could not provide more α-glucosidase inhibition sites. As a result, the inhibitory activity was decreased. Among the methods used to treat diabetic patients, one involves the inhibition of α-glucosidase secretion by attaching to the inhibitor through electrostatic interactions and hydrogen bonds. As a consequence, the disaccharides are not degraded into monosaccharides in the small intestine [19]. González-Montoya et al. [20] stated that peptides which contain amino acids with hydroxyl side groups were effective in inhibiting α-glucosidase. When ultrasound was applied as pretreatment, the Tenebrio molitor peptides released by trypsin was able to inhibit α-glucosidase (∼ 50%) [4]. Ultrasound coupled with subcritical water (USW) technology was employed to extract anti-diabetes peptides from S. platensis [21].
The results of α-amylase inhibitory activity of post-treated US and ACP hydrolysates indicated that all hydrolysates had an inhibitory effect but the highest inhibitory activities toward α-amylase were observed in HGH-U25, HGH-U75, SGH-U50 and SGH-U75, while the control showed the lowest inhibitory activity (2.28 ± 0.06%) (Table 1). The peptides post-treated with APC showed less effect on inhibition of α-amylase activity than ultrasound post-treatment. Another method used to treat diabetic patients is the use of α-amylase inhibitors that delay the digestion of carbohydrates [22]. Hu et al. [21] reported that ultrasound treatment (200 W) can release α-amylase inhibitory peptide from S. platensis (∼ 37%).
The highest anti-diabetic activity (HGH-U25) was separated into different fractions with varying Mws including (MW < 1 kDa) and (MW 1–10 kDa), (MW 10–100 kDa), and (MW > 100 kDa). The highest inhibition (37.56 ± 1.12 and 14.25 ± 0.11) of α-amylase and α-glucosidase activities was HGH-U25 (MW < 1 kDa), while the fractions HGH-U25 (MW ≥ 100 kDa) exhibited little or no inhibitory effect (6.81 ± 1.33 & N.D.) (Table 2). The result indicated that the peptide's molecular size determined the α-glucosidase inhibitory activity. This might be the result of the presence of amino acids with sulfur that dramatically increased α-glucosidase inhibitory activity. Peptides with proline residues close to the C-terminal, and amino acids with hydroxyl groups at their N-terminal accounting for 3.78, 10.88, and 13.57 g/100 g, respectively showed α-glucosidase inhibitory activity [19, 20]. Wang et al. [13] suggested an anti-diabetic peptide (< 5 kDa) from fermented soybean protein. The highest and lowest α-amylase inhibitory activity of Pinto bean peptides were attributed to peptide with MW < 3 kDa (62.10 ± 3.49%) and MW > 100 kDa (18.11 ± 0.55%), respectively [23]. The hydrolysate (MW < 1 kDa) of buckwheat protein exhibited significantly (p < 0.05) stronger α-glucosidase (90%) and α-amylase (79%) inhibition activity compared to others [24]. Although the relationship between the structure and the activity of these peptides is not well known, it can be claimed that gluten peptides produced by sonication after enzymatic digestion (SGH-U25) had anti-diabetic activity.
Micellar solubilization of cholesterol of hydrolysates as affected by post-treatments
To be absorbed by the intestinal epithelium, cholesterol and triglycerides must be transported in micelles and remained non-aggregated. Human bile is frequently modeled using phosphatidylcholine and bile salts suspensions. The detergent effect of bile salts and the amphiphilic character of phospholipids are combined and sonicated to create small micelles similar to those found in the digestive tract. This process is crucial for solubilizing digested products and allowing cholesterol to be dissolved and absorbed. Several precursor structures (cholesterol crystals) are created when cholesterol is too high in the body. The bile is responsible for removing these crystals from the body [25]. Other sterols including phytosterols, can reduce cholesterol concentration because micelles have limited space. One mechanism investigated for explaining the effect of food on lowering plasma cholesterol is how protein hydrolysates modulate cholesterol absorption [26].
