
Abstract
Chickpea protein isolate was hydrolyzed batchwise using Alcalase as an endopeptidase and Flavourzyme as an exopeptidase, by either individual or sequential treatment. Secondary structure, SDS-PAGE molecular weight profile, functional properties, and antioxidant activity of the hydrolysates were investigated. Alcalase was more effective than Flavourzyme to cleave the peptide bonds, and the degree of hydrolysis (DH) of Alcalase-treated hydrolysate was 25.8% compared to Flavourzyme-treated counterpart with a DH of 11.9%. Sequential treatment increased the DH up to 50%. The hydrolysis process significantly changed the protein’s secondary structure characterized by decreased ordered structures and increased disordered structures. The more notable changes occurred for those that were treated sequentially. Protein banding patterns of the hydrolysates were also markedly changed, especially for those treated by Alcalase in which no visible band was observed. Furthermore, hydrolysates had a significant (P < 0.05) increase in the solubility, emulsifying capacity, and DPPH· scavenging capacity. The changes in DH and protein secondary structure significantly correlated with functional properties and DPPH· scavenging capacity. DH presented a significant inverse association with emulsifying stability (r = − 0.940), but a positive correlation with DPPH· scavenging activity (r = 0.951). β-sheet was found to be negatively correlated with both solubility (r = − 0.979) and DPPH· scavenging activity (r = − 0.979). However, an opposite trend was observed for β-turn which positively correlated with both solubility (r = 0.881) and DPPH· scavenging activity (r = − 0.909). The results reveal that hydrolysis enhanced the functional properties and antioxidant activity of chickpea protein which may be beneficial for potential functional food ingredient applications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Growing interest in plant-based diets has taken the protein market toward plant-based proteins. Legumes, including chickpea, are anticipated to be among the next new “hot” protein sources [1]. Chickpeas (Cicer arietinum), the third most important grain legume in the world, have been used for human consumption and nutrition throughout history [2]. They have an average protein content of 16–21% and are recognized as a good protein source because of their balanced amino acid composition and high bioavailability [3].
Recently, there is a great interest in the hydrolysis of protein to generate hydrolysates and peptides to improve protein’s functional quality and bioactivities for potential health benefits [4, 5]. Protein hydrolysates are produced by either enzymatic or chemical hydrolysis to form shorter chain peptides and amino acids. Compared to chemical treatment which requires harsh environments, enzymatic hydrolysis could be performed under mild conditions and yields hydrolysates with high specificity and minimal by-products. Therefore, enzymatic hydrolysis presents a good potential in modifying protein structure to improve their functional properties without comprising their nutritional value [6, 7]. The properties of enzymatic hydrolysates are governed by many factors, including the choice of enzyme and processing conditions (pH, temperature, time, and enzyme concentration) [8]. Of various proteases used, Alcalase and Flavourzyme are two of the most common enzymes for protein hydrolysis. Alcalase, an endopeptidase derived from Bacillus licheniformis, has a broad catalytic activity and cleaves the peptide bonds in the interior of polypeptide chains, while Flavourzyme, a mixture of endoprotease and exopeptidase derived from Aspergillus oryzae, mainly cleaves amino acids from the end of polypeptide chains [5, 9, 10]. Several studies on utilization of Alcalase and Flavourzyme to produce chickpea protein hydrolysates with bioactive potential have been reported [3, 4, 11,12,13,14].
Globulins are the major protein fractions present in chickpea and account for 60–80% of the extractable protein. However, its globular structure might be a limiting factor for hydrolysis action of a single protease [8]. Therefore, use of a sequential hydrolysis by combining an endopeptidase (Alcalase) and an exopeptidase (Flavourzyme) would be a better alternative because the cleavage of peptide bonds by Alcalase increases the number of end peptide sites for the action of Flavourzyme. The objectives of this study are to investigate effects of individual and sequential enzymatic hydrolysis using Alcalase and Flavourzyme on secondary structure, functional properties (solubility and emulsifying capacity), and antioxidant activity of chickpea protein hydrolysates, and understand the relationship between structure and property of the hydrolysates.
