
Abstract
A simple analytical method was developed and validated in chilli, tomato, grape and mango fruits using liquid chromatography tandem mass spectrometry. The method comprised of extraction with ethyl acetate and cyclohexane mixture followed by d-SPE cleanup employing modified quick, easy, cheap, effective, rugged and safe extraction method and quantified in LC–MS/MS using gradient elution. The method was validated in concentration ranging from 0.01 to 0.1 µg g−1. The recovery of azoxystrobin in different crops was ranging from 84.36 to 95.64 % at three different concentration levels of analytes with relative standard deviation (HorRat < 20 %) of 4–14 %. The global uncertainty was calculated at limit of quantification level i.e. 0.01 µg g−1. In order to evaluate in safety use in India, a field study was conducted with the following extraction method. The calculated half life periods of azoxystrobin were ranging from 3.10–3.46, 3.64–3.46, 1.65–1.96 and 1.32–1.36 days respectively for experimental substrates. The PHI values of azoxystrobin in chilli, tomato, grape and mango fruits were determined as 4.76, 3.90, 4.06 and 10.74 days respectively.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Tomato (Solanum lycopersicum) is an important vegetable fruit in India, it occupy about 8.23 % of our total vegetable production. As chilli (Capsicum annum L.) is the most important and highest producing commercial spice crop named as “wonder spice” in India [1]. Grapes (Vitis vinifera L.) belong to the world’s largest fruit crops with a global production of around 69 million tons in 2006 [2]. Mango (Mangifera indica L.) is the major fruit of India and is known as “King of fruits”. These all crops contain a large medicinal aspect which offers health benefits.
Azoxystrobin [methyl(E)-2-{2-[6-(2-cyanophenoxy)pyrimidin-4-yloxy]Phenyl} 3methoxy-acrylate)] is a broad-spectrum fungicide with protectant, erradicant, translaminar, and systemic properties and is used on a wide range of crops. It belongs to the subclass Strobilurins may extract from the fungus Strobilurus tenacellus. They have a suppressive effect on other fungi, reducing competition for nutrients. They inhibit electron transfer in mitochondria, disrupting metabolism and preventing growth of the target (ambiguous) fungi. It can provide protection against downy mildew and powdery mildew [3]. The established acceptable daily intake (ADI) is 0.2 mg kg−1 body weight per day [4] and maximum residue limits (MRLs) in Indian subtropical climate are 1.0, 1.0, 0.5 and 0.01 mg kg−1 for chilli, tomato, grape and mango respectively [5]. So, persistence and degradation of azoxystrobin in/on these widely used crops is essential to understand its behavior in tropical climatic condition in India and to validate an efficient analytical method.
In general, pesticide residue analysis is carried out in a sequence of steps, viz. extraction of target compounds from sample matrix, cleanup and pre-concentration, followed by chromatographic analysis [6–10]. A large number of research work were carried out with azoxystrobin in different substrate viz. orange (LOD and LOQ was 0.001, 0.004 µg g−1 using QuEChERS in LC–MS/MS) [6], tomato (detected LOD and LOQ was 0.001, 0.0029 µg g−1) [7], white wine and rose wine (experimental LOD and LOQ was 0.00025, 0.00396 µg mL−1 using single-drop microextraction in GC–MS) [8], melon (experimental LOD and LOQ was 0.001, 0.01 µg g−1 using QuEChERS in LC–MS/MS) [9], apple (found LOD and LOQ was 0.001, 0.01 µg g−1 in GC-ECD & NPD using QuEChERS method of extraction) [10], solid-state Raman spectroscopic method was introduced for the quantitative determination of multiresidual treatments of fungicides viz. trifloxystrobin, tebuconazole, and propiconazole (LOD, LOQ was ranging from 0.003–0.014 to 0.01–0.046 µg g−1 respectively for three fungicides) [11]. To understand the fate and degradation behaviour of target fungicide under field condition, Wilkinson et al. [12] gather the informations on azoxystrobin metabolism studied in wheat, grapes, peanuts, rice, and cotton. 2-((6-(2-(3-oxoprop-1-en-2-yl)phenoxy)pyrimidin-4-yl)oxy)benzonitrile (m/z = 343.10) and methyl 2-(2-((6-(2-cyanophenoxy)pyrimidin-4-yl)oxy)phenyl)acrylate (m/z = 373.36) was found as daughter ions for confirmative analysis in mass spectrometer. Boudina et al. [13] studies the photochemical degradation of azoxystrobin under aqueous medium of different pH equipped with high-pressure mercury lamp. Degradation of azoxystrobin involves photo-isomerization, photo-hydrolysis of the methyl ester and of the nitrile group, cleavage of the acrylate double bond, photohydrolytic ether cleavage between the aromatic ring giving phenol and oxidative cleavage of the acrylate double bond. Banerjee et al. [14–16] were explained the chemical and photochemical transformations of other strobilurin fungicides viz. trifloxystrobin.