As illustrated in Table 1, both hydrolysates with ACP and US post-treatments inhibited cholesterol solubility into micelles significantly (p < 0.05). It was proven that SGH-CP-60/30 (60 kV, 30 s) effectively reduced the solubility of cholesterol (34.32 ± 2.75%) as compared to the control (9.28 ± 0.45%). One of the main reasons for the low solubilization of micellar cholesterol in the control was the low hydrophobic amino acid composition (Table 3). The control also had the lowest surface hydrophobicity (Fig. 2A). After sonication, inhibitory activity was significantly (p < 0.05) improved (25.11 ± 1.41%), which revealed that sonication could change both the structure and functional properties of hydrolysates. Hydrophobic peptides have also been demonstrated to have the ability to lower the critical micellar concentration by binding to bile salts, thereby decreasing the micellar lipid-carrying capacity [26]. It has been demonstrated that the voltage applied and time of ACP processing play a significant role in the biological activities. The generation of peptide with lowering solubilization of the micellar cholesterol can be achieved by moderate ACP treatment and had a high potential for industrial production [7]. As demonstrated by Won et al. [27] low and limited exposure to ACP increased the inhibitory activity, whereas extended exposure at high flow rates decreased the inhibitory capacity. Ashraf et al. [8] found that the composition and hydrophobicity of amino acids in both mung bean and white kidney bean sonicated hydrolysates might be involved in (p < 0.05) inhibition of cholesterol absorption (45.00% ± 1.99; 31.90% ± 0.77).
Applications of enzymolysis for the production of biopeptide possessing the lowering blood cholesterol activity have also been reported in the literature for white rice (10 mg/mL, 5–15%) [26], chickpea (2 mg/mL, 50%) [28], soybean (5 mg/mL 56.9%) [7] and sunflower (1 mg/mL, 60%) [25] hydrolysates.
As the SGH-CP60/30 had the strongest cholesterol-lowering activities, four molecular weight cut-off membranes were used for fractionation (Table 2). The results showed that all fractions were able to lower the solubility of cholesterol but the strongest (53.26 ± 1.91%) inhibitory effect was achieved for the medium molecular weight (10–100 kDa) fraction. The finer and larger MW fractions showed the lower inhibitory activity 31.44 ± 1.64% and 13.85 ± 0.12%, respectively. The exact mechanisms responsible for the cholesterol-lowering effects are still unclear. The amino acid composition of hydrolysates might influence their cholesterol-lowering effects. Our results coincided well with the former observation that hydrophobic peptides derived from soy protein [7] and mung bean [29] could bind bile acids, thereby lowering cholesterol levels. The in vitro bile acid binding capacity of citrus pectin was higher with mono-frequency ultrasound than with dual-frequency ultrasound treatment [30, 31]. ACP might also have modulated the physicochemical properties of the peptides in hydrolysate, leading to an increase in the cholesterol-lowering activity.
Fractionation via RP-HPLC
Two major proteins in wheat gluten were gliadins (α-, β-, γ- and ω) and glutenins (high molecular weight (HMW), and low molecular weight (LMW) [10, 32]. Actinidin with MW of ~ 29 kDa was present [1, 2]. Glutenin showed diverse bands over the range of 67–88 kDa as HMW and MW of 28–39 kDa as LMW. GLY was also composed of the α-β and λ-GLY with MW of 28–39 kDa and ω-GLY with MW of 39–55 kDa [1]. Based on preliminary study, actinidin was effectively degraded and high-molecular-weight bands (≥ 10 kDa) disappeared. US and ACP treatments did not induce major changes in the protein electrophoretic patterns (data not shown). Since the sample fractions with MW below 1 kDa in HGH-U25 and SGH-CP60/30 having MW in the range of 10–100 kDa had stronger anti-diabetic activity and inhibition of micellar solubilisation of cholesterol, respectively, it was chosen for further fractionation using RP-HPLC, one of the most important and rapid techniques for purification and separation of bio-peptides [7].