Materials and methods
Materials
Kabuli chickpea, provided by a local food processor, were grounded using a micro-mill (Bell-Art Products, Wayne, NJ) to pass through a size-60 mesh sieve prior to isolating protein. Alcalase 2.4 L, an endopeptidase from Bacillus Licheniform, has a specific activity of 2.4 Anson Units (AU)/g and density of 1.18 g/mL. Flaourzyme 500 L, an exopeptidase and endoprotease complex from Aspergillus oryzae, has a specific activity of 500 Leucine aminopeptidase unit (LAPU)/g and density of 1.30 g/mL. Both enzymes were purchased from Sigma-Aldrich (St. Louis, MO). All other chemicals were of analytical grades and purchased from Fisher Scientific (Hampton NH).
Isolation of protein
Protein was isolated from ground raw chickpea powders using the method described by Stone et al. [15] with some modifications. Chickpea flour was defatted by mixing with hexane (1:3, w/v) for 1 h. The hexane was then decanted, and residual hexane was allowed to evaporate overnight. Defatted flour was dispersed in distilled water and pH was adjusted to 9.5 using 0.1 N NaOH. The dispersion was stirred for 1 h and then centrifuged at 4500×g for 20 min. After centrifuging, the pH was adjusted to 4.5 using 1 N HCl to precipitate proteins isoelectrically. The precipitate was recovered by centrifugation at 4500×g for 20 min and the supernatant was discarded. The precipitate was washed twice using distilled water (1:10 w/v), recovered by centrifugation at 4500×g for 10 min, and then freeze-dried. All isolated protein preparations were combined and mixed thoroughly prior to further processing. The isolations have an extraction yield of 20.4% (w/w) and protein content of 85.9% (w/w) for calculating the ratio of enzymes used in hydrolysis step.
Enzymatic hydrolysis of protein isolate
The protein isolate was hydrolyzed using either Alcalase or Flavourzyme or by sequential treatment as described by Clemente et al. [3] and Yust et al. [13] with some modifications. Hydrolysis was carried out in a beaker placed in a temperature-controlled water bath. During the reaction, both the water bath and the hydrolysis mixture were stirred to maintain uniform temperature throughout the system, and the mixture was maintained at constant pH by titration using 1 N NaOH. Total hydrolysis time was set to 3 h because a stationary phase hydrolysis after 3 h was observed in our preliminary experiments. Upon completion of hydrolysis, the enzymes were inactivated by heating the hydrolysate mixture to 80 °C for 15 min after which the mixture was cooled to room temperature, neutralized, and centrifuged at 12,000×g for 20 min. Hydrolysate supernatants were freeze-dried and stored at − 20 °C prior to analyses. Hydrolysis parameters are presented in Table 1, the hydrolysates are designated as HA (Alcalase only), HF (Flavourzyme only), HA1F2 (Alcalase-Flavourzyme (1 h:2 h), and HA2F1 (Alcalase-Flavourzyme (2 h:1 h) based on the hydrolysis conditions.
Characterization of protein hydrolysates
Degree of hydrolysis
Degree of hydrolysis (DH), defined as the proportion of cleaved peptide bonds in a protein hydrolysate, was determined using the pH stat method as described by Ghribi et al. [4] based on the amount of NaOH consumed to maintain constant pH during the hydrolysis. DH is expressed as
where B is the amount of NaOH (mL); N is the normality of the NaOH; M is the initial mass (g) of protein being hydrolyzed; htot is the total number of peptide bonds in the protein substrate and is assumed to be 7.22 mmol/g [4, 16]; α represents the average degree of dissociation of the a-NH2 groups in the protein substrate and expressed as:
where pH and pK are the values at which the proteolysis was conducted with pK of 7.5 at 25 °C.