The object of the present study was to optimize and validate an analytical method based on matrix solid phase dispersion (MSPD; modified QuEChERS approach) followed by dispersive solid phase extraction (d-SPE) clean up. After validation of proposed method, four field trials were conducted to determine the degradation kinetics of azoxystrobin on chilli, tomato, grape and mango matrices by LC–MS/MS. The study also estimated the significant advantages of MSPD over liquid–liquid extraction (LLE) method of analysis and focused on the risk assessment of azoxystrobin on human health under the tropical humid climatic condition of West Bengal.
Materials and method
Reagents and chemicals
Azoxystrobin reference standards (purity > 99.9 % m/m) and azoxystrobin formulation (23 % SC) was supplied by M/S Indofil Industries Ltd., Mumbai. HPLC grade organic solvents as methanol, acetonitrile, ethyl acetate (JT Baker, Phillipsburg, USA), purified water (Millipore, Bedford, MA) were used. Analytical reagent grade formic acid, anhydrous sodium sulfate and sodium chloride, ammonium acetate crystal pure (Merck India Ltd., Mumbai, India), Primary secondary amine (PSA; 40 µm particle size), graphitized carbon black (GCB; United Chemical Technology, Bellefonte, PA), 0.2 µm ultipor N66 nylon 6, 6 membrane filter from Pall Corporation, were used for sample analysis. Activated Na2SO4 was prepared by heated in a muffle furnace at 400–450 °C for 5 h before use and kept in desiccator.
Field trial details
To validate the method and sample analysis for quantification of azoxystrobin, a field experiment was conducted at Experimental Research Farm, B.C.K.V., Mohanpur, Nadia, West Bengal for chilli, tomato, mango and at Malancha Farm, Birbhum, West Bengal for Grape (Variety: Suryamukhi, Patharkuchi, Amrapali and Pusa Navarang respectively). The fungicide, azoxystrobin 23 % SC was applied twice (15 days interval) in the field at the doses of 125 g a.i. h−1 and 250 g a.i. h−1 during Mar.’11 to May’11, Dec.’11 to Apr.’12, Apr.’12 to May’12 and Mar.’12 to May’12 respectively. The average temperature range was 9.70–34.50, 12.38–34.50, 24.20–36.70 and 18.09–38.60 °C with average relative humidity 40.57–94.77, 37.00–95.58, 49.00–90.40 and 29.90–83.65 %. Total rainfall was 9.77, 2.23, 4.20 and 56.40 mm respectively.
Preparation of standard solution
Stock solution of azoxystrobin standard was prepared by accurately weighing 10 ± 0.001 mg of the fungicide in volumetric flasks (certified “A” class) with a digital balance (Sartorius, CP 225D, Germany) and dissolving it in 10 mL methanol. This stock solution was stored in a refrigerator at 4 °C. A working standard of 1.0 mg L−1 was prepared by diluting the intermediate stock standard solution. From freshly prepared working standards, the calibration standards within the range 0.001–1.0 µg mL−1 (ten levels) were prepared by serial dilution [17, 18] with CH3OH:H2O (9:1, v/v) + 0.1 % formic acid (Fig. 1).

Calibration curve of analytical standard (0.001–1 µg g−1; 8 levels) of azoxystrobin (solvent only)
A simple dilution is one in which a unit volume of a liquid material of interest is combined with an appropriate volume of a solvent liquid to achieve the desired concentration. The dilution factor is the total number of unit volumes in which solute material will be dissolved. The dilute material must then be thoroughly mixed to achieve the true dilution.
