
Abstract
The drivers of population differentiation in oceanic high dispersal organisms, have been crucial for research in evolutionary biology. Adaptation to different environments is commonly invoked as a driver of differentiation in the oceans, in alternative to geographic isolation. In this study, we investigate the population structure and phylogeography of the bottlenose dolphin (Tursiops truncatus) in the Mediterranean Sea, using microsatellite loci and the entire mtDNA control region. By further comparing the Mediterranean populations with the well described Atlantic populations, we addressed the following hypotheses: (1) bottlenose dolphins show population structure within the environmentally complex Eastern Mediterranean Sea; (2) population structure was gained locally or otherwise results from chance distribution of pre-existing genetic structure; (3) strong demographic variations within the Mediterranean basin have affected genetic variation sufficiently to bias detected patterns of population structure. Our results suggest that bottlenose dolphin exhibits population structures that correspond well to the main Mediterranean oceanographic basins. Furthermore, we found evidence for fine scale population division within the Adriatic and the Levantine seas. We further describe for the first time, a distinction between populations inhabiting pelagic and coastal regions within the Mediterranean. Phylogeographic analysis suggests that current genetic structure, results mostly from stochastic distribution of Atlantic genetic variation, during a recent post-glacial expansion. Comparison with Atlantic mtDNA haplotypes, further suggest the existence of a metapopulation across North Atlantic/Mediterranean, with pelagic regions acting as source for coastal environments.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Despite an apparent lack of physical barriers to dispersal, many marine organisms exhibit population structure over scales smaller than their dispersal potential (Norris 2000; Bierne et al. 2003). The Mediterranean Sea in particular is a global biodiversity hotspot (Almada et al. 2001; Coll et al. 2010), and several marine species exhibit complex population structure patterns over relatively short geographic distances (e.g. Ascids: Perez-Portela and Turon 2008; Echinoderms: Zulliger et al. 2009; Molluscs: Perez-Losada et al. 2007, Calvo et al. 2009; Marine turtles: Carreras et al. 2006; Cetaceans: Natoli et al. 2005; Gaspari et al. 2007a, b; Fish: Carreras-Carbonell et al. 2006; Charrier et al. 2006; Domingues et al. 2007). The Mediterranean is thus a particularly interesting region to investigate the drivers of population structure in marine organisms.
Population structure in the Mediterranean Sea is often suggested to result from differential adaptation to the environmental complexity of the basin (Borsa et al. 1997; Naciri et al. 1999; Bahri-Sfar et al. 2000, Domingues et al. 2005; Galarza et al. 2009; Zulliger et al. 2009). However, alternative mechanisms such as isolation-by-distance (Zulliger et al. 2009; Casado-Amezúa et al. 2012), or strong regional demographic variations (Rolland et al. 2006) have also been proposed. This complexity is further emphasized by the lack of consistent patterns of differentiation across the Strait of Gibraltar for various marine taxa (Patarnello et al. 2007).
The bottlenose dolphin (Tursiops truncatus) represents a good model to test different hypotheses regarding the drivers of genetic differentiation. In European waters (Mediterranean, Eastern North Atlantic, and North Sea), its population structure appears to correlate strongly with environmental differences (Natoli et al. 2005), consistent with suggestions that differences in habitat requirements drive population structure in cetaceans (Mendez et al. 2011; Amaral et al. 2012). In the Atlantic, populations typically segregate between lineages inhabiting pelagic and coastal environments (Hoelzel et al. 1998, Natoli et al. 2004), and mitogenomic analysis showed that in European waters, these two ecotypes show incomplete lineage sorting (Moura et al. 2013b), suggesting recent establishment of the observed population structure patterns. Several communities inhabiting estuarine/bay environments, are genetically differentiated from individuals sampled in open waters, both coastal and pelagic (Parsons et al. 2002; Nichols et al. 2007; Fernández et al. 2011; Mirimin et al. 2011; Louis et al. 2014a), which is thought to result from occupation of newly formed habitats after the Last Glacial Maxima (LGM) (Louis et al. 2014b).
It has also been suggested that the Mediterranean populations of bottlenose dolphin have recently occupied the area from Atlantic populations (Natoli et al. 2005). However, preliminary data shows evidence of fine scale population structure within the Mediterranean basin (Gaspari et al. 2013), consistent with morphological variation described between basins (Sharir et al. 2011), and the existence of groups with different levels of site fidelity (Bearzi et al. 2005, 2009). Previous analyses have suffered from low power, both in terms of the markers used, and the number and geographic representation of samples. To date, no study has compared fine scale genetic structure within the Mediterranean, with the well described genetic structure in the North Atlantic (Natoli et al. 2004; Tezanos-Pinto et al. 2008; Louis et al. 2014a). This information can thus allow to distinguish between genetic structure that results from random distribution of ancestral variation, from that established within the Mediterranean due to local adaptation.