Secondary structure
Secondary structure of chickpea protein isolate (CPI) and its hydrolysates was measured using a Varian 640 Fourier-transform Infrared Spectrometer (FTIR, Agilent Technologies, Santa Clara, CA) as described by Carbonaro et al. [17] with some modifications. Freeze-dried samples were compressed in the sample compartment of a universal ATR platform (Pike Technologies MIRacle™ ATR, Madison, WI) and were scanned from 400 to 4000 cm−1 at a resolution of 2 cm−1. Amide I band (1600–1700 cm−1) was then evaluated using PeakFit v4.12 curve-fitting and analysis software (Systat Software, San Jose, CA). The spectral data were deconvolved and individual component peaks were identified by fitting a Gaussian fixed-band model using the 2nd derivative method. Iterative curve-fitting was performed until the r2 and F-values of the fitted model stabilized. Peak centers were used to assign peaks to the corresponding features of protein secondary structure.
SDS-PAGE molecular weight profile
SDS-PAGE molecular weight profile of CPI and its hydrolysates was determined by a method described by Xu et al. [18]. Freeze-dried samples were mixed with Tricine sample buffer and the concentration of the solutions was standardized to 4 mg/mL. Electrophoresis was conducted using pre-cast 1 mm × 12 well, 10–20% acrylamide Tricine-SDS gel with Tricine-SDS running buffer (Invitrogen, Thermo Fisher Scientific, Waltham, MA). Approximately 6 μg of protein or hydrolysate was loaded per sample well and high range (10–245 kDa) molecular weight ladder was added to a separate well (Thermo Scientific Spectra Multicolor Low Range Protein Ladder, Waltham, MA). Electrophoresis was performed at a constant voltage (125 V), and the run was stopped when the tracking dye was within about 1 cm of the gel edge. The gel was fixed for 2 h using 50% (v/v) MeOH, 10% (v/v) acetic acid, and 40% water, stained for 1 h by fixative plus 0.1% (w/v) Coomassie Blue R250, and then destained using 5% (v/v) methanol, 7.5% (v/v) acetic acid, and 87.5% water to visualize protein bands. Gels were preserved in 7% acetic acid and photographed using an EC3 Imaging System with VisionWorks LS Imaging Acquisition and Analysis software, v6.4.3 (UVP BioImaging Systems, Upland, CA).
Functional properties
Protein solubility (PS) of CPI and its hydrolysates was determined at pH 2, 4, 7 and 9 according to Kaur et al. [19] with some modifications. Each freeze-dried sample (0.5 g) was dispersed in 20 mL of distilled water and the pH was adjusted to one of the pre-determined values. The solutions were stirred for 1 h at room temperature followed by centrifuging at 8000×g for 15 min. The protein content of the supernatants was quantified using the combustion method (AOAC 990.03) [20] using a Vario MAX CN Protein Analyzer (Elementar Americas, Inc., Mt. Laurel, NJ). Protein solubility is expressed as percentage ratio of supernatant protein to total protein contents.
Emulsifying properties of CPI and its hydrolysates were determined by the method of Klompong et al. [21]. Protein and hydrolysate solutions (1%, pH 7, 10 mL) were mixed with vegetable oil (2 mL) and the mixture was homogenized at 10,000 rpm for 2 min. An aliquot of emulsion (50 μL) pipetted from near the bottom of the container at 0 and 10 min after homogenization was dispensed into 0.1% sodium dodecyl sulphate (SDS) solution (5 mL). The absorbance of diluted samples was measured at 500 nm (Thermo Fisher Scientific Evolution 60S spectrophotometer, Waltham, MA). The emulsifying activity index (EAI) and the emulsion stability index (ESI) were calculated as described by Molina et al. [22].
where A0 is the absorbance of the diluted emulsion immediately after homogenization, dilution factor = 100; Ө is the fraction of oil to form the emulsion (0.17); ΔA is the change in the absorbance between 0 and 10 min (A0 − A10), and t is the time interval between initial and final measurements.
Antioxidant activity
Antioxidant activity of CPI and its hydrolysates were determined by DPPH free-radical scavenging capacity as described by Xu et al. [23]. DPPH· solution was prepared by dissolving DPPH in 75% DMSO and diluting to a final concentration of 0.2 mM DPPH·. CPI and its hydrolysates (1.0 mL) were mixed with 1.0 mL of fresh DPPH· solution, incubated in the dark for 1 h, then measured at 515 nm using an Evolution 60S spectrophotometer (Thermo Scientific, Pittsburgh, PA) against a 75% DMSO blank. DPPH· radical scavenging activity was calculated by the following equation:
where AS is the sample absorbance and AC is the absorbance for a control without sample.
