
Abstract
Three water-soluble polysaccharides fractions (PFP1a, PFP2b and PFP3c) were isolated and purified from Pleurotus ferulae by DEAE-Sepharose CL-6B and Sepharose CL-6B column chromatography. Their chemical characterization was conducted by chemical methods, chromatography and Fourier transform infrared spectroscopy. Antioxidant activities of crude polysaccharides from P. ferulae (CPFP) and its purified fractions were investigated on the basis of superoxide anion, DPPH, hydroxyl radical scavenging activity and reducing power. The result indicated PFP1a, PFP2b and PFP3c had obvious characteristic peaks of polysaccharides and were mainly composed of six kinds of monosaccharides including rhamnose, arabinose, glucose, mannose, xylose and galactose. The average molecular weight of PFP1a, PFP2b and PFP3c were approximately 5.9 × 105 Da, 3.2 × 105 Da and 2.6 × 104 Da, respectively. PFP1a, PFP2b and PFP3c exhibited antioxidant activities in a concentration-dependent manner. The results demonstrated that the three polysaccharides had the potential to be new natural antioxidant in the food and pharmaceutical industry, especially the PFP2b and PFP3c containing more uronic acid.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Oxidation is an essential biological process in which many living organisms produce energy. However, the uncontrolled production of oxygen-derived free radicals is hostile and harmful to cells. It can cause a chain reaction resulting in the multiplication of new free radicals and some damage such as interference and manipulation of proteins, tissue loosening, genetic damage and promotion of disease and aging [1, 2]. In the last decade, huge interests had received great attention and extensive research had been conducted to determine free radical scavengers or antioxidants that can reduce the risk of oxidative damage and several cardiovascular diseases. Although synthetic antioxidants such as propyl gallate (PG) and butylated hydroxytoluene (BHT) can effectively inhibit oxidative damage, their potential toxicity to humans is being considered and worried [3]. In order to protect the human body from free radicals and reduce risk of various diseases such as heart disease, cancer, arthritis and aging, many efforts have been made to find new and safe antioxidants from natural sources [4].
Recently, many bioactive polysaccharides obtained from different natural sources have been demonstrated to play an important role as free radical scavengers in preventing oxidative damage in living organism and can be explored as novel potential antioxidants [5, 6]. Polysaccharides have attracted much attention in the field of biochemistry and pharmacology [7].
The popularity of edible mushrooms has increased because they are highly nutritious food and also considered to have nourishing and medicinal properties, especially in Chinese folk or traditional medicine. Some mushroom polysaccharides have been demonstrated to play an important role as a dietary free radical scavenger in the prevention of oxidative damage in living organisms [8,9,10]. Therefore, discovery and evaluation of polysaccharides extracted from edible mushrooms as new safe compounds for functional food or medicine has become a hot research spot.
Pleurotus ferulae is an edible mushroom that belongs to the family of pleurotaceae and agaricales and grows on the roots of Ferula communis [11]. Nowadays, P. ferulae is commonly planted in China. However, up to now, no detailed investigation has been carried out on purification, characterization and antioxidant activities of different polysaccharides isolated from the fruiting bodies of P. ferulae.
Therefore, the aim of the present study was mainly to investigate purification and characterization of the major polysaccharides of the fruiting bodies of P. ferulae and evaluate their antioxidant activities in vitro for seeking a new natural antioxidant used in food and pharmaceutical industry.
Materials and methods
Materials and chemicals
The fruiting bodies of P. ferulae were obtained from Altay (Xinjiang province, China). It was dried at 50 °C in an oven for 48 h and ground to pass through a 1 mm screen. DEAE-Sepharose CL-6B and Sepharose CL-6B were purchased from Pharmacia Chemical Co. Dextrans with different molecular weights, standard monosaccharide, sodium salicylate, 1,1-diphenyl-2-picrylhydrazyl (DPPH), phenazine methosulfate (PMS), nitroblue tetrazolium chloride (NBT), β-nicotinamide adenine dinucleotide (NADH), and potassium ferricyanide were purchased from Sigma Chemical Co. (St. Louis, USA). All other chemicals and solvents used were of analytical grade.
Extraction and purification of polysaccharides
The powder of fruiting bodies of P. ferulae was extracted with distilled water at 90 ℃ for 5 h, then filtered through a vacuum filter and centrifuged at 2795×g for 30 min. The supernatant was concentrated in a rotary evaporator, and precipitated with cold 95% ethanol (1:4, V/V) at 4 ℃ for 24 h and then lyophilized to get crude water-soluble polysaccharides from P. ferulae (CPFP) (Fig. 1).