Furthermore, several studies suggest that strong demographic history has confounded population structure patterns in other North Atlantic cetaceans, including common dolphin (Delphinus delphis) (Natoli et al. 2008; Moura et al. 2013a), white-beaked dolphin (Lagenorhynchus albirostris) (Banguera-Hinestroza et al. 2010), white-sided dolphin (Lagenorhynchus acutus) (Banguera-Hinestroza et al. 2014), bowhead whale (Balaena mysticetus) (Foote et al. 2013), and the killer whale (Orcinus orca) (Hoelzel et al. 2002; Moura et al. 2015). Given recent suggestions of population size changes in bottlenose dolphin for some Mediterranean regions (Bearzi and Fortuna 2006; Pleslić et al. 2013), further detailed analysis of the patterns and processes of population structure is required to advise appropriate conservation measures, and enable an accurate appreciation of the potential for recolonization of local populations by vagrant individuals.
In this study we investigate population structure and phylogeography of the bottlenose dolphin within the Mediterranean Sea, using both nuclear and mitochondrial markers. We employed a comprehensive sample set that covered most of the main Mediterranean basins, namely the Tyrrhenian, Adriatic, Ionian, Aegean and Levantine seas (largely unexplored). These basins all represent unique oceanographic features, characterized by differences in bathymetry, temperature, salinity, and productivity, among others. We evaluated the following key hypotheses: (1) bottlenose dolphins show fine scale population structure within the environmentally complex Eastern Mediterranean Sea; (2) Population structure was gained locally or otherwise results from chance distribution of pre-existing genetic structure; (3) Strong demographic variations within the Mediterranean Sea have affected genetic variation sufficiently to bias detected patterns of population structure. Because the bottlenose dolphin is a top predator in the Mediterranean Sea (Bearzi et al. 2009), phylogeographic patterns of this species will reflect wider changes in the environment, and thus be crucial in understanding the biogeographic history of the basin.
Materials and Methods
Sample Collection and DNA Extraction and Amplification
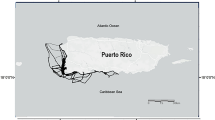
Tissue samples from 194 adult common bottlenose dolphins were collected between 1992 and 2011 from the five main eastern Mediterranean basins (Fig. 1), through biopsies of free-ranging animals (fr) and stranded specimens (str). Samples numbers are as follows: Adriatic Sea (Adriatic north: 7 fr and 50 str; Adriatic central-south: 21 fr and 9 str), Ionian Sea (14 str), Aegean Sea (10 str), Tyrrhenian Sea (16 str), and Levantine basin (68 str). One important consideration, is that the Ionian Sea is considerably deeper than all other basins and is, in this respect, similar to pelagic regions. DNA was extracted with phenol/chloroform and ethanol precipitation from tissue samples preserved in salt saturated 20 % DMSO or 95 % ethanol.

Map of the study area. Samples were obtained from stranded and free-ranging Tursiops truncatus from the main Mediterranean basins (dashed circles), namely: Tyrrhenian, Adriatic, Ionian, Aegean, and Levantine. Shaded areas represents the two main topographical discontinuities of the Adriatic Sea floor

PCA plot reflecting the relation between geographic origin of sample and cluster assignment from structure K = 9. Plot made using Obstruct (Gayevskiy et al. 2014)
Samples were genotyped at 12 microsatellite loci, namely EV37Mn, EV14Pm (Valsecchi and Amos 1996), TtruGT6 (Caldwell et al. 2002), D08 (Shinohara et al. 1997) and Ttr04, Ttr11, Ttr19, Ttr34, Ttr58, Ttr63, TtrRH1 and TtrRC12 (Rosel et al. 2005). Genotypes were determined using an ABI 3100 genetic analyser with Genotyper (Applied Biosystems). A binning procedure was performed to ensure that all alleles were identified correctly across populations. Genotyping accuracy was assessed by randomly re-amplifying 30 % of the samples as controls. Gender was determined through differential amplification of the zinc finger gene regions present in the X and Y chromosomes (ZFX and ZFY, respectively), as described by Bérubé and Palsbøll (1996).
The mitochondrial DNA (mtDNA) control region (920 bp) was amplified and sequenced using the primers TURCRL5483 (5′-GGTCTTGTAAACCGGAAAAGG-3′) and TURCRH6379 (5′-GCAGACTTACACATGCAAGCA-3′) designed in this study, as described in Gaspari et al. (2013).
Raw sequence chromatographs from both strands were edited and aligned using CodonCode Aligner (CodonCode Corporation).
Summary Statistics
Duplicate samples were identified using the Excel Microsatellite Toolkit (Park 2008), and by calculating probabilities of identity P(ID) and P(ID)sib for each basin (Fig. 1) using GenAlEx (Peakall and Smouse 2006). In the presence of population substructure or in small populations where related individuals may remain in proximity and be sampled, P(ID)sib provides a more conservative estimator of the probability of finding the same multi locus genotype at random within the population.
