
Abstract
Foodborne pathogens Listeria monocytogenes, Salmonella Typhimurium and Escherichia coli O157:H7 continue to be leading causes of foodborne illness. Herein, we describe a sensitive and reliable lateral flow immunoassay for the simultaneous detection of these three bacteria via multiplex PCR and test strip by using gold nanoparticles (AuNPs) as label. Biotin, FITC and digoxin were used as tags for Listeria monocytogenes, Salmonella Typhimurium and Escherichia coli O157:H7, respectively. AuNPs were conjugated with thiolated probes and detected by multiplex lateral flow strip with three test lines corresponding to the three bacteria. The proposed method exhibited excellent specificity (no cross-reactivity with other tested foodborne bacteria were observed), good precision with the coefficient of variation less than 8.73%, and satisfactory recoveries (82.34–98.34%). The limits of detection were from 0.3 × 101 to 3.5 × 102 CFU mL−1 for the three bacteria in chicken breast sample, without culture enrichment, revealing the feasible and reliable of this method. The developed process, including sample preparation, PCR amplification, AuNPs conjugation and test strip detection, can be completed within 4 h. In light of the high level of targets specificity and sensitivity, this method provides a novel application potential for the simultaneous detection of foodborne pathogenic microorganisms.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Foodborne illnesses are an important public health problem worldwide. WHO estimates that unsafe food causes 600 million foodborne illnesses and 420,000 deaths per year in the world [1]. Foodborne pathogens are the leading cause of foodborne disease outbreaks, they can cause severe diarrhoea or debilitating infections including meningitis. Salmonella, Enterohaemorrhagic Escherichia coli (EHEC) and Listeria monocytogenes are the most common foodborne pathogens that affect millions of people annually. Salmonella Typhimurium (S. Typhimurium), as one of the most frequent serotypes of Salmonella enterica species, is the leading cause of gastroenteritis and bacteraemia worldwide [2]. Escherichia coli O157:H7 (E. coli O157:H7) is a predominant serotype of EHEC that can cause bloody diarrhea and haemolytic uraemic syndrome, it produces type I and/or type II shiga toxin [3]. Listeria monocytogenes (L. monocytogenes) are widely disturbed in nature and can cause listeriosis, which is one of the most serious foodborne diseases. Listeriosis primarily afflicts pregnant women and peoples with immuno-compromised status, the mortality rate can be as high as 30% [4]. A highly sensitive and accurate detection of these foodborne pathogens is of great importance to prevent foodborne illnesses.
The current gold standard for the detection of foodborne pathogens is conventional culture-based methods that need long time to complete (more than 2 days) and labor intensive. Polymerase chain reaction (PCR)-based methods have been widely used for the rapid, sensitive and specific detection of pathogenic bacteria in foods [5]. Furthermore, multiplex PCR allows to amplify more than one gene in one reaction tube [6]. It provides a simple and sensitive tool for the simultaneous detection of pathogenic bacteria, which is time-saving and needs less sample [7]. However, the PCR-based assays require complicated gel electrophoresis and gel imaging systems to obtain the results, it greatly limits its application in easy-to-use detection [8].
In recent years, lateral flow immunoassay (LFIA) is a promising point-of-care diagnostic method, owing to its advantages of user-friendly operation, rapid, cost-effective, visualized and does not rely on professional techniques or instruments. It has been widely studied in various fields such as food safety detection, clinical diagnosis, environmental analysis, drug residue analysis [9]. Among the variety of materials, gold nanoparticles (AuNPs) are one of the most commonly used labels in LFIA for their excellent optical and physical properties, and good biocompatibility [10]. However, the sensitivity of the AuNP-based assay for the detection of foodborne bacteria is not high (105 CFU/mL) [11]. The combination of PCR-based method can be used to improve the sensitive of LFIA [12, 13]. It is necessary to develop a rapid, portable and sensitive method for simultaneously detecting multiple foodborne pathogenic bacteria in food.