In HGH-U25 (< 1 kDa), two fractions (S1 and S2) were isolated, and their α-amylase and α-glucosidase inhibition activities were calculated (Fig. 3A). The results showed that they possessed different inhibition activities. The S1 and S2 sub-fractions showed high potential for inhibition of α-glucosidase and α-amylase at 12.33 − 22.17% and 15.12 − 37.16%, respectively. Significant differences in inhibition activities were observed among the two fractions and S2 exhibited the strongest inhibitory activities. The cholesterol-lowering activity in the SGH-CP60/30 having peptide with MW in the range of 10–100 kDa was measured. The results showed (Fig. 3B) that they possessed different inhibition activities. The P1, P2 and P3 sub-fractions showed cholesterol-lowering activities around 11.27, 39.81 and 55.26%, respectively. These results suggested that P2 and P3 contributed more to the cholesterol-lowering activities. The cholesterol-lowering activity in P3 were significantly higher (p < 0.05) than those of the remaining fractions. Fractions S2 and P3 had relatively long retention times, indicating the presence of peptides with low polarity or non-polar peptides.

RP-HPLC profile and anti-diabetic (HGH-U25 < 1 kD) (A) and cholesterol-lowering (SGH-CP60/30, 10–100 kD) (B) activities of peptides obtained from chromatogram
Amino acid profile
Table 3 illustrates the amino acid profile of antidiabetic sub-fractions (S1 and S2) and cholesterol-lowering sub-fractions (P1, P2, and P3) obtained by prep-RP-HPLC of HGH-U25 (MW ≤ 1 kDa) and SGH-CP60/30 (Mw of 10–100 kDa), respectively. There was a significant difference (p < 0.05) in the proportion of hydrophobic amino acids in the sub-fractions (S1, S2, P1, P2 and P3), which was 18.85, 40.91, 18.53, 36.47, and 54.11%, respectively. The major amino acids found in sub-fractions were aspartic acid (Asp), cysteine (Cys), glutamic acid (Glu), glutamine (Gln), serine (Ser), arginine (Arg), proline (Pro), asparagine (Asn) and theronine (Thr). All the sub-fractioned were found to be rich in aspartic acid (9.90–15.40 g/100g) and glutamine (24.71–27.05 g/100g). The high amounts of aspartic acid and proline (1.51–29.11 g/100 g) and serine (3.41–21.50 g/100 g) are essential for various bodily functions. Memory, learning, and neurotransmitter production are affected by glutamic acid and glycine. Blood circulation, erectile dysfunction treatment, wound healing, and immunity are all attributed to arginine’s function [6, 23].
There was a considerable difference (p < 0.05) between the sub-fractions in the composition of hydrophobic amino acids, which might explain why S2 showed a slightly better inhibitory action (both glucose and amylase inhibitory) and P3 showed a cholesterol-lowering effect (Sect. 3.5). The sub-fractions had an essential amino acid (EAA)/total amino acid (TAA) and EAA/non-essential amino acid (non-EAA) of 10.32–20.28% and 12.29–27.09%, respectively. In adults, FAO/WHO joint committee recommends that the ratio of EAA to non-EAA should not be less than 60% and the ratio of EAA to TAA should not be lower than 40%. Based on the results, none of the sub-fractions had nutritionally desirable characteristics. Therefore, enrichment of these peptides with EAA is still needed. Ultrasound and cold plasma post-treatments did not help yield bioactive peptides with desirable nutritional properties.
Although many of these techniques can be very efficient in the lab-scale, their high manufacturing costs usually make them ineffective for large-scale applications. Identified challenges to the commercialization of peptide-based products include the lack of large-scale technologies and the high cost of purifying methods. In industrial biotechnology processes, the main operating cost (70%) of a project is related to the separation and purification process [1]. In this study, the gluten hydrolysates were post-treated by US and ACP, isolated, and identified. Ten gram of wheat gluten produced 3.4 g (34% w/w) of partially purified peptides with α-amylase (37.56%), α-glucosidase (14.25%) inhibition, and cholesterol-lowering (47.08%) activity. The anti-diabetic and cholesterol-lowering effectiveness of post-treated (US and ACP) gluten hydrolysate was still needed to be improved via the combined methods or other approaches.