Scheme for extraction and purification of polysaccharides of the fruiting bodies of P. ferulae mushroom
CPFP was dissolved in distilled water and passed through a DEAE-Sepharose CL-6B anion-exchange chromatography column (2.6 cm × 30 cm). The column was first eluted with 0.1 M sodium acetate buffer, followed with gradient solution of NaCl (0–1.5 M) at a flow rate of 1.0 mL/min. After that, all eluted fractions (each for 10 mL) were collected and quantified for the polysaccharide content by the phenol–sulfuric acid method using glucose as standard [12]. Based on the quantified curve, three polysaccharide fractions were collected and denoted as PFP1, PFP2 and PFP3, and then further purified on a Sepharose CL-6B gel-filtration column (2.6 cm × 160 cm) with 0.05 mol/L NaCl. Three main new fractions were collected and denoted as PFP1a, PFP2b and PFP3c (Fig. 1).
Analytical methods
Total carbohydrate was determined by the phenol–sulphuric acid colorimetric method with d-glucose as the standard at 490 nm [12]. The uronic acid content was determined by measuring the absorbance at 525 nm using m-hydroxybiphenyl colorimetric procedure and with glucuronic acid as the standard [13]. The protein content was measured according to Lowry’s method using bovine serum albumin as the standard [14].
Analysis of molecular weights
Molecular weights of PFP fractions were determined by high performance gel permeation chromatography (HPGPC) with a Waters HPLC apparatus equipped with two serially linked UltrahydrogelTM Linear (Ø7.8 mm × 300 mm) columns, a Waters 2410 interferometric refractometer detector and UV detector connected in series with a Millennium32 workstation. The molecular weights were estimated by reference to the calibration curve made under the conditions described above from dextran t-series standards of known molecular weights.
Analysis of monosaccharide composition
Monosaccharide composition of polysaccharides was analyzed by gas chromatography. The PFP fractions (10 mg) dissolved in 2 M trifluoroacetic acid (TFA, 2 mL) were hydrolyzed at 121 °C for 3 h in a sealed glass tube. The hydrolyzate was repeatedly co-concentrated with methanol to remove the excess acid at 50 °C, and then the hydrolyzed products were prepared for acetylation. Acetylation was carried out with 10 mg hydroxylamine hydrochloride and 0.5 mL pyridine for 30 min at 90 °C. After the incubation, the tubes were removed from the heat block, allowed to cool to room temperature, and then 0.5 mL of acetic anhydride was added and mixed thoroughly by vortexing. The tubes were sealed and incubated in a water bath shaker set at 90 °C for 30 min again. After cooling, approximately 0.1 mL of clear supernatant was added to the autosampler vials with inserts for injection into the gas chromatograph on a GCMS-QP2010 Plus (SHIMADZU, JAP) instrument equipped with a hydrogen flame ionization detector, using a DB-1 column (30 m × 0.25 mm × 0.25 µm). The chromatographic conditions were as follows: high-purity helium was used as the carrier gas at a flow rate of 1 mL/min. The temperature of the injector and detector was 250 °C. An initial column temperature held at 100 °C followed by 10 °C/min to 280 °C. Injections were made in the splitless mode. The temperature of mass spectrometer ion source was 230 °C. The sample (1 µL) was injected into the column with the split ratio of 10:1.
FT-IR spectra
Fourier-transform infrared spectra were recorded three times from different polysaccharide fractions powder (1 mg) in KBr pellets on a Nicolet Nexus FT-IR spectrometer in the range of 4000–400 cm−1.
Superoxide anion scavenging assay of PFP fractions
The scavenging activity of the samples toward superoxide anion radical was investigated according to the reported method [15]. Briefly, each 1.0 mL of NBT solution (156 µmol/L of NBT in 0.1 M phosphate buffer, pH 7.4), NADH solution (468 µmol/L of NADH in 0.1 M phosphate buffer, pH 7.4) and PFP solution were mixed. 1.0 mL of PMS solution (60 µmol/L PMS in 0.1 M phosphate buffer, pH 7.4) was added to the mixture. Then the reaction started. The reaction mixture was incubated at 25 ℃ for 5 min, and the absorbance at 560 nm was measured against a blank (water and 0.1 M phosphate buffer instead of the samples and NBT solution, respectively). The scavenging activity of the samples on the superoxide radical was calculated according to the following equation:
where A0 is the absorbance of the control (water instead of PFP solution), A1 is the absorbance of the sample and A2 is the absorbance of the samples under identical conditions as A1 with 0.1 M phosphate buffer instead of NBT solution.