Herein, a novel method based on multiplex PCR and LFIA test strip for simultaneous detection of S. Typhimurium, E. coli O157:H7 and L. monocytogenes was developed. The sensitivity of PCR system was improved by optimizing the concentration of primers and annealing temperature. AuNPs were conjugated with PCR products via thiol (-SH) group on probes in an acid environment, and visualized detected by test strip. The existence of S. Typhimurium, E. coli O157:H7 and L. monocytogenes shows red color on the corresponding test line of test strip. No bands will appear on the strip if there is no target in sample, which is more convenient and accurate than the indirect competitive method. This study offers a promising approach for simultaneous detection of foodborne pathogens.
Materials and methods
Materials
Salmonella. Typhimurium (CICC 21484) was employed from China Center of Industrial Culture Collection (CICC), L. monocytogenes (ATCC 15313), E. coli O157:H7 (ATCC 43895), P. aeruginosa (ATCC 10145), V. parahaemolyticus (ATCC 17802) and S. aureus (ATCC 6538) were employed from American Type Culture Collection (ATCC), Thiol labeled primer LM-hlyF, biotin labeled primer LM-hlyR specifically to L. monocytogenes; Thiol labeled primer ST-hutF, FITC labeled primer ST-hutR specifically to S. Typhimurium; and thiol labeled primer EC-rfbEF, digoxin labeled primer EC-rfbER specifically to E. coli O157:H7, were purchased from BGI Technology Co., Ltd (Shenzhen, China). Sodium citrate, Taq PCR Master Mix, Ezup column bacteria genomic DNA isolation kit, DNA marker, and 4S Green Plus nucleic acid stain were bought from Sangon Biotechnology Co., Ltd (Shanghai, China). Agarose was procured from Solarbio Life Sciences (Beijing, China). Anti-biotin antibody was procured from Abcam (Shanghai, China); Anti-FITC antibody was purchased from BioMag beads (Wuxi, China); Anti-digoxin was bought from Jackson (USA). Backing card, sample pad, nitrocellulose (NC) membrane and absorbent pad were obtained from Shanghai JieYi Biotechnology Co., Ltd (Shanghai, China). Chloroauric acid was purchased from Macklin (Shanghai, China). Trypticase soy broth (TSB) was bought from Qingdao Hope Bio-Technology Co., Ltd (Qingdao, China). Commercial test kit was purchased from Meizheng Bio (Beijing, China).
The detection principle of multiplex LFIA
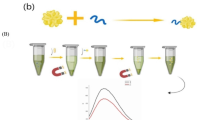
The principle of the developed detection method of bacteria is shown in Fig. 1. AuNPs in red wine color was prepared by using sodium citrate as reducing and stabilizing agent. Bacterial DNA was extracted by boiling for 10 min, which is simple, rapid, economic and environmental friendly as compare with traditional solvent extraction and commercial kits. For multiplex PCR, three pairs of primers specific for hut gene of S. Typhimurium, rfbE gene of E. coli O157: H7 and hly gene of L. monocytogenes were prepared. The forward primers for hut, rfbE and hly gene were labeled with thiol group, and the reverse primers for hut, rfbE and hly gene were labeled with FITC, digoxin and biotin tags, respectively. As a result, PCR products of S. Typhimurium contained thiol group and FITC tag, PCR products of E. coli O157: H7 contained thiol group and digoxin tag, PCR products of L. monocytogenes contained thiol group and biotin tag. The multiplex PCR products were conjugated with AuNPs in citric acid buffer (pH 3), and the remaining sites on AuNPs were blocked by BSA. The AuNP-PCR product-BSA was detected by LFIA strip by adding it to sample pad and migrated along the LFIA toward absorbent pad. When it reaches NC membrane, biotin tag will be captured by anti-biotin antibody coated on test line 1 (T1), FITC tag will be captured by anti-FITC antibody coated on test line 2 (T2), and digoxin tag will be captured by anti-digoxin antibody coated on test line 3 (T3). If sample contains target bacteria, the corresponding test lines will appear red color, which attributed to the immune binding between antigens on AuNPs and antibodies on test line. With the accumulation of PCR products, the color on the test line will be gradually deepened. On the contrary, no band will appear on the test line if target bacteria absent in the sample.