Statistical analyses
Three replications were used to obtain average values and standard deviations for all tests. All results were analyzed with SAS version 9.4 statistical software (SAS Institute Inc., Cary, NC, USA). ANOVA with Duncan’s Multiple Range test was used to evaluate the statistical significance of differences and probability (P) ≤ 0.05 indicates significance. Pearson’s correlation coefficient (r) was used to evaluate covariance relationships between structure and properties. Graphical plots were prepared using Graphpad Prism7, except for those of the deconvolved Amide I peaks that were prepared using Peak Fit 4.12.
Results and discussion
Degree of hydrolysis
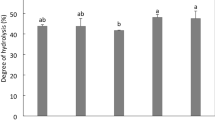
Degree of hydrolysis (DH) is a key parameter for monitoring the extent of protein hydrolysis reaction [24]. DH of four different hydrolysates produced by either individual enzyme (Alcalase or Flavourzyme) or sequential hydrolysis are shown in Fig. 1. DH ranged from 11.9% for HF to 50.2% for HA2F1. In comparison to sequential hydrolysis, hydrolysis using individual enzyme resulted in lower DH values. Of the two enzymes applied, Alcalase is more efficient at cleaving peptide bonds and a significantly (P < 0.05) higher DH was observed for HA than HF. This is in agreement with previous studies where Alcalase-treated cowpea, rice dreg and okra protein hydrolysates exhibited higher DH values than their Flavourzyme-treated counterparts [8, 25, 26]. Alcalase has a broad catalytic activity and expects to be more efficient in cleaving larger peptides to produce extensive hydrolysis, while Flavourzyme mainly targets terminal peptide bonds [27]. However, it is worth noting that extensive hydrolysis in a reasonable time could not be achieved in a one-step reaction where only one enzyme was applied. When the two enzymes were applied sequentially, the resulting hydrolysates had significantly (P < 0.05) higher DH, 45.0% for HA1F2 and 50.2% for HA2F1, respectively. Initial Alcalase hydrolysis predigests the protein and increases the available number of N-terminal sites to facilitate Flavourzyme hydrolysis. Prolonged Alcalase treatment led to more effective hydrolysis and DH of HA2F1 was significantly higher than that of HA1F2. Our results are in the range of previous studies on chickpea protein hydrolysates which had a DH greater than 50% when a sequential Alcalase/Flavourzyme hydrolysis was applied [3, 11].

Degree of hydrolysis of different protein hydrolysates
Secondary structure
Of a number of current techniques, FTIR spectroscopy has been widely used to study protein secondary structure. Figure 2 shows characteristic bands found in the spectra including Amide I band centered at 1642 cm−1 associated with C=O stretching and Amide II band centered at 1536 cm−1 related to NH bending and CN stretching [28]. The amide I absorption zone is the most important for elucidating protein secondary structure, as it is the sum of overlapping component bands: α-helix, β-sheet (parallel and anti-parallel), β-turn and random coils [29]. Gaussian-shaped deconvolved amide I bands curves of CPI and its hydrolysates are shown in Fig. 3, while assignment of amide I peak frequencies to protein secondary structure and their average summative percentage areas are summarized in Tables 2 and 3. CPI had an ordered secondary structure and mainly consist of β-sheet (60.4%), which is in agreement with previous findings showing that proteins in raw common bean and lentil flours had a high content in β-sheet structures [17]. Furthermore, α-helix and β-turn structures accounted for 21.7% and 18.0% of CPI secondary structure, respectively. The hydrolysis process changes appreciably the protein’s secondary structure characterized by decreased ordered structures (β-sheet and α-helix), increased β-turn structures, and appeared new random coils. All hydrolysates had a new peak centered at 1643–1645 cm−1 which was assigned to random coil structure, and β-sheet peak centered at 1692 cm−1 was completely disappeared in both HA1F2 and HA2F1 (Table 2). HA1F2 had the biggest change with the highest β-turn content and the lowest ratio of β-sheet and α-helix, and HF showed the least changes in both β-turn and β-sheet (Table 3). A significant negative correlation was observed between DH and β-sheet (r = − 0.908), while DH had positive relationships with both random coil (r = 0.907) and β-turn (r = 0.894) (Table 5). Hydrolysis significantly destroyed inter-molecular hydrogen bonds in β-sheet to form a disordered structure. An inverse correlation of β-sheet with β-turn (r = − 0.953) and of β-turn with random coil (r = − 0.971) was found, suggesting that β-sheet structure was first converted to β-turn and further converted to disordered structure during the hydrolysis. This implied that partial unfolding process dominates protein structural change, since the turn structure was considered to be a product of protein unfolding of any higher order structures [30]. Our results agree with previous studies of casein or soybean protein showing that hydrolysis causes a pronounced loss of ordered structures [31, 32].