Hydroxyl radical scavenging assay of PFP fractions
The scavenging activity of the samples toward hydroxyl radical was investigated according to the reported method [16] with a little modification. The sample was dissolved in distilled water (10 mL) at the concentration of 250, 500, 1000, 2000 and 4000 µg/mL, respectively. 0.1 mL sample solution was mixed with 0.8 mL of 1.75 mM deoxyribose, 0.1 mM ferrous ammonium sulfate, 0.2 M phosphate buffer (pH 7.4) and 0.1 mM EDTA, then 0.1 mL of 1.0 mM ascorbic acid and 0.1 mL of 10 mM H2O2 were added to the reaction solution. The reaction solution was incubated for 15 min at 37 °C and then 0.5 mL of thiobarbituric acid (1%) and 1 mL of trichloroacetic acid (2.8%) were added to the mixture. The mixture was boiled for 10 min and then cooled on ice. The absorbance of the mixture was measured at 532 nm. The scavenging activity of the samples on the hydroxyl radical was calculated according to the following equation:
where A0 is the absorbance of the control (without polysaccharide sample) and A1 is the absorbance of the polysaccharide sample.
DPPH radical scavenging assay of PFP fractions
DPPH radical is a stable free radical, which has been widely used to evaluate the free radical scavenging activity of natural compounds. Alcohol solution of DPPH has a characteristic absorption maximum at 517 nm. When DPPH ethanol solution is reduced, absorbance decreases and the solution changes from purple to light yellow. The scavenging activity of DPPH radical was carried out according to the reported method [17] with some modifications. Briefly, 3.0 mL various concentrations of CPFP and its purified fractions (250–1000 µg/mL) was added to 1 mL of DPPH solution (0.1 mM, in 95% ethanol). The mixture was shaken vigorously and left to stand for 30 min at room temperature and kept in the dark. The absorbance of the solution was measured at 517 nm with a UV–Vis spectrophotometer. The DPPH radical scavenging activity was calculated according to the following equation:
where A0 is the absorbance of DPPH solution without the tested samples and A1 is the absorbance in the presence of the tested samples with DPPH solution.
Reducing power
The reducing power was determined based on the reported method [18] with some modification. Samples with different concentrations (250–4000 µg/mL) were mixed with 2.5 mL of a phosphate buffer (pH 6.6, 0.2 M) and 2.5 mL of potassium ferricyanide (1%, w/v). The mixture was incubated at 50 ℃ for 20 min. Then, 2.5 mL of 10% trichloroacetic acid was added to to the mixture and centrifuged at 4000×g for 10 min. A 2.5 mL sample of the supernatant was collected and mixed with 2.5 mL of distilled water and 2.5 mL of ferric chloride (0.1%, w/v). After incubating at room temperature for 15 min, the absorbance of the mixture was measured at 700 nm using ascorbic acid (250–4000 µg/mL) as a positive control. The increased absorbance of the reaction mixture indicated an increased reducing power.
Statistical analysis
All the measurements were performed in triplicate and all the values were presented as means of three determinations ± SD (standard deviation). The Statistical Analysis Systems (SAS, version 8.1) software was used for statistical analysis. One-way analysis of variance (ANOVA) was used to determine the differences between the sample results. All values with p < 0.05 were considered statistically significant.
Results and discussion
Isolation, purification and characterization of different polysaccharides fractions
CPFP were isolated and purified to obtain three major new homogeneous fractions denoted as PFP1a, PFP2b, PFP3c, which accounted for 82.6%, 75.2% and 67.5%, respectively. The average molecular weight (Mw) of PFP1a, PFP2b and PFP3c analyzed by HPGPC were approximately 5.9 × 105 Da, 3.2 × 105 Da and 2.6 × 104 Da, respectively (Table 1).