Schematic illustration of detection principle of this study, a Preparation of AuNPs, b Multiplex PCR assay, c LFIA strip detection
Bacterial culture
Salmonella. Typhimurium, E. coli O157: H7, L. monocytogenes, S. aureus, V. parahaemolyticus and P. aeruginosa were cultured in TSB and incubated at 37 °C with shaking (150 rpm). The logarithmic phase cultures were collected through centrifugation at 7512 Xg for 2 min, followed by washing and resuspending in sterile phosphate buffer (PBS, 0.01 M, pH 7.4). Then, the bacteria cultures were diluted with sterile PBS to make different concentrations. The number of bacteria was determined by plate count method and bacteria were cultured on agar plate.
Genomic DNA isolation and multiplex PCR
Genomic DNA of the bacteria was extracted by simple boiling at 100 °C for 10 min, after cooling down to room temperature, and centrifuged at 7512 Xg for 2 min, the supernatant of bacterial culture which contained DNA was used as template for multiplex PCR. Meanwhile, DNA was extracted and purified according to the manufacturer’s protocol of Ezup column bacteria genomic DNA isolation kit. The limit of detection of proposed method for the purified DNA was determined in order to provide the basis for the performance of bacterial DNA detection. The quantity and quality of the obtained DNA were evaluated according to A260 and A260/A280, respectively, with a NanoReady Ultramicro spectrophotometer (Life Real, Hangzhou, China).
The multiplex PCR was performed with reaction solution contained 2 µL of each genomic DNA, 2 µL of each primer (at a final concentration of 0.8 µM), 25 µL 2 × Taq PCR Master Mix buffer and sterilized ddH2O to make final volume of 50 µL. The specific primers were listed in Table S1. PCR reactions were carried out using a S1000TM thermal cycler (BioRad Laboratories, USA), the PCR condition included initial denaturation for 5 min at 95 °C, followed by 32 cycles of 95 °C for 30 s, 53 °C for 30 s and 72 °C for 30 s, and a final extension at 72 °C for 10 min. PCR products were stored at -20 °C for further detection.
Conjugation of AuNPs with multiplex PCR products
AuNPs was prepared by the Turkevich method using sodium citrate as reducing agent with slightly modifications [14]. Briefly, 100 µL of 0.1 mM HAuCl4 was added to 99.9 mL Milli Q water and stirred vigorously. After boiling, 1 mL of 1% sodium citrate was added quickly to the HAuCl4 solution under stirring and boiled for another 10 min. The obtained red color solution was cooled to room temperature and stored at 4 °C in dark. The synthesized AuNPs were characterized by UV–vis spectrum, TEM and FT-IR analysis.
PCR products were conjugated with AuNPs according to the report of Zhang et al. [15]. Specifically, 5 µL of 1 M citric acid buffer (PH 3) was added to 50 µL PCR products, 50 µL AuNPs was added to the above solution and incubated for 3 min to make AuNP-PCR product conjugation. Afterward, the mixture was centrifuged at 4694 Xg for 5 min, the pellet was resuspended in 100 µL of 10% BSA for blocking, the AuNP-PCR product-BSA composite was obtained.
Fabrication of the test strip
The test strip was prepared according to previous study [16], it consisted of four parts: sample pad (1.5 cm × 3 mm), NC membrane (2.5 cm × 3 mm), absorbent pad (3 cm × 3 mm) and backing pad (7 cm × 3 mm). There are three test lines on the NC membrane, T1 was spotted with anti-biotin antibody, T2 was spotted with anti-FITC antibody and T3 was spotted with anti-digoxin antibody at a rate of 1 μL cm−1, respectively. The prepared test strip was dried at 37 °C for 4 h, cut into 3 mm width, and stored in a desiccator kept in dark.
The detection of S. Typhimurium, E. coli O157: H7 and L. monocytogenes
Different concentrations of S. Typhimurium, E. coli O157: H7 and L. monocytogenes ranged from 109 to 101 CFU mL−1 were prepared to determine the limit of detection of the assay. After DNA extraction and PCR amplification, 20 µL of the AuNP-PCR product-BSA suspension were detected by test strip and results were observed in 10 min. As control, PCR products were analyzed using 2% agarose gel electrophoresis and monitored under a Gel Imager System (geldoc EZ, USA).
In order to confirm the specificity of this assay, three common foodborne pathogens at a high concentration (107 CFU mL−1) were detected by the test strip, including S. aureus, V. parahaemolyticus and P. aeruginosa. All experiments were repeated three times to prove credibility.