On the other hand serial dilution is simply a series of simple dilutions which amplifies the dilution factor quickly beginning with a small initial quantity of material. The source of dilution material for each step comes from the diluted of the previous. In a serial dilution the total dilution factor at any point is the product of the individual dilution factor in each step up to it.
For the preparation of calibration curve, standard solutions are prepared by serial dilution method (In calibration curves, correlation coefficient (R2) values are 0.8973 and 0.9997 for simple dilution and serial dilution respectively).
Fortifications and recovery study
Chilli, tomato, grape and mango samples were collected from the control plots of the trial field. The collected samples were used in fortification experiments and as matrix blanks for matrix-matched calibration standards. By using matrix solid phase dispersion (MSPD) method of extraction, the recovery study of azoxystrobin was carried out by using the control samples (all four matrices) spiked at desired concentration level, viz. 0.01, 0.05 and 0.10 µg g−1 (LOQ, fivefold of LOQ and tenfold of LOQ). As for comparison, conventional LLE method was studied, where the fortified concentration levels are 0.02, 0.05 and 0.1 µg g−1. The fortified samples were mixed well and left standing for 2 h to distribute pesticides evenly and given time to interact with the matrix before extraction.
Extraction and cleanup
Extraction procedure by conventional LLE method
Fifty grams of tomato, chilli, grape and mango samples was blended separately using Polytron homogenizer (PT-MR-3100, Kinemetica AG, Switzerland) → The homogenized sample (5 g) was taken in a 500 mL separatory funnel + 100 mL of 5 % of aqueous NaCl solution was added \(\mathop{\longrightarrow}\limits^{{{\text{Shake\,for\,15}}\,{\text{min}}.}}\) Subsequently extracted with 200 mL (100 + 50 + 50) of ethyl acetate → The organic layer was passed through activated Na2SO4 + concentrated using rotary vacuum evaporator (EYELA) at 40 °C → The extract was evaporated and reconstituted in 6 mL ethyl acetate.
Extraction procedure by MSPD method
Fifty grams tomato, chilli, grape and mango samples \({\mathop{\longrightarrow}\limits^{\text{homogenized}} }\) The homogenized sample (5 g) was taken in a 50 mL centrifuge tube 10 mL Ethyl Acetate + cyclohexane (9 + 1; v/v) mixture was added \(\mathop{\longrightarrow}\limits^{{{\text{Vortex\,for\,2}}\,{\text{min}}.}}\) Adding 5 g of activated Na2SO4 \(\mathop{\longrightarrow}\limits^{{ {\text{Vortex\,for\,1}}\,{\text{min}}.}}\) Aliquot was centrifuged by refrigerated centrifuge; Model Avanti J-30I (Beckman coulter, USA) with rotor heads JA-30.50 for 5 min at 5000 rpm → 6 mL supernatant extract was taken in 10 mL centrifuge tube.
Dispersive solid phase extraction for clean up
6 mL supernatant extract + 75 mg PSA + 75 mg GCB + 450 mg of activated Na2SO4 was added to it \(\mathop{\longrightarrow}\limits^{{{\text{Vortex\,for\,2}}\,{\text{min}}.}}_{{{\text{centrifuged\,for\,5}}\,{\text{min}}@{ 5}000\,{\text{rpm}}}}\) 2 mL supernatant liquid was collected → evaporated to dryness under a gentle stream of N2 in Turbo Vap LV instrument from Caliper Life Science (Hopkinton, MA, USA).
All the residue was then reconstituted with [MeOH:H2O (9:1, v/v) +0.1 % Formic acid]. The sample was then filtered through 0.2 µ membrane filter. Now the sample is ready for the final analysis in LC–MS/MS.