The content of total carbohydrate, protein and uronic acid of PFP1a, PFP2b and PFP3c was presented in Table 1. The content of proteins of PFP3c was determined as 8.3%, while no proteins existed in the PFP1a and PFP2b. No uronic acid existed in the PFP1a and the content of uronic acid determined in PFP2b and PFP3c increased gradually from 6.6 to 27.2%. The monosaccharide composition was presented in Table 2, PFP1a was composed of rhamnose, arabinose, glucose and galactose in a molar ratio of 1:3.6:2.3:3.1. PFP2b was composed of rhamnose, mannose and galactose in a molar ratio of 2:1.6:5.2. PFP3c was composed of xylose, mannose and glucose in a molar ratio of 2.3:1.2:3.5.
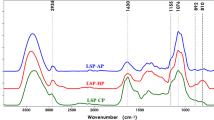
IR spectra of PFP1a, PFP2b and PFP3c was shown in Fig. 2. As shown in Fig. 2, a broadly stretched intense peak at 3250–3500 cm−1 was attributed to characteristic of hydroxyl (O–H) groups. A weak peak at 2750–3000 cm−1 indicated the presence of C–H group. A absorption peak at 1600 cm−1 in PFP2b and PFP3c attributed to carboxyl (C=O) group in uronic acid. However, no absorption peak at 1600 cm−1 in PFP1a revealed that PFP1a had no uronic acid, which agreed with our previous result of uronic acid measurement. A peak at 1024.2 cm−1 was attributed to the sugar ring and glycosidic bond C–O stretching vibrations. Based on the above analysis, PFP1a, PFP2b and PFP3c had characteristics of the functional groups of polysaccharides.

IR spectra of PFP1a, PFP2b and PFP3c of P. ferulae mushroom
Superoxide anion scavenging assay of CPFP and its purified fractions
Scavenging activity of CPFP and its purified fractions (PFP1a, PFP2b and PFP3c) on superoxide anion were presented in Fig. 3. As shown in Fig. 3, scavenging activity CPFP and its purified fractions correlated positively well (p < 0.05) with the increase of polysaccharide concentration ranging from 250 to 4000 µg/mL. In addition, scavenging activity of CPFP and its purified fractions on superoxide anion followed the order: CPFP, PFP3c, PFP2b, PFP1a and were 81.2, 51.8, 46.8 and 12.5% at the concentration of 2000 µg/mL, respectively. Among three purified fractions, PFP3c was observed to posses the strongest scavenging ability, which had the highest content of proteinous substances and uronic acid in the polysaccharide.

Scavenging activity of CPFP and its purified fractions on superoxide anion radical
Among different reactive oxygen species (ROS), superoxide anion is generated first. Although superoxide anion is a relatively weak oxidant, it may decompose to form stronger ROS, such as singlet oxygen and hydroxyl radical, which initiate peroxidation of lipids. superoxide anion is also known to initiate indirectly the lipid peroxidation as a result of the formation of H2O2, creating precursors of hydroxyl radical [19]. The excellent antioxidant activity of PFP3c might be attributed to its special characterization and composition. Higher scavenging activity was found when the content of proteinous substances and uronic acid increased. This result agreed with the findings that the content of proteinous substances in the polysaccharide molecules potentiate their free radical scavenging activity [20]. In addition, PFP3c had the lowest molecular weight (2.6 × 104 Da) among three purified fractions. It had been reported that the molecular weight of polysaccharides was an important parameter influencing antioxidant activity and the low molecular weight showed the high antioxidant activities [21].
Hydroxyl radical scavenging assay of CPFP and its purified fractions
The hydroxyl radical scavenging activity of CPFP and its purified fractions (PFP1a, PFP2b and PFP3c) was shown in Fig. 4. All the samples exhibited weak scavenging ability at low concentration with only about 10% at the concentration of 250 µg/mL. In addition, all samples showed hydroxyl radical scavenging activity in a concentration-dependent manner. Among all samples, the order of CPFP and its purified fractions on hydroxyl radical scavenging activity was CPFP > PFP3c > PFP2b > PFP1a. There was no significant difference on scavenging activity between PFP2b and PFP1a at the concentration ranging from 250 to 500 µg/mL. At the concentration of 2000 µg/mL, the hydroxyl radical scavenging activity of CPFP, PFP3c, PFP2b and PFP1a was 75.5, 62.9, 23.2 and 19.2%, respectively. The results demonstrated that CPFP and its purified fractions possessed hydroxyl scavenging activity. The highest scavenging activity of CPFP may be due to the presence of some other chemical substances such as tocopherol, pigments as well as the synergistic effects among them, which also contribute to the total scavenging activity.