Detection of artificially contaminated food samples
Chicken breast were obtained from a local supermarket and analyzed within 1 h. Samples were pretreatment according to the culture-based method. Briefly, chicken breast was divided into three portions of 25 g and washed twice with deionized water, soaked in 75% ethanol for 10 min, and then sterilized with UV light for 30 min. Bacteria in varying concentrations were spread evenly each 25 g chicken breast and incubated overnight. Then, the chicken breast was placed in 225 mL PBS, and treated with flapped and concussed to obtain the bacterial suspension. The suspension was detected by the proposed method. At the same time, the plate count method was performed to verify the detection.
Results and discussion
Optimization of multiplex PCR assay and antibody concentration on test line of LFIA
In order to ensure the specificity of the multiplex PCR and obtain each specific PCR products, annealing temperature (48.8 °C, 50 °C, 53 °C, 55.5 °C, 56.2 °C, 58.8 °C, 60 °C, 62.2 °C) and concentration of primer pairs (Table S2) were optimized in this study. The multiplex PCR products were 360 bp, 495 bp and 678 bp, corresponding to specific genes of L. monocytogenes, S. Typhimurium and E. coli O157:H7, respectively (Figs. 2a and 3a). The gray value of the band was measured and analyzed by image J software. It was found that intensity of bands reached maximum when the annealing temperature was 53 °C (Fig. 2b) and the concentrations of the primer pairs was 0.8 µM respectively (Fig. 3b). The antibody concentration on test lines is critical for the detection and the cost of test strip. The anti-biotin antibody, anti-FITC antibody and anti-digoxin antibody at the concentration of 0.4 M, 0.6 M, 0.8 M and 1 M were optimized respectively. The intensity of bands reached the maximum when the concentrations of all three antibodies were 1 M (Fig. 4).

Optimization of multiplex PCR annealing temperature

Optimization of concentrations of the primer pairs

Optimization of antibody concentration
Characterization of AuNPs and AuNP@PCR products
The AuNPs were synthesized by HAuCl4 and sodium citrate through heating stirring process. The AuNPs were uniformly elliptical spherical and the size was approximately 50–75 nm (Fig. S1), with maximum absorption peak at 525 nm (Fig. 5b). The size of AuNP@PCR products significant increased compared with AuNPs (Fig. 5a). Furthermore, the zeta potential of AuNPs and AuNP@PCR products were -26.4 ± 1.2 mV and -37.75 ± 1.5 mV, respectively (Fig. 5d), indicating the AuNPs were successfully conjugated with PCR products. According to the FTIR spectra of AuNPs, peaks at 1249 cm−1, 1452 cm−1 and 3240 cm−1 were due to C-CH3 tensile vibration and the bending vibration of methylene C–H, O–H stretching vibration of hydroxyl functional groups, respectively [17,18,19,20,21,22], the FTIR spectra of AuNP@PCR products showed more peaks than AuNPs, due to the existent of DNA, biotin, FITC digoxin, etc., its functional groups are more complex.

Size distribution (a), UV–visible absorption spectra (b), FT-IR spectra (c) and zeta potential (d) of AuNPs and AuNP@PCR products
Sensitivity of the assay
The limit of detection of genomic DNA by the LFIA test strip was firstly studied. Serial dilutions (70, 35, 17.5, 8.8, 4.4, 2.2, 1.1, 0.6, 0.3, 0.15 and 0.075 ng/µL) of each bacteria's DNA were prepared. The DNA was subjected to multiplex PCR assay, and sterilized ddH2O served as control in the same manner. After multiplex PCR amplification, PCR products were conjugated with AuNPs and then detected by the strip. The PCR products were analyzed using 2% agarose gel electrophoresis as control. The limit of detection of genomic DNA were found to be 150, 150, 600 pg/µL for L. monocytogenes, S. Typhimurium and E. coli O157:H7, respectively (Fig. 6). The limit of detection of DNA examined by traditional agarose gel method were 1.1 ng/µL for the three bacteria (Fig.S2A), which is less sensitive than the proposed LFIA by this study.