LC–MS/MS analysis
The HPLC separation was performed on a Agilent 1200 Series separation module liquid chromatography (Agilent Technologies) equipped with a quaternary solvent delivery system by 10 µL via autosampler on a reversed phase Zorbax Eclipse XDB-C18 (5 µm; 4.6 × 150 mm) column (Agilent Technologies, USA) and a Agilent 6410 triple-quadruple spectrometer equipped with an electrospray source (ESI) was used for detection and quantification. Injection volume was 10 µL, column temperature was 25 ± 2.0 °C and the analysis performed with a flow rate of 0.5 mL/min. The mobile phase was composed of (A) water, 5 mM ammonium acetate and 0.1 % acetic acid and (B) methanol, 5 mM ammonium acetate and 0.1 % acetic acid. Gradient: 0.0–2.0 min—5.0 % B to 95 % B, 2.0–10.0 min—back to the initial condition of 5 % B, at 15.0 min, it ends with 5 % B.
Estimation of the fungicide was performed in positive mode by a single multireaction monitoring (MRM) with mass transition from parent ion 403.90 to daughter ion 371.90. A second mass transition was used 403.90 > 343.90 for confirmation. The collision energy was 5 V, gas flow was 8 mL min−1 with temperature 300 °C and the fragmenter, nebulizer was 60 V and 20 psi respectively. The ratio of the peak area of these two daughter ions for azoxystrobin was 28.1. The corresponding ratio in the positive samples was determined and confirmed in accordance with European Union guidelines [21].
Preparation of matrix-matched calibration standards
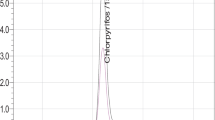
For calibration in LC–MS/MS, ten concentration levels (0.001, 0.002, 0.005, 0.01, 0.02, 0.05, 0.10, 0.25, 0.50 and 1.00 µg L−1) were prepared. The standard calibration curve is reported in Fig. 1. Chromatograms of qualifier, quantifier ion ratio as well as mass transition of azoxystrobin analytical standard in limit of detection (LOD) level (0.001 µg g−1) are reported in Fig. 2. To evaluate the matrix effect, matrix-matched calibration curve was prepared by using the extracts of blank matrix.

Chromatograms of azoxystrobin standard (conc. 0.001 µg g−1) with qualifier, quantifier ion transition and ion ratio
Method validation
The analytical method was validated as per the single laboratory validation (SLV) approach [20, 21]. A brief comparison was done between conventional LLE and MSPD method of extraction. The performance of the method was evaluated considering different validation parameters that include the precision, accuracy-recovery experiment, matrix effect (%ME) and uncertainty measurement.
The calibration curve for azoxystrobin in pure solvent and matrix were obtained by plotting the peak area against the concentration of the corresponding calibration standards at ten calibration levels ranging between 0.001 and 1.0 µg g−1 (Fig. 1).
In the single-laboratory validation (SLV) studies precision were determined from the reproducibility (R) and repeatability (r). The Horwitz ratio (HorRat) pertaining to within-laboratory precision, which indicates the acceptability of a method with respect to precision [19], was calculated for all of the pesticides by Eq. ( 1 ). The percentage of matrix effect (ME %) was assessed by employing matrix matched standards. Global uncertainty was determined for azoxystrobin at the level of 0.01 µg g−1 (for MSPD) and 0.02 µg g−1 (for LLE) as per the statistical procedure of the EURACHEM/CITAC Guide CG 4 [22]. Seven individual sources of uncertainty were taken into account. The combined uncertainty (UC) was calculated as in Eq. (2 ). The uncertainty values, %ME and HorRat values for azoxystrobin are reported as relative uncertainties in Table 1.
The limit of detection (LOD = 0.001 µg g−1) was determined by considering a signal-to-noise ratio of 3 with reference to the background noise obtained from blank sample, whereas the limits of quantification (LOQ = 0.02 µg g−1 for LLE and 0.01 µg g−1 for MSPD) were determined by considering a signal-to-noise ratio of 10 irrespective of the chilli, tomato, grape and mango matrices by using matrix-matched standards.
Results and discussion
Two extraction techniques were MSPD using QuEChERS approach and conventional LLE method of residual fungicide from widely used crops were compared and analyzed by liquid chromatography coupled with triply quadrupole mass spectrometry for conformation.