Scavenging activity of CPFP and its purified fractions on hydroxyl radical
The hydroxyl radical is considered to be a highly potent free radical in biological tissues which can easily react with most cellular molecules such as amino acids, proteins, and DNA, thus resulting in severe damage to the adjacent biomolecules [8]. It is also believed to be an active initiator for peroxidation of lipids [22]. Hydroxyl radicals were generated by reaction of iron–EDTA complex with H2O2 in the presence of ascorbic acid, attack deoxyribose to form products upon heating with 2-thiobarbituaric acid under acid conditions, yield a pink tint. Added hydroxyl radical scavengers compete with deoxyribose for the resulted hydroxyl radicals and diminish tint formation [23]. Hydrogen peroxide and superoxide molecules can lead to oxidative injury in the biomolecules indirectly by producing hydroxyl radical via fenton reaction and/or iron-catalyzed Haber–Weiss reaction, which can be prevented and/or inhibited by antioxidants [24]. The hydroxyl radical scavenging ability was attributed to various mechanisms, such as suppression against hydroxyl radical generation, decomposition of peroxides and prevention of continued hydrogen abstraction. The possibility of scavenging hydroxyl radical of CPFP, PFP3c, PFP2b and PFP1a may be due to the supply of hydrogen by polysaccharides, which combine with radicals and forms a stable radical to terminate the radical chain reaction [25]. The other possibility is that polysaccharide can combine with the radical ions which are necessary for radical chain reaction, and the reaction is terminated [1]. Our results showed that PFP3c and PFP2b containing uronic acid had the stronger scavenging activity than PFP1a containing no uronic acid, which suggested that uronic acid might be an effective indicator of antioxidant activity of the samples. Specific reasons need further study in future.
DPPH radical scavenging assay of CPFP and its purified fractions
The scavenging activity of CPFP and its purified fractions on the DPPH radical was shown in Fig. 5. The scavenging activity of CPFP and its purified fractions on DPPH radical was correlated positively well with increasing concentrations. In addition, the DPPH scavenging activity of CPFP increased significantly with the increasing concentrations and were stronger than that of its purified fractions at every concentration point. Interestingly, PFP2b exhibited higher radical scavenging activity than PFP3c at the lower concentrations (250–1000 µg/mL). However, the radical scavenging activity of PFP2b was lower than that of PFP3c at the higher concentrations (2000–4000 µg/mL). At the concentration of 2000 µg/mL, the scavenging activity of CPFP, PFP3c, PFP2b and PFP1a on the DPPH radical were 83.6, 62.1, 49.8 and 17.5%, respectively. The mechanism of scavenging DPPH radical is caused by the fact that natural compounds can transfer either an electron or a hydrogen atom to DPPH [26]. The results indicated that CPFP and its purified fractions might act as electron or hydrogen donator to scavenge DPPH.

Scavenging activity of CPFP and its purified fractions on DPPH radical
Reducing power of CPFP and its purified fractions
Reducing power of CPFP and its purified fractions was shown in Fig. 6. As shown in Fig. 6, at the concentration range of 250–4000 µg/mL, reducing power of CPFP and its purified fractions increased with the increase of sample concentration, which was weaker than ascorbic acid. At the concentration of 2000 µg/mL, reducing power of CPFP, PFP3c, PFP2b and PFP1a was 0.67, 0.34, 0.31 and 0.25, respectively. The results indicated that the order of CPFP and its purified fractions on reducing power was CPFP > PFP3c > PFP1a. It has been reported that there is a direct correlation between antioxidant activity and reducing capacity [27]. Reductones are also reported to react with certain precursors of peroxide, thus preventing peroxide formation [28]. The oxidation and reduction are dynamic antithesis in the body. Reducing power assay is widely used to assess the potential antioxidant activity of many chemicals [29]. In the reaction system, the addition of the antioxidant substances could reduce the Fe3+ in the potassium ferricyanide to the Fe2+ form monitored by formation of prussian blue at 700 nm [30]. The reducing properties were generally associated with the presence of electron-donating groups or hydrogen atoms, which could react with free radicals to stabilize and block radical chain reactions [31]. Our results indicated that the reductone and hydroxyl groups of CPFP and its purified fractions could act as electron donors and reacted with free radicals to convert them to more stable products, thereby terminating the radical chain reaction. CPFP and its purified fractions were likely to contribute to the observed antioxidant effect and their reducing capacity may serve as a significant indicator of its potential antioxidant activity.