Sensitivity of multiplex LFIA for DNA detection (a) and intensity analysis of test lines (b)
The sensitivity of the developed method for the detection of bacteria was studied under the optimal conditions. Different concentrations of L. monocytogenes, S. Typhimurium and E. coli O157:H7 (101–109 CFU mL−1) were prepared by diluting the logarithmic phase bacteria with sterile PBS, and quantified by plate count method. As shown in Fig. 7a, the limit of detection of E. coli O157:H7, S. Typhimurium and L. monocytogenes were 1.6 × 102 CFU mL−1, 1.0 × 102 CFU mL−1, 1.0 × 101 CFU mL−1 by the LFIA with naked eye. The limit of detection of L. monocytogenes, S. Typhimurium and E. coli O157:H7 by traditional agarose gel were 0.9 × 107, 1.2 × 107, 1.5 × 107 CFU mL−1, respectively (Fig.S2B). The detection sensitivity of the LFIA proposed in this study improved 106 CFU mL−1 for L. monocytogenes and 105 CFU mL−1 for S. Typhimurium and E. coli O157:H7, as compared with traditional agarose gel assay. Moreover, agarose gel is time-consuming, toxic, and requires gel imager equipment, whereas the test strip is portable and visible. The intensity of test lines showed good linear relationship with the corresponding bacteria. The linear equation of L. monocytogenes concentration in the range of 1.0 × 101 to 0.8 × 109 CFU mL−1 was Y = 33.29 + 32.50 X with correlation coefficient (R2) of 0.941 (Fig. 7b), S. Typhimurium concentration in the range of 1.0 × 102 to 1.1 × 109 CFU mL−1 was Y = − 74.08 + 57.02 X with correlation coefficient (R2) of 0.996 (Fig. 7c), E. coli O157:H7 concentration in the range of 1.6 × 102 to 1.5 × 109 CFU mL−1 was Y = − 64.36 + 43.91X with correlation coefficient (R2) of 0.962 (Fig. 7d).

Sensitivity for target bacteria cultures (a), the linear plot between T1 intensity and different concentrations of L. monocytogenes concentration (b), the linear plot between T2 intensity and different concentrations of S. Typhimurium (c), and the linear plot between T3 intensity and different concentrations of E. coli O157:H7 (d)
The multiplex LFIA employed PCR amplification and AuNPs labeling to improve the detection sensitivity of the three foodborne pathogenic bacteria. By comparing with some recently reported PCR-based methods and AuNP-based methods for detection of foodborne pathogenic bacteria, the proposed method showed better performance in terms of sensitivity and speed (Table 1). The developed multiplex strips are faster, more sensitive and less time consuming than single test line strip when detecting of three species of bacteria.
Specificity of the assay
The multiplex LFIA was tested for the specificity against three other foodborne pathogen bacteria including S. aureus, V. parahaemolyticus and P. aeruginosa. The traditional agarose gel verified the accurate and specific of multiplex PCR (Fig.S2C). As shown in Fig. 8, the appearance of band on the test line was only observed when corresponding target bacteria is presence. No significant changes were observed in the presence of other foodborne pathogen bacteria, and no cross-reactivity with each other of the three bacteria were observed. Therefore, the multiplex LFIA is highly specific and ensures the accurate detection of target bacteria.

Specificity of multiplex LFIA (a) and intensity analysis of test lines (b)
Detection of bacteria in food samples
The prepared chicken breast samples spiked with E. coli O157:H7 (101–108 CFU mL−1), S. Typhimurium (101–108 CFU mL−1) and L. monocytogenes (101–108 CFU mL−1) were tested by the multiplex LFIA strip, the recovery rates and their coefficients of variation (CV) were calculated, and the accuracy of the proposed method was evaluated by plate count method. Traditional agarose gel method was also used to detect the multiplex PCR products as control, and the limit of detection turned out to be 0.5 × 107, 1.9 × 107, 3.7 × 107 CFU mL−1 of L. monocytogenes, S. Typhimurium and E. coli O157:H7, respectively (Fig.S2D). For comparison, a commercial L. monocytogenes LFIA strip, S. Typhimurium LFIA strip and E. coli O157:H7 LFIA (Meizheng Bio, China) were employed to detect L. monocytogenes, S. Typhimurium and E. coli O157:H7, respectively. The limit of detection were 9.2 × 107 CFU mL−1, 1.2 × 106 CFU mL−1, 1.6 × 106 CFU mL−1 of L. monocytogenes, S. Typhimurium and E. coli O157:H7, respectively (Fig.S3).