Conventional liquid–liquid extraction method
With pure ethyl acetate, the recovery was (considering three levels as 0.02, 0.05, 0.1 µg g−1) 52–81, 85–96, 90–93 and 86–92 % for chilli, tomato, grape and mango respectively in neutral (at pH ≈ 7.0) medium with the HorRat values of 35–59 % (>20 %) and percentage of relative standard deviation (%RSD) were in the range of 9–35 % as considered 20 ng g−1 level for all matrices with low matrix interferences (Table 1).
Matrix solid phase dispersion (QuEChERS approach) method
For ethyl acetate + cyclohexane (9:1, v/v) solvent mixture in neutral medium (at pH ≈ 7.0) the recoveries were in between (considering three levels as 0.01, 0.05, 0.1 µg g−1) 89–96, 87–95, 84–88, 88–95 % respectively for all matrices with good precision when quantified with matrix-matched standards. Precision in terms of HorRat (SLV) at 10 ng g−1 level was 13–19 % (≤ 20 %) for azoxystrobin (Table 1) and percentage of RSD in the range of 5–13 %, indicating satisfactory repeatability and ruggedness of the methodology. This method was linear over the range assayed, 0.001–1.0 (ten levels) µg g−1 in matrix match calibration curve.
From the entire study it clearly revealed that at neutral medium the mixture of ethyl acetate and cyclohexane gave higher recovery percentage (84–96 % for all matrices), low matrix effect (suppression or enhancement), good sensitivity (LOD = 0.001 µg g−1, LOQ = 0.01 µg g−1) with satisfactory uncertainty parameters used in matrix solid phase extraction (MSPD) with d-SPE clean up for regular analysis of azoxystrobin residue in chilli, tomato, grape and mango matrices. Although, pure ethyl acetate produce good (52–96 % for all substrates) recoveries using LLE with d-SPE clean up but LLE method requires long time, large quantity of solvent, less sensitive (LOQ = 0.02 µg g−1) which is not sufficient and useful for regular analysis. From the entire experiment it can be conclude that the very much appropriate solvent was ethyl acetate + cyclohexane mixture for extraction and PSA, GCB and Na2SO4 for d-SPE clean up.
Analysis of real sample
After validation of the proposed method, we perform a real field trial of azoxystrobin on chilli, tomato, grape and mango as mentioned in field trial details. The T1/2 of azoxystrobin was calculated using Hoskins formula [23]. The half-life period (T1/2) was presented in Table 2 and Fig. 3. Quantity of azoxystrobin in all the experimental samples was below detection limit (BDL) at 5–7 days from the date of application. In the present study no residues of azoxystrobin were detected at the time of harvest in chilli, tomato, grapes, mango and respective field soil samples irrespective of any doses, which refers that azoxystrobin does not possess any background contamination property in chilli, tomato, grapes and mango when applied at the recommended dose. There was no residue of azoxystrobin was found in the untreated field sample.

Degradation kinetic study of azoxystrobin in field samples
Risk assessment study of azoxystrobin
Risk assessment is crucial to the process of making decisions about pesticides, both new and existing. New pesticides must be evaluated before they can enter the market. Existing pesticides must be re-evaluated periodically to ensure that they continue to meet the appropriate safety standard. Generally a pesticide remains in soil, water and plant for some time after its application and before it breaks down. So, proper risk assessment study is required accordingly so that a minimum amount of pesticide is left in environment as well as the food chain. Residues of pesticides may remain in treated products and get into human food chain. These residues should not exceed a limit above which they may pose risks to human health. The concepts of persistence, maximum residue limit (MRLs), acceptable daily intake (ADI) for pesticides has been devised to keep a check on the pesticide residues in food chain and keep them within safe limits.