FTIR spectra of chickpea protein isolate and its hydrolysates

Gaussian-shaped deconvolved amide I bands of chickpea protein isolate and its hydrolysates
Molecular weight profiles
SDS-PAGE analysis of CPI and its hydrolysates is shown in Fig. 4. CPI exhibited multiple bands with molecular weight (MW) distribution ranging from ~ 7 to ~ 70 kDa and the major bands between 22–24 and 37–41 kDa. Chang et al. [33] studied CPI and identified the bands of ~ 9–10, ~ 22–24 and ~ 39–41 kDa as legumin subunit 11S protein, bands of ~ 15, ~ 18, ~ 33–35, and ~ 70 kDa as vicilin subunit 7S protein, and a band of 7.0 kDa as subunits of 2S albumin. Their results suggest that legumin subunit 11S protein is the dominant protein in our CPI samples. Hydrolysis, especially Alcalase treatment, markedly changed the protein molecular weight distribution pattern. HF exhibited a significant reduction in the 50–70 kDa and 37–45 kDa bands with a concomitant increase in intensities of 7–9 and ~ 28–36 kDa bands, respectively. The loss of these specific bands suggests that the respective CPI subunits are the most susceptible to enzymatic hydrolysis [34]. Furthermore, all hydrolysates, treated by Alcalase either alone (HA) or in combination with Flavourzyme (HA2F1 and HA1F2), had no visible protein band. Our results are consistent with a previous study for lentil which was hydrolyzed by Alcalase and indicating a thorough hydrolysis [35]. The differences in the band profiles between Alcalase- and Flavourzyme-treated hydrolysates reflect associated enzyme specificity for available catalytic sites. Alcalase cleaved peptide bonds in the globulins, while Flavourzyme had limited effect of cleavage, suggesting that more Flavourzyme-susceptible catalytic sites are buried within the globulins structure rather than presented on the surface [8]. These changes align with our observations on changes in DH and secondary structure and affected functional properties and radical scavenging capacity.

Molecular weight profiles of chickpea protein isolate and its hydrolysates using SDS-PAGE (L for Ladder)
Functional properties
Protein solubility
Protein solubility (PS) is one of the most important protein functionalities. PS profiles of CPI and its hydrolysates at different pH values are shown in Table 4. PS of CPI was pH-dependent with minimum solubility of 1.73% at pH 4.0, close to the isoelectric point (pH 4.3) for chickpea protein [36]. The lack of net charge at the isoelectric point minimizes repulsive forces and enhances attractive forces, thereby causing insolubility, aggregation and precipitation [37]. Hydrolysis significantly increased PS at all tested pH values, especially at pH 4 where PS increased 50 times. This is supported by Wouters et al. [38] who summarized in their review that enzymatic hydrolysis could strongly promote plant protein solubility over a range of pH values. HF exhibited the least increase of PS compared to the other hydrolysates (HA, HA1F2 and HA2F1) at each pH level examined. The same trend was observed by Meinlschmidt et al. [39] in their study of soy protein isolates prepared using different enzymes. The improvement in PS by enzymatic hydrolysis could be attributed to changes in the secondary structure of proteins associated with decreased ordered structure and increased disordered structure. This is confirmed by a significant negative correlation between PS and β-sheet (r = − 0.979) and a positive relation between PS and β-turn (r = 0.881) (Table 5). In addition, formation of smaller peptides as demonstrated in above SDS molecular weight profile, and increasing ionizable amino and carboxyl groups and their interaction with water molecules also contributed to the increase in PS of hydrolysates [7, 39].