As shown in Fig. 9a, the limit of detection of the developed multiplex LFIA strip of L. monocytogenes, S. Typhimurium and E. coli O157:H7 was 0.3 × 101 CFU mL−1, 2.0 × 101 CFU mL−1 and 3.5 × 102 CFU mL−1, respectively. As compare with commercial LFIA strips, sensitivity of developed multiplex LFIA strip was improved 106 CFU mL−1of L. monocytogenes, 105 CFU mL−1of S. Typhimurium and 104 CFU mL−1of E. coli O157:H7, and the result can be read in one strip, which is convenient and low cost. The intensity of band on test lines were gradually increased with the increasing of bacteria concentration (Fig. 9b-d). The recoveries of this study were ranging from 92.7% to 112.1%, with the CV less than 8.7%, and the recoveries of plate count were in the range of 96.6% to 102.9% with the CV less than 7.3% (Table 2). Therefore, the developed multiplex LFIA is sensitive and efficient, shows a promising application in simultaneous detection of foodborne pathogen bacteria in food samples.

Sensitivity of chicken breast samples contained target bacteria (a), the linear plot between T1 intensity and L. monocytogenes concentration (b), the linear plot between T2 intensity and S. Typhimurium (c), and the linear plot between T3 intensity and E. coli O157:H7 (d)
Conclusions
In the present study, a novel multiplex LFIA was developed for simultaneous detection of L. monocytogenes, S. Typhimurium and E. coli O157:H7. The highly sensitive of this method was take advantages of PCR amplification and AuNPs labeling. The reliable of the method was benefit by the specific of the primers and antibodies. The limit of detection of L. monocytogenes, S. Typhimurium, E. coli O157:H7 cultures was 1.0 × 101 CFU mL−1, 1.0 × 102 CFU mL−1 and 1.6 × 102 CFU mL−1, respectively. Thus, the developed method was rapid, sensitive, specific, cost-effectiveness, and suitability for the simultaneous detection of the three target bacteria in chicken breast. The potential for application and adaptation of this method in various bacteria and food products needs further exploration and development. By providing a reliable and sensitive approach for simultaneous detection of foodborne pathogens, this research contributes to ensuring food safety and protecting public health.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
F. Fung, H.S. Wang, S. Menon, Food safety in the 21st century. Biomed. J. 41(2), 88–95 (2018). https://doi.org/10.1016/j.bj.2018.03.003
H.H. Sun, Y.P. Wang, P.C. Du, L. Bai, The epidemiology of monophasic Salmonella Typhimurium. Foodborne Pathog. Dis. 17(2), 87–97 (2020). https://doi.org/10.1089/fpd.2019.2676
W. Cha, P.M. Fratamico, L.E. Ruth, A.S. Bowman, J.M. Nolting, S.D. Manning, J.A. Funk, Prevalence and characteristics of Shiga toxin-producing Escherichia coli in finishing pigs: implications on public health. Int. J. Food Microbiol. 264, 8–15 (2018). https://doi.org/10.1016/j.ijfoodmicro.2017.10.017
L. Radoshevich, P. Cossart, Listeria monocytogenes: towards a complete picture of its physiology and pathogenesis. Nat. Rev. Microbiol. 16, 32–46 (2018). https://doi.org/10.1038/nrmicro.2017.126
J.Q. Chen, S. Healey, P. Regan, P. Laksanalamai, Z.L. Hu, PCR-based methodologies for detection and characterization of Listeria monocytogenes and Listeria ivanovii in foods and environmental sources. Food Sci. Human Wellness 6(2), 39–59 (2017). https://doi.org/10.1016/j.fshw.2017.03.001
B. Sahu, S.D. Singh, B.K. Behera, S.K. Panda, A. Das, P.K. Parida, Rapid detection of Salmonella contamination in seafoods using multiplex PCR. Brazilian J. Microbiol. 50(3), 807–816 (2019). https://doi.org/10.1007/s42770-019-00072-8