The persistence of any chemical is generally expressed in terms of half-life (T1/2) or DT50 i.e. time for disappearance of pesticide to 50 per cent of its initial concentration. T1/2 values are often obtained by fitting first-order kinetics to observed degradation patterns as: C t = C 0 e −kt, where Ct is chemical concentration (µg g−1) at time t (h), C0 is initial concentration (µg g−1), and k is the first order rate constant (h−1) independent of Ct and C0. The calculated T1/2 of azoxystrobin was (using Hoskins’ formula) [23] varied between 3.10–3.46, 2.64–3.46, 1.32–1.37 and 1.65–1.97 days in chilli, tomato, grape and mango respectively. In the present study no residues of azoxystrobin were detected in harvest chilli, tomato, grapes and mango samples irrespective of any doses, which refers that azoxystrobin does not possess any background contamination property in those matrices when applied at the recommended dose. The initial deposits (2 h after spraying) of azoxystrobin in those trials of each matrices were 0.84 & 1.30, 0.58 & 1.01, 0.58 & 0.96 and 0.68 & 1.24 µg g−1 respectively for T1 & T2 doses which was found to be less than its Indian and European Union maximum residue limit (1.0, 1.0, 0.5 and 0.01 µg g−1 for four matrices) [24]. The residues dissipated gradually to 0.17 & 0.31 (7 days), 0.09 & 0.25 (7 days), 0.069 (5 days) & 0.08 (7 days) and 0.048 (5 days) & 0.03 (7 days) µg g−1 and 0.06–0.14 µg g−1 (for four matrices respectively) and were below the quantifiable limit of 0.01 µg g−1 at 10 days after the application. Based on these observations, it may be concluded that residual azoxystrobin concentrations at the time of harvest will be below those associated with toxicological risk when applied at the recommended dose. Acceptable daily intake (ADI) for azoxystrobin has been observed to be 0.20 µg g−1 body weight per day. Accordingly, an adult of 60 kg can tolerate an intake of 12,000 µg day−1 without any appreciable exposure risks. A consumption of 200 g of test substrates (viz. chilli, tomato, grape and mango) by an adult with initial deposit as observed at double dose (1.30, 1.01, 0.96 and 1.24 µg g−1 for chilli, tomato, grape and mango respectively) will lead to intake of only 260, 202, 192 and 248 µg day−1 which is toxicologically acceptable and quite safe from health point of view of the consumer.
Conclusion
The proposed simple MSPD method of extraction with ethyl acetate + cyclohexane (9 + 1; v/v) mixture, cleanup by d-SPE and quantified by LC–MS/MS using MRM provided high cleanup efficiency and low matrix effects thus enabling adaptation of this sensitive and selective method for routine residue analysis of azoxystrobin on Chilli, tomato, grape and mango matrices with satisfactory recovery (84.36–95.64 %). The method offers low cost of analysis, short time as well as satisfactory uncertainty parameters. From the field data of residue analysis we can calculate the pre-harvest interval (PHI) and half life period of the target molecule, as from the established MRLs and ADI (0.2 µg g−1) data of azoxystrobin (Table 1) we can demand that azoxystrobin is toxicologically acceptable and quite safe from health point of view of the consumer. The proposed method is sensitive enough (i.e. LOD = 0.001 µg g−1 and LOQ = 0.01 µg g−1 for all matrices) for the analysis of azoxystrobin.
References
R.R. Karpate, R. Saxena, Post harvest profile of chilli. (Dept. of Ag. and Cooperation, Ministry of Agriculture, Nagpur, 2009), MRIN-2
FAO Statistical Database, FAOSTAT, (2007), http://www.fao.org. Accessed 29 June 2013
P.C. Adsule, Good Agricultural Practices for Production of Quality Table Grapes. (N.R.C for Grapes, Pune, 2013), pp. 38–57. www.drs.nio.org/drs/bitstream/2264/4073/1/Mar_Geol_307-310_88a.pdf. Accessed 18 Feb 2013