Emulsifying properties
Emulsifying capacity, quantified as emulsifying activity index (EAI) and emulsifying stability index (ESI), is an important functional attribute in a food system such as salad dressings and mayonnaise where oil separation may be deleterious to product quality or appearance. CPI had an EAI of 85.5 m2/g and ESI of 15.7 min, respectively. Hydrolysis processing significantly increased both EAI and ESI, ranging from 136.1 to 213.7 m2/g and 26.5 to 38.6 min, respectively (Table 4). However, these increases depended on enzyme type and the corresponding hydrolysis condition. Hydrolysates produced by individual enzyme had significantly higher EAI and ESI compared to their counterparts produced by sequential treatment. HA had a higher EAI but a lower ESI than HF. In general, many factors including DH, enzyme specificity, protein solubility and molecular flexibility, are considered to govern emulsifying properties of the hydrolysates [40]. In our study, a significant inverse association between DH and EAI of the hydrolysates was observed (r = − 0.940) (Table 5). Higher emulsifying capacity in the lower DH hydrolysates could be attributed to the partial unfolding of protein molecules increasing their molecular flexibility, therefore, enhancing rearrangement of protein at the oil–water interface and preventing coalescence [30]. Decreased EAI and ESI associated with higher DH appears to result from smaller peptides produced during excessive hydrolysis losing their ability to interact with both aqueous and non-aqueous phases [7]. No significant correlation was found between emulsifying capacity with either secondary structure or solubility. Similar results was reported by Zhao et al. [25] who found that EAI and ESI of rice dreg protein hydrolysates did not correlate directly with their solubility.
Antioxidant activity
Because of its stability and capacity to act as a free radical scavenger, DPPH· radical has been widely used to evaluate the antioxidant activity of natural compounds [12]. The radical scavenging ability of DPPH· is due to its hydrogen-donating ability, thereby terminating the radical chain reaction [41]. CPI has a lower DPPH· scavenging activity of 8.5% compared to all hydrolysates which had an activity ranging from about 25% for HA and HF to 44.3% for HA1F2 and 48.4% for HA2F1, respectively (Fig. 5). This could be attributed to the peptides generated during the hydrolysis more effectively reacting with free radicals and converting them to stable products than the original protein [42]. The hydrolysates produced from sequential treatment had significantly higher DPPH· scavenging activity compared to their counterparts produced by individual enzymes. This could be explained that sequential hydrolysis favorably enhanced protein unfolding and subsequent release of peptides with DPPH· scavenging activity. Our results agree with those reported on the hydrolysis of chickpea protein and Nile tilapia using a two-step enzymatic process [13, 33]. DPPH· scavenging activity was found to positively correlated with both DH (r = 0.951) and β-turn (r = 0.909), and negatively correlated with β-sheet (r = − 0.899) (Table 5). An increase in DPPH· scavenging activity with increasing DH indicated that more bioactive peptides which were encrypted in original protein structure were released in the hydrolysates with a higher DH. Our result is consistent with previous studies for peanut protein and camel milk casein hydrolysates [43, 44]. As far as seconary structure, a positive correlation between DPPH· scavenging activity and β-turn (r = 0.909) was confirmed by a previous study in which β-turn and random coils were found to be dominant structures in bioactive peptides [45]. Although there was a negative correlation between β-sheet and DPPH· scavenging activity (r = − 0.899), β-sheet structure was still considered to be responsible for release of bioactive peptides during enzymatic digestion [45].