H. Zhang, S. Morrison, Y.W. Tang, Multiplex polymerase chain reaction tests for detection of pathogens associated with gastroenteritis. Clin. Lab. Med. 35(2), 461–86 (2015). https://doi.org/10.1016/j.cll.2015.02.006
X. Yang, X. Zhou, M. Zhu, Sensitive detection of Listeria monocytogenes based on highly efficient enrichment with vancomycin-conjugated brush-like magnetic nano-platforms. Biosensors & Bioelectronics 91, 238–245 (2017). https://doi.org/10.1016/j.bios.2016.11.044
Y.M. Tian, T. Bu, M. Zhang, Metal-polydopamine framework based lateral flow assay for high sensitive detection of tetracycline in food samples. Food Chem. 339, 127854 (2021). https://doi.org/10.1016/j.foodchem.2020.127854
S.L. Hou, J.J. Ma, Y.Q. Cheng, One-step rapid detection of fumonisin B-1, dexyonivalenol and zearalenone in grains. Food Control 117, 107107 (2020). https://doi.org/10.1016/j.foodcont.2020.107107
S. Ledlod, S. Areekit, S. Santiwatanakul, K. Chansiri, Colorimetric aptasensor for detecting Salmonella spp., Listeria monocytogenes, and Escherichia coli in meat samples. Food Sci. Technol. Int. 26(5), 430–443 (2020). https://doi.org/10.1177/1082013219899593
B. Jin, B. Ma, J.L. Li, Simultaneous detection of five foodborne pathogens using a mini automatic nucleic acid extractor combined with recombinase polymerase amplification and lateral flow immunoassay. Microorganisms 10(7), 1352 (2022). https://doi.org/10.3390/microorganisms10071352
A.N. Baker, G.W. Hawker-Bond, P.G. Georgiou, S. Dedola, R.A. Field, M.I. Gibson, Glycosylated gold nanoparticles in point of care diagnostics: from aggregation to lateral flow. Chem. Soc. Rev. 51(16), 7238–7259 (2022). https://doi.org/10.1039/d2cs00267a
J. Kimling, M. Maier, B. Okenve, V. Kotaidis, H. Ballot, A. Plech, Turkevich method for gold nanoparticle synthesis revisited. J. Phys. Chem. B 32(110), 15700–15707 (2006). https://doi.org/10.1021/jp061667w
X. Zhang, M.R. Servos, J.W. Liu, Instantaneous and quantitative functionalization of gold nanoparticles with thiolated DNA using a pH-Assisted and surfactant-free route. J. Am. Chem. Soc. 134(17), 7266–7269 (2012). https://doi.org/10.1021/ja3014055
S.J. Wu, J. Du, Y.H. Bai, Solvothermal synthesis of α-Fe2O3 polyhedrons and its application in an immunochromatographic strip test for the detection of foodborne pathogen Listeria monocytogenes. Nanotechnology 32, 085502 (2021). https://doi.org/10.1088/1361-6528/abcb30
S.M. Sivakumar, N. Sukumaran, Induction of immune response of hepatitis B vaccine using polyester polymer as an adjuvant. Procedia Vaccinol. 1(1), 164–173 (2009). https://doi.org/10.1016/j.provac.2009.07.028
P. Sharma, P.J. Babu, U. Bora, Sapindus mukorossi aqueous fruit extract as reducing, capping and dispersing agents in synthesis of gold nanoparticles. Micro & Nano Lett. 7(12), 1296–1299 (2012). https://doi.org/10.1049/mnl.2012.0684
B. Ganapuram, M. Alle, R. Dadigala, A. Dasari, V. Maragoni, V. Guttena, Catalytic reduction of methylene blue and Congo red dyes using green synthesized gold nanoparticles capped by Salmalia malabarica gum. Int. Nano Lett. 5(4), 215–222 (2015). https://doi.org/10.1007/s40089-015-0158-3
A.H. Tanzil, S.T. Sultana, S.R. Saunders, A.C. Dohnalkova, L. Shi, E. Davenport, P. Ha, H. Beyenal, Production of gold nanoparticles by electrode-respiring Geobacter sulfurreducens biofilms. Enzyme Microbial Technol. 95, 69–75 (2016). https://doi.org/10.1016/j.enzmictec.2016.07.012
J.B. Punuri, P. Sharma, M.C. Kalita, U. Bora, Green synthesis of biocompatible gold nanoparticles using Fagopyrum esculentum leaf extract. Front. Mater. Sci. 5(4), 379–387 (2011). https://doi.org/10.1007/s11706-011-0153-1