Conclusions
Innovative technologies (US and ACP) could be applied for the post-treatments of hydrolysate to obtain bio-functional peptides derived from hard and soft wheat gluten. GPC analysis showed that the US and ACP-treated peptides had MW in the range of 451–1002 and MW of 451 and > 12,400 Da, respectively. With the increase of US amplitude in US, voltage and time in ACP, the hydrophobic value and free S–H content of the hydrolysates were modified. HGH-U25 fraction with MW < 1 and SGH-CP60/30 fraction with MW of 10–100 kDa had the highest anti-diabetic and cholesterol-lowering activity, respectively which mainly due to the presence of hydrophobic amino acids. However, additional in vivo studies (animal experiments) are required to ascertain whether α-glucosidase and α-amylase inhibitory peptides, and cholesterol-lowering activities can be employed as drugs for the treatment of type II diabetes and hyperlipidemia advantage.
Data availability
All the resulting data are available and will be provided to any reader upon request.
References
B. Mousavi, M.H. AzizI, S. Abbasi, Antidiabetic bio-peptides of soft and hard wheat gluten. Food Chem: Mol. Sci. 4, 100104 (2022). https://doi.org/10.1016/j.fochms.2022.100104
L. Kaur, M. Shane, P. Rhutherfurd, J. Moughan, D. Lynley, M.J. Boland, Actinidin enhances gastric protein as assessed using an in vitro gastric digestion Model. J. Agric. Food Chem. 58, 5068–5073 (2010). https://doi.org/10.1021/jf903332a
T. Guimarães, E.K. Silva, V.O. Alvarenga, A.L. Costa, R.L. Cunha, A.S. Sant’Ana, M.Q. Freitas, M.A. Meireles, A.G. Cruz, Physicochemical changes and microbial inactivation after high-intensity ultrasound processing of prebiotic whey beverage applying different ultrasonic power levels. Ultrason. Sonochem. 44, 251–260 (2018). https://doi.org/10.1016/j.ultsonch.2018.02.012
F. Rivero-Pino, F.J. Espejo-Carpio, R. Pérez-Gálvez, A. Guadix, E.M. Guadix, Effect of ultrasound pretreatment and sequential hydrolysis on the production of tenebrio molitor antidiabetic peptides. Food Bioprod. Process. 123, 217–224 (2020). https://doi.org/10.1016/j.fbp.2020.07.003
E. Takai, T. Kitamura, J. Kuwabara, S. Ikawa, S. Yoshizawa, K. Shiraki, H. Kawasaki, R. Arakawa, K. Kitano, Chemical modification of amino acids by atmospheric-pressure cold plasma in aqueous solution. J. Phys. D. Appl. Phys. 47, 285403 (2014). https://doi.org/10.1088/0022-3727/47/28/285403
G.J. Fadimu, A. Farahnaky, H. Gill, T. Truong, Influence of ultrasonic pretreatment on structural properties and biological activities of lupin protein hydrolysate. Int. J. Food Sci. 57(3), 1729–1738 (2022). https://doi.org/10.1016/j.foodchem.2022.132457
S. Huang, C. Li, S. Li, S.R. Ruan, N.O. Azam, H. Yang, Effects of ultrasound-assisted sodium bisulfite pretreatment on the preparation of cholesterol-lowering peptide precursors from soybean protein. Int. J. Biol. Macromol. 183, 295–304 (2021). https://doi.org/10.1016/j.ijbiomac.2021.04.125
L. Ashraf, M. Liu, T. Awais, L. Xiao, X. Wang, L. Zhou, S. Tong, S. Zhou, Effect of thermosensation pre-treatment on mung bean (Vigna radiata) and white kidney bean (Phaseolus vulgaris) proteins: enzymatic hydrolysis, cholesterol-lowering activity, and structural characterization. Ultrason. Sonochem. 66, 105121 (2020). https://doi.org/10.1016/j.ultsonch.2020.105121