Reducing power of CPFP and its purified fractions
Conclusions
Three main polysaccharide fractions (PFP3c, PFP2b and PFP1a) were isolated and purified successfully from CPFP by DEAE-Sepharose CL-6B and Sepharose CL-6B column chromatography. Antioxidant activity assay indicated that radical scavenging activity of PFP3c, PFP2b and PFP1a was in a concentration-dependent pattern. PFP3c and PFP2b exhibited greater capacity in scavenging free radicals. The results implied that some factors such as the content of proteins and uronic acid and molecular weight may affect their antioxidant activities. However, the antioxidant mechanisms of the three polysaccharides are not fully understood. Therefore, it is suggested that possible antioxidant mechanisms of the three polysaccharides should be clarified in our further research work. In addition, the structural feature, relationship between chemical characteristics and antioxidant activities and the identification of toxic and safety of the three polysaccharides need further research as new natural antioxidant in pharmaceutical and food industries in the future.
References
M. Shi, Z.Y. Zhang, Y.G. Yang, Antioxidant and immunoregulatory activity of Ganoderma lucidum polysaccharide (GLP). Carbohydr. Polym. 95, 200–206 (2013)
J.W. Li, Y.F. Liu, L.P. Fan, L.Z. Ai, L. Shan, Antioxidant activities of polysaccharides from the fruiting bodies of Zizyphus jujuba cv. Jinsixiaozao. Carbohydr. Polym. 84, 390–394 (2011)
W.C. Zeng, Z. Zhang, L.R. Jia, Antioxidant activity and characterization of antioxidant polysaccharides from pine needle (Cedrus deodara). Carbohydr. Polym. 108, 58–64 (2014)
L. Karre, K. Lopez, K.J. Getty, Natural antioxidants in meat and poultry products. Meat Sci. 94, 220–227 (2013)
J.F. Yuan, Z.Q. Zhang, Z.C. Fan, J.X. Yang, Antioxidant effects and cytotoxicity of three purified polysaccharides from Ligusticum chuanxiong Hort. Carbohydr. Polym. 74, 822–827 (2008)
A. Matkowski, P. Tasarz, E. Szypula, Antioxidant activity of herb extracts from five medicinal plants from Lamiaceae subfamily Lamioideae. J. Med. Plants Res. 11, 321–330 (2008)
H. Cao, Polysaccharides from Chinese tea: recent advance on bioactivity and function. Int. J. Biol. Macromol. 62, 76–79 (2013)
W.Q. Zhong, N. Liu, Y.G. Xie, Y.M. Zhao, X. Song, W.M. Zhong, Antioxidant and anti-aging activities of mycelial polysaccharides from Lepista sordida. Int. J. Biol. Macromol. 60, 355–359 (2013)
R. Jia, L. Cao, P. Xu, G. Jeney, G. Yin, In vitro and in vivo hepatoprotective and antioxidant effects of Astragalus polysaccharides against carbon tetrachloride induced hepatocyte damage in common carp (Cyprinus carpio). Fish Physiol. Biochem. 38, 871–881 (2012)
B. Liang, M.L. Jin, H.B. Liu, Water-soluble polysaccharide from dried Lycium barbarum fruits: isolation, structural features and antioxidant activity. Carbohydr. Polym. 83, 1947–1951 (2011)
N. Alam, K.N. Yoon, J.S. Lee, H.J. Cho, T.S. Lee, Consequence of the antioxidant activities and tyrosinase inhibitory effects of various extracts from the fruiting bodies of Pleurotus ferulae. Saudi J. Biol. Sci. 19, 111–118 (2012)
M. Dubois, K.A. Gilles, J.K. Hamilton, P.A. Rebers, F. Smith, Colorimetric method for determination of sugars and related substances. Anal. Chem. 28, 350–356 (1956)
N. Blumenkrantz, H.G. Asboe, New method for quantitative determination of uronic acids. Anal. Biochem. 54, 484–489 (1973)
O.H. Lowry, N.J. Rosebrough, A.L. Farr, R.J. Randall, Protein measurement with the folin phenol reagent. J. Biochem. 193, 265–275 (1951)
X. Li, A. Zhou, Y. Han, Anti-oxidation and anti-microorganism activities of purification polysaccharide from Lygodium japonicum in vitro. Carbohydr. Polym. 66, 34–42 (2006)