DPPH· scavenging capacity of chickpea protein isolate and its hydrolysates
Conclusion
Enzymatic hydrolysis of chickpea protein isolate by either Alacalase or Flavourzyme individually or by sequential treatment was found to effectively alter protein structure, protein profile banding pattern, and improve their functional and antioxidant properties. Enzyme type and treatment conditions affected the efficacy of the hydrolysis. Sequential hydrolysis appeared to have more pronounced effects as evidence by more pronounced changes in protein secondary structure and profile banding patterns along with a higher degree of hydrolysis and DPPH· scavenging capacity. The changes in protein secondary structure and molecular weight distribution patterns affected functional properties and DPPH· free radical scavenging capacity. The results provide the useful information on choosing appropriate hydrolysis methods and conditions to enhance the functional and antioxidant properties of chickpea protein. Therefore, this study may be of interest for the food industry to develop new functional ingredients for a wide range of uses and formulations. A study of amino acid composition and surface hydrophobicity of these hydrolysates is ongoing.
References
L.M. Ohr, Food Tech. 10, 70–75 (2018)
S. Abbo, D. Shtienberg, J. Lichtenzveig, S. Lev-Yadun, A. Gopher, Q. Rev, Biology 78, 435–448 (2003)
A. Clemente, J. Vioque, R. Sanchez-Vioque, J. Pedroche, F. Millán, J. Agric. Food Chem. 47, 3776–3781 (1999)
A.M. Ghribi, I.M. Gafsi, A. Sila, C. Blecker, S. Danthine, H. Attia, A. Bougatef, S. Besbes, Food Chem. 187, 322–330 (2015)
B.H. Sarmadi, A. Ismail, Peptides 31, 1949–1956 (2010)
V. Muhamyankaka, C.F. Shoemaker, M. Nalwoga, X.M. Zhang, Int. Food Res. J. 20, 2227–2240 (2013)
O.L. Tavano, J. Mol. Catal. B: Enzym. 90, 1–11 (2013)
M.R. Segura-Campos, L. Espinosa-García, L.A. Chel-Guerrero, D.A. Betancur-Ancona, Int. Food Prop. 15, 770–780 (2012)
X. Kong, H. Zhou, H. Qian, Food Chem. 102, 759–763 (2007)
M. Merz, T. Eisele, P. Berends, D. Appel, S. Rabe, I. Blank, T. Stressler, L. Fischer, J. Agric. Food Chem. 63, 5682–5693 (2015)
A. Clemente, J. Vioque, R. Sánchez-Vioque, J. Pedroche, J. Bautista, F. Millán, Food Chem. 67, 269–274 (1999)
Y. Li, B. Jiang, T. Zhang, W. Mu, J. Liu, Food Chem. 106, 444–450 (2008)
M.D.M. Yust, M.D.C. Millán-Linares, J.M. Alcaide-Hidalgo, F. Millán, J. Pedroche, J. Sci. Food Agric. 92, 1994–2001 (2012)
C. Torres-Fuentes, M.D.M. Contreras, I. Recio, M. Alaiz, J. Vioque, Food Chem. 180, 194–202 (2015)
A.K. Stone, A. Karalash, R.T. Tyler, T.D. Warkentin, M.T. Nickerson, Food Res. Int. 76, 31–38 (2015)
J. Adler-Nissen, J. Agric. Food Chem. 27, 1256–1262 (1979)
M. Carbonaro, P. Maselli, P. Dore, A. Nucara, Food Chem. 108, 361–368 (2008)
Y. Xu, M. Obielodan, E. Sismour, A. Arnett, S. Alzahrani, B. Zhang, Int. J. Food Sci. Technol. 52, 1147–1154 (2017)
M. Kaur, N. Singh, Food Chem. 102, 366–374 (2007)
Association of Official Analytical Chemists (AOAC). Official Methods of Analysis, 17th edn. (Association of Official Analytical Chemists, Washington DC, 2000)
V. Klompong, S. Benjakul, D. Kantachote, F. Shahidi, Food Chem. 102, 1317–1327 (2007)