S.K. Pankaj, N.N. Misra, P.J. Cullen, Kinetics of tomato peroxidase inactivation by atmospheric pressure cold plasma based on dielectric barrier discharge. Innov. Food Sc. Emerg. Technol. 19, 153–157 (2013). https://doi.org/10.1016/j.ifset.2013.03.001
I.A. Jayawardana, M.J. Boland, K. Higgs, M. Zou, T. Loo, W.C. Mcnabb, C.A. Montoya, The kiwifruit enzyme actinidin enhances the hydrolysis of gluten proteins during simulated gastrointestinal digestion. Food Chem. 341(1), 128239 (2020). https://doi.org/10.1016/j.foodchem.2020.128239
H. Mahdavian-Mehr, A. Koochaki, Effect of atmospheric cold plasma on structure, interfacial and emulsifying properties of Grass pea (Lathyrus sativus L.) protein isolate. Food Hydrocoll. 106, 105899 (2020). https://doi.org/10.1016/j.foodhyd.2020.105899
B. Zhang, Q. Sun, H.J. Liu, S.Z. Li, Z.Q. Jiang, Characterization of actinidin from Chinese kiwifruit cultivars and its applications in meat tenderization and production of angiotensin I converting enzyme (ACE) inhibitory peptides. LWT Food Sci. Technol. 78, 1–7 (2017). https://doi.org/10.1016/j.lwt.2016.12.012
R. Wang, H. Zhao, X. Pan, C. Orfila, W. Lu, Y. Ma, Preparation of bioactive peptides with antidiabetic, antihypertensive, and antioxidant activities and identification of α-glucosidase inhibitory peptides from soy protein. Food Sci. Nutr. 7, 1848–1856 (2019). https://doi.org/10.1002/fsn3.1038
N. Jin, Y. Wang, F. Crawford, N-terminal additions to the WE14 peptide of chromogranin A create strong autoantigen agonists in type 1 diabetes. Proc. Natl. Acad. Sci. USA 112(43), 13318–13323 (2015). https://doi.org/10.1073/pnas.1517862112
J. Chandrapala, B. Zisu, M. Palmer, S. Kentish, M. Ashokkumar, Effects of ultrasound on the thermal and structural characteristics of proteins in reconstituted whey protein concentrate. Ultrason. Sonochem. 18(5), 951–957 (2011). https://doi.org/10.1016/j.ultsonch.2010.12.016
R. Saleem, R. Ahmad, Effect of low-frequency ultrasonication on biochemical and structural properties of chicken actomyosin. Food Chem. 205, 43–51 (2016). https://doi.org/10.1016/j.foodchem.2016.03.003
J. Zhao, Y.L. Xiong, D.H. McNear, Changes in structural characteristics of antioxidative soy protein hydrolysates resulting from scavenging of hydroxyl radicals. J. Food Sci. 78(2), 152–159 (2013). https://doi.org/10.1111/1750-3841.12030
A. Segat, N.N. Misra, A. Fabbro, F. Buchini, G. Lippe, P.J. Cullen, Effects of ozone processing on chemical, structural and functional properties of whey protein isolate. Food Res. Int. 66, 365–372 (2014). https://doi.org/10.1016/j.foodres.2014.10.002
M.A. Ibrahim, M.J. Bester, A.W. Neitz, A.R.M. Gaspar, Rational in silico design of novel α-glucosidase inhibitory peptides and in vitro evaluation of promising candidates. Biomed. Pharmacother. 107, 234–242 (2018). https://doi.org/10.1016/j.biopha.2018.07.163
M. González-Montoya, B. Hernández-Ledesma, R. Mora-Escobedo, C. Martínez-Villaluenga, Bioactive peptides from germinated soybean with anti-diabetic potential by inhibition of dipeptidyl peptidase-IV, α-amylase, and α-glucosidase enzymes. Int. J. Mol. Sci. 19(10), 2883 (2018). https://doi.org/10.3390/ijms19102883
F. Hu, A.T. Ci, H. Wang, Y.Y. Zhang, J.G. Zhang, K. Thakur, Identification and hydrolysis kinetic of a novel antioxidant peptide from pecan meal using alcalase. Food Chem. 261, 301–310 (2018). https://doi.org/10.1016/j.foodchem.2018.04.025