A. Ghiselli, M. Nardini, A. Baldi, C. Scaccini, Antioxidant activity of different phenolic fractions separated from an Italian red wine. J. Agric. Food Chem. 46, 361–367 (1998)
A.Q. Zhang, X.Q. Li, C. Xing, J.H. Yang, P.L. Sun, Antioxidant activity of polysaccharide extracted from Pleurotus eryngii using response surface methodology. Int. J. Biol. Macromol. 65, 28–32 (2014)
M. Oyaizu, Studies on products of browning reaction: antioxidative activities of products of browning reaction prepared from glucose amine. Jpn. J. Nutr. 44, 307–315 (1986)
A.S. Meyer, A. Isaksen, Application of enzymes as food antioxidants. Trends Food Sci. Technol. 6, 300–304 (1995)
F. Liu, V.E. Ooi, S.T. Chang, Free radical scavenging activities of mushroom polysaccharide extracts. Life Sci. 60, 763–771 (1997)
J.L. Wang, J. Zhang, B.T. Zhao, X.F. Wan, Y.Q. Wu, J. Yao, A comparison study on microwave-assisted extraction of Potentilla anserine L. polysaccharides with conventional method: molecule weight and antioxidant activities evaluation. Carbohydr. Polym. 80, 84–93 (2010)
B. Halliwell, S. Chirico, Lipid peroxidation: its mechanism, measurement, and significance. Am. J.Clin. Nutr. 57, 715S–724S (1993)
Z.Y. Cheng, J. Ren, Y.Z. Li, W.B. Chang, Z.D. Chen, Study on the multiple mechanisms underlying the reaction between hydroxyl radical and phenolic compounds by qualitative structure and activity relationship. Bioorg. Med. Chem. 10, 4067–4073 (2002)
O. Erel, A novel automated method to measure total antioxidant response against potent free radical reactions. Clin. Biochem. 37, 112–119 (2004)
R.J. He, J.M. Ye, Y.J. Zhao, W.K. Su, Partial characterization, antioxidant and antitumor activities of polysaccharides from Philomycusbilineatus. Int. J. Biol. Macromol. 65, 573–580 (2014)
G.H. Naik, K.I. Priyadarsini, J.G. Satav, M.M. Banavalikar, D.P. Sohoni, M.K. Biyani, Comparative antioxidant activity of individual herbal components used in ayurvedic medicine. Phytochemistry 1, 97–104 (2003)
R. Amarowicz, R.B. Pegg, P. Rahimi-Moghaddam, B. Barl, J.A. Weil, Free radical scavenging capacity and antioxidant activity of selected plant species from the Canadian prairies. Food Chem. 84, 551–562 (2004)
G. Hikmet, A. Burhan, D. Gokhan, E. Selim, Y. Ismet, Antioxidant, free radical scavenging and metal chelating characteristics of propolis. Am. J. Biochem. Biotechnol. 1, 27–31 (2005)
Z.P. Ye, W. Wang, Q.X. Yuan, H. Ye, Y. Sun, H.C. Zhang, X.X. Zeng, Box-Behnken design for extraction optimization, characterization andin vitro antioxidant activity of Cicer arietinum L. hull polysaccharides. Carbohydr. Polym. 147, 354–364 (2016)
X.Y. Li, L. Wang, Y. Wang, Z.H. Xiong, Effect of drying method on physicochemical properties andantioxidant activities of Hohenbuehelia serotina polysaccharides. Process Biochem. 51, 1100–1108 (2016)
X.J. Du, H.M. Mu, S. Zhou, Y. Zhang, X.L. Zhu, Chemical analysis and antioxidant activity of polysaccharides extracted from Inonotus obliquus sclerotia. Int. J. Biol. Macromol. 62, 691–696 (2013)
Acknowledgements
The authors are grateful for financial sponsored by the national spark program of China (2015GA690021).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
There are no conflicts of interest to declare.
Rights and permissions
About this article

Cite this article
Chen, Y., Gao, L. & Gao, R. Purification, chemical characterization and antioxidant activities of polysaccharides of fruiting bodies of Pleurotus ferulae mushroom. Food Measure 13, 222–230 (2019). https://doi.org/10.1007/s11694-018-9935-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11694-018-9935-9