E. Molina, A. Papadopoulou, D.A. Ledward, Food Hydrocolloid 15, 263–269 (2001)
Y. Xu, A. Cartier, A. Porter, K. Lalancette, Z. Abraha-Eyob, E. Sismour, H. Si, X. Wang, N. Rehmani, L. Githinji, J. Food Meas. Charact. 12, 2591–2597 (2018)
C. Kamnrdpetch, M. Weiss, C. Kasper, T. Scheper, Enzym. Microb. Technol. 40, 508–514 (2007)
Q. Zhao, H. Xiong, C. Selomulya, X. Chen, H. Zhong, S. Wang, W. Sun, Q. Zhou, Food Chem. 134, 1360–1367 (2012)
F. Sbroggio, M.S. Montilha, M.R.G. Figueiredo, S.R. Georgetti, L. Kurozawa, Food Sci. Technol. 36, 375–381 (2016)
C.M. Silver, R. Aline, C. Prentice, Int. Food Res. J. 21, 1751–1761 (2014)
W. Gallagher, (2018). https://www.chem.uwec.edu/chem455_S05/Pages/Manuals/FTIR_of_proteins.pdf. Accessed 26 July 2019
J. Ahmed, S.K. Varshney, H.S. Ramaswamy, LWT Food Sci. Tech. 42, 1538–1544 (2009)
M.A.M. Mune, D.S. Sogi, Int. J. Food Prop. 19, 371–384 (2016)
J. Wang, Y. Su, F. Jia, H. Jin, Chem. Cent. J. 7, 62 (2013)
J. Zhao. Y.L. Xiong, D.H. McNear, J. Food Sci. 78, C152–C159 (2013)
Y.-W. Chang, I. Alli, A.T. Molina, Y. Konishi, J.I. Boye, Food Bioprocess Technol. 5, 618–625 (2012)
A.M. Ghribi, A. Sila, R. Przybylski, N. Nedjar-Arroume, I. Makhlouf, C. Blecher, H. Attia, P. Dhulster, A. Bougatef, S. Besbes, J. Funct. Foods 12, 516–525 (2015)
B. Cabanillas, M.M. Pedrosa, J. Rodríguez, A. González, M. Muzquiz, C. Cuadrado, J.F. Crespo, C. Burbano, Mol. Nutr. Food Res. 54, 1266–1272 (2010)
R. Sánchez-Vioque, A. Clemente, J. Vioque, J. Bautista, F. Millán, Food Chem. 64, 237–243 (1999)
N. Singh, M. Kaur, K.S. Sandhu, Dry. Technol. 23, 975–988 (2005)
A.G.B. Wouters, I. Rombouts, E. Fierens, K. Brijs, J.A. Delcour, Compr. Rev. Food Sci. Food Saf. 15, 786–800 (2016)
P. Meinlschmidt, D. Sussmann, U. Schweiggert-Weisz, P. Eisner, Food Sci. Nutr. 4, 11–23 (2016)
S.D.D. Santos, V.G. Martins, M. Salas-Mellado, C. Prentice, Food Bioprocess Technol. 4, 1399–1406 (2011)
S. Yarnpakdee, S. Benjakul, H. Kristinsson, H. Kishimura, J. Food Sci. Technol. 52, 3336–3349 (2015)
C. Pazinatto, L.G. Malta, G.M. Pastore, M.F. Netto, Food Sci. Technol. 33, 485–493 (2013)
S.N. Jamdar, V. Rajalakshmi, M.D. Pednekar, F. Juan, V. Yardi, A. Sharma, Food Chem. 121, 178–184 (2010)
D. Kumar, M.K. Chatli, R. Singh, N. Mehta, P. Kumar, Dairy Sci. Technol. 96, 391–404 (2016).
M. Carbonaro, M. Nardini, P. Maselli, A. Nucara, Org. Agric. 16, 334–335 (2015)
M. Carbonaro, A. Nucara, Amino Acids 38, 679–690 (2010)
Acknowledgements
Funding was provided through the USDA Evans-Allen program and the research was conducted at Virginia State University Agricultural Research Station (Journal Series number 361).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human or animal subjects.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Xu, Y., Galanopoulos, M., Sismour, E. et al. Effect of enzymatic hydrolysis using endo- and exo-proteases on secondary structure, functional, and antioxidant properties of chickpea protein hydrolysates. Food Measure 14, 343–352 (2020). https://doi.org/10.1007/s11694-019-00296-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11694-019-00296-0