J.B. Punuri, P. Sharma, S. Saranya, R. Tamuli, U. Bora, Piper betle mediated green synthesis of biocompatible gold nanoparticles. Int. Nano Lett. 2(1), 18–27 (2012). https://doi.org/10.1186/2228-5326-2-18
Y.J. Sung, H.J. Suk, H.Y. Sung, T. Li, H. Poo, M.G. Kim, Novel antibody/gold nanoparticle/magnetic nanoparticle nanocomposites for immunomagnetic separation and rapid colorimetric detection of Staphylococcus aureus in milk. Biosensors & Bioelectronics 43, 432–439 (2013). https://doi.org/10.1016/j.bios.2012.12.052
Y.X. Wang, B. Suo, A new 7-plex PCR assay for simultaneous detection of shiga toxin-producing Escherichia coli O157 and Salmonella Enteritidis in meat products. J. fur Verbraucherschutz und Lebensmittelsicherheit J. Consum. Prot. Food Saf. 6(4), 441–447 (2011). https://doi.org/10.1007/s00003-011-0696-1
M. Magliulo, P. Simoni, M. Guardigli, E. Michelini, M. Luciani, R. Lelli, A. Roda, A rapid multiplexed chemiluminescent immunoassay for the detection of Escherichia coli O157:H7, Yersinia enterocolitica, Salmonella typhimurium, and Listeria monocytogenes pathogen bacteria. J. Agric. Food Chem. 55(13), 4933–4939 (2007). https://doi.org/10.1021/jf063600b
B. Suo, Y.X. Wang, Evaluation of a multiplex selective enrichment broth SEL for simultaneous detection of injured Salmonella, Escherichia coli O157:H7 and Listeria monocytogenes. Brazilian J. Microbiol. 44(3), 737–742 (2013). https://doi.org/10.1590/S1517-83822013000300011
Y. Yang, F. Xu, H. Xu H, Z.P. Aguilar, R. Niu, Y. Yuan, J. Sun, X. You, W. Lai, Y. Xiong, C. Wan, H. Wei, Magnetic nano-beads based separation combined with propidium monoazide treatment and multiplex PCR assay for simultaneous detection of viable Salmonella Typhimurium, Escherichia coli O157:H7 and Listeria monocytogenes in food products. Food Microbiol. 34(2), 418–424 (2013). https://doi.org/10.1016/j.fm.2013.01.004
Y. Zhang, X.Z. Hu, Q.J. Wang, Sensitive and specific detection of Escherichia coli, Listeria monocytogenes, and Salmonella enterica serovar Typhimurium in milk by microchip electrophoresis combined with multiplex PCR amplification. Microchemical Journal 157, 104876 (2020). https://doi.org/10.1016/j.microc.2020.104876
J. Du, S.J. Wu, L.Y. Niu, J.G. Li, D.B. Zhao, Y.H. Bai, A gold nanoparticles-assisted multiplex PCR assay for simultaneous detection of Salmonella typhimurium, Listeria monocytogenes and Escherichia coli O157:H7. Analytical Methods 12, 212–217 (2020). https://doi.org/10.1039/c9ay02282a
M. BlažkoVá, M. KoetS, J.H. Wichers, A. van Amerongen, L. Fukal, P. Rauch, Nucleic acid lateral flow immunoassay for the detection of pathogenic bacteria from food. Czech J. Food Sci. 27, S350-353 (2009). https://doi.org/10.17221/959-cjfs
Funding
This work was supported by the Major Special Project on Public Welfare of Henan Province [Grant Number 201300110100].
Author information
Authors and Affiliations
Contributions
DZ: Conception, Methodology, Writing-review. JL: Material preparation, Data collection, Writing-original draft. JD: Analysis and Writing-editing. KL: Material preparation. YB: Funding acquisition, Resources, Supervision. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhao, D., Liu, J., Du, J. et al. A highly sensitive multiplex lateral flow immunoassay for simultaneous detection of Listeria monocytogenes, Salmonella Typhimurium and Escherichia coli O157:H7. Food Measure 17, 6577–6587 (2023). https://doi.org/10.1007/s11694-023-02116-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11694-023-02116-y