FAO database, Azoxystrobin: 229, (FAO/WHO Plant Production and protection 215, Ministry of Agriculture, Forestry and Fisheries, Tokyo, Japan, 2012), pp. 55–95. http://www.fao.org/fileadmin/templates/agphome/documents/Pests_Pesticides/JMPR/Report12/JMPR_2012_Report.pdf. Accessed 18 June 2014
The gazette of India: Extraordinary (2009) Part II-Sec. 3(i): 8. (Dept. of Health and Public welfare, New Delhi), www.drugscontrol.org/GSR%20157%20(E)%20dtd%204.3.09.pdf. Accessed 30 July 2013
O. Golge, B. Kabak, J. Food Compos. Anal. 41, 86–97 (2015). doi:10.1016/j.jfca.2015.02.007
O. Golge, B. Kabak, Food Chem. 176, 319–332 (2015). doi:10.1016/j.jfca.2015.02.007
J.P. dos Anjos, J.B. de Andrade, Microchem. J. 120, 69–76 (2015). doi:10.1016/j.microc.2015.01.009
M.D.H. Prodhan, E.-N. Papadakis, E. Papadopoulou-Mourkidou, Int. J Environ. Anal. Chem. 1–11 (2015). doi:10.1080/03067319.2015.1025227
E. Szpyrka, A. Kurdziel, A. Matyaszek, M. Podbielska, J. Rupar, M. Słowik-Borowiec, Food Control 48, 137–142 (2015). doi:10.1016/j.foodcont.2014.05.039
B. Ivanova, M. Spiteller, Analyst 137(14), 3355–3364 (2012). doi:10.1039/C2AN35174A
M. Wilkinson, (1994) ICIA5504: Metabolism in winter wheat, Jealott’s Hill Research Station, Zeneca Agrochemicals, UK. FAO Report No. RJ1682B. Syngenta File No. ICI5504/0286
A. Boudina, C. Emmelin, A. Baaliouamer, O. Païssé, J.M. Chovelon, Chemosphere 68(7), 1280–1288 (2007). doi:10.1016/j.chemosphere.2007.01.051
K. Banerjee, A.P. Ligon, M. Spiteller, Anal. Bioanal. Chem. 382(7), 1527–1533 (2005). doi:10.1007/s00216-005-3336-8
K. Banerjee, A.P. Ligon, M. Spiteller, Agric. Food Chem. 54(25), 9479–9487 (2006). doi:10.1021/jf0620214
K. Banerjee, A.P. Ligon, M. Spiteller, Anal. Bioanal. Chem. 388(8), 1831–1838 (2007). doi:10.1007/s00216-007-1382-0
G. Anderson, M.E. Lewiston, Preparing solutions and making dilutions. (Mississippi Genome Exploration Laboratory, Mississippi State, MS) http://www.mgel.msstate.edu/pdf/solutions.pdf. Accessed 10 July 2014
A. Ben-David, C.E. Davidson, J. Microbiol. Methods 107, 214–221 (2014). doi:10.1016/j.mimet.2014.08.023
W. Horwitz, L.R. Kamps, K.W. Boyer, J. Assoc. Off. Anal. Chem. 63(6), 1344–1354 (1980)
Method validation and quality control procedures for pesticide residues analysis in food and feed (2009) Document No. SANCO/10684/2009. 6th EU AQC
European Union Commission Directorate of General Health and Consumer Protection (2000) Guidance document on residue analytical methods. Document No. SANCO/825/00 rev. 6
Quantifying uncertainty in analytical measurement, (2000) Document no. EURACHEM/CITAC Guide CG 4, 2nd ed., http://www.measurementuncertainty.org.html. Accessed 15 July 2013
W.M. Hoskins, FAO Plant Prot. Bull. 9, 163–168 (1961)
EFSA, Reasoned opinion on the review of the existing maximum residue levels (MRLs) for azoxystrobin according to Article 12 of Regulation (EC) No 396/20051, European Food Safety Authority (EFSA), Parma, Italy, EFSA Journal, 11(12): 3497–3594 (2013). http://www.efsa.europa.eu/en/efsajournal/doc/3497.pdf. Accessed 25 Sep 2014
Acknowledgments
Authors are thankful to Indofil Industries Limited, Mumbai for financial assistance and grateful to Department of Agril. Chemicals, Bidhan Chandra KrishiViswavidyalaya, Mohanpur, WB, India, for all instrumental facilities. They are also thankful to Department of Chemistry, Visva-Bharati, Santiniketan, WB, India for the general facilities to continue the research.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Mukherjee, S., Mukherjee, S., Das, G.K. et al. Analytical method validation and comparison of two extraction techniques for screening of azoxystrobin from widely used crops using LC–MS/MS. Food Measure 9, 517–524 (2015). https://doi.org/10.1007/s11694-015-9260-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11694-015-9260-5