J. Ren, S. Chen, C. Li, Z. Gu, L. Cheng, Y. Hong, A two-stage modification method using 1, 4-α-glucan branching enzyme lowers the in vitro digestibility of corn starch. Food Chem. 305, 125441 (2020). https://doi.org/10.1016/j.foodchem.2019.125441
Y.Y. Ngoh, C.Y. Gan, Enzyme-assisted extraction and identification of antioxidative and alpha-amylase inhibitory peptides from Pinto beans (Phaseolus vulgaris cv. Pinto). Food Chem. 190, 331–337 (2016). https://doi.org/10.1016/j.foodchem.2015.05.120
T. Tao, D. Pan, Y.Y. Zheng, T.J. Ma, Optimization of hydrolyzed crude extract from Tartary buckwheat protein and analysis of its hypoglycemic activity in Vitro. IOP. Conf. Ser: Earth Environ. Sci. 295, e032065 (2019). https://doi.org/10.1088/1755-1315/295/3/032065
C. Megias, J. Pedroche, M. Yust, M. Alaiz, J. Girón-Calle, F. Millán, Sunflower protein hydrolysates reduce cholesterol micellar solubility. Plant Foods Hum. Nutr. 64(2), 86–93 (2009). https://doi.org/10.1007/s11130-009-0108-1
H. Zhang, W.H. Yokoyama, H. Zhang, Concentration-dependent displacement of cholesterol in micelles by hydrophobic rice bran protein hydrolysates. J. Sci. Food Agric. 92, 1395–1401 (2012). https://doi.org/10.1002/jsfa.4713
M.Y. Won, S.J. Lee, S.C. Min, Mandarin preservation by microwave-powered cold plasma treatment. Innov. Food Sci. Emerg. Technol. 39, 25–32 (2017). https://doi.org/10.1016/j.ifset.2016.10.021
A. Gharibi, M. Gafsi, A. Sila, C. Blecker, S. Danthine, H. Attia, Effects of enzymatic hydrolysis on conformational and functional properties of chickpea protein isolate. Food Chem. 187, 322–330 (2015). https://doi.org/10.1016/j.foodchem.2015.04.109
C. Sonklin, M.A. Alashi, N. Laohakunjit, O. Kerdchoechuen, R.T. Aluko, Identification of antihypertensive peptides from mung bean protein hydrolysate and their effects in spontaneously hypertensive rats. J. Funct. Foods 64, 103635 (2020). https://doi.org/10.1016/j.jff.2019.103635
B. Mousavi, S. Ghaderi, Effect of varying levels of acorne flour on antioxidant, staling and sensory properties of Iranian Toast. Int. J. Food Stud. 10, 322–333 (2021). https://doi.org/10.7455/ijfs/10.2.2021.a4
S. Cheng, M. Tu, H. Liu, G. Zhao, M. Du, Food-derived antithrombotic peptides: preparation, identification, and interactions with thrombin. Crit. Rev. Food Sci. Nutr. 59(1), S81–S95 (2019). https://doi.org/10.1080/10408398.2018.1524363
B. Mousavi, M. Kadivar, Effect of brine solution as a wheat conditioner, on lipase, amylase, and lipoxygenase activities in flour and its corresponding dough rheological properties. J. Food Process. Preserv. (2018). https://doi.org/10.1111/jfpp.13631
Author information
Authors and Affiliations
Contributions
BM: Conceptualization, Investigation, Writing—Original Draft. SB: Writing—Review & Editing. GH: Investigation. SP: Resources. SG: Supervision, Funding acquisition.
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Mousavi, B., Benjakul, S., Hassani, G. et al. Enhancement of in vitro cholesterol-lowering and anti-diabetic activities of gluten hydrolysate prepared using kiwifruit protease as affected by ultrasound and atmospheric cold plasma post-treatments. Food Measure 17, 6268–6280 (2023). https://doi.org/10.1007/s11694-023-02118-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11694-023-02118-w