Genetic diversity in each basin (Fig. 1) was assessed by calculating number of alleles, mean number of private alleles, observed and expected heterozygosity, autocorrelation coefficient (r) and inbreeding coefficient (F IS) using GenAlEx (Peakall and Smouse 2006). Allelic Richness was calculated in FSTAT (Goudet 2001) based on the minimum sample size. Departure from Hardy–Weinberg equilibrium (HWE) was tested for each microsatellite locus in each population using the Fisher exact test with 1000 permutations, as implemented by Arlequin (Excoffier et al. 2005).
For mtDNA, unique haplotypes were identified with Arlequin. Genetic diversity in each basin was assessed by calculating number of polymorphic sites, number of haplotypes, pairwise identity (π) and haplotype diversity (H).
Analysis of Genetic Differentiation
The presence of fine scale population structure among the main Mediterranean basins (Fig. 1) was investigated through the analysis of molecular variance (AMOVA), carried out using Arlequin for both mtDNA and microsatellites using F ST as an estimator.
For mtDNA, genetic structure was further analysed by constructing a minimum-spanning network using Network (Bandelt et al. 1999). A second network was constructed including sequences from previous studies, in order to assess the relationships between Mediterranean and North Atlantic mtDNA haplotypes. GenBank was queried using the expressions Tursiops + ”control region” and Tursiops + D-Loop, while retaining only those entries that were over 500 bp and corresponded to animals sampled in the North Atlantic (accession numbers in Table S1; Western North Atlantic Coastal ecotype was excluded).
For microsatellites, we used two methods to determine the most likely number of distinct genetic clusters, without a priori assignment of individuals to populations. We used the Bayesian clustering methods implemented in Structure (Pritchard et al. 2000), using the admixture model with correlated allele frequencies, without specifying sampling locations or geographic origin of samples. The model was run for cluster number (K) from 1 to 15, using a burn-in period of 150,000 Markov Chain Monte Carlo (MCMC) iterations followed by 1,000,000 iterations. Five independent runs were conducted for each value of K to check for convergence of results, analysed using Structure-Harvester (Earl and vonHoldt 2012).
Correlation between genetic structures and basin of origin, was assessed using the software Obstruct (Gayevskiy et al. 2014). This method analyses how much a given pattern of inferred population structure is explained by a predetermined population assignment, by calculating the statistic R2, based on the sum of squares. The levels of K with the highest support from the Structure runs were used as input, and the five different oceanographic basins were used as the predetermined population assignment.
In addition, we used the spatially explicit method implemented in TESS to investigate fine scale population structure within the Adriatic and the Levantine basins, by running the conditional autoregressive admixture model, using burn-in of 20,000 steps followed by 120,000 MCMC steps (Tyrrhenian samples were not used to avoid biases resulting from the rectangular shape of the Italian Peninsula; this means that the linear distance between Tyrrhenian and Adriatic is much shorter than the oceanic route separating these two basins, which involves travelling the entire coastline distance). The number of clusters (K) to test was set from 1 to 10, with 10 replicates run for each K. The spatial interaction parameter was set to 0.6 and the degree of trend to linear (which are the default parameters). The most likely number of clusters was selected by plotting deviance information criterion (DIC) values against K, and by examining plots of individual assignment probabilities. When K was defined, the run with the lowest DIC was used, and individuals were assigned to clusters based on maximum assignment probabilities.
Analysis of Gene Flow Within the Mediterranean
Recent and asymmetric migration rates among the five main basins were estimated using the Bayesian method implemented in BayesAss (Wilson and Rannala 2003). Preliminary runs were performed to adjust the MCMC mixing parameters of migrations rates, allele frequencies and inbreeding coefficients, to ensure proposed acceptance rates around 30 %. We then performed 10 runs with a burn in of 1 × 106 iterations followed by 2 × 107 MCMC iterations and a sampling frequency of 1000. Consistency of the results between the runs was also checked. In addition, sex-biased dispersal was analysed in GenAlEx, by calculating gender-specific Assignment Index correction (AIc) and testing difference for statistical significance using a Mann–Whitney U test (Mossman and Waser 1999).
Historical Demography
For microsatellite data, Fu’s Fs and Tajima’s D neutrality tests were carried out in GenAlEx, as well as tests for recent reduction in population size using the software Bottleneck (Cornuet and Luikart 1996). Tests were carried out using both the stepwise mutation model (SMM) and the infinite allele model, as well as a combined model assuming 70 % SMM and variance set to 30. This was complemented by testing for a shift in the mode of allele frequency distribution, which is more adequate for identifying recent bottlenecks.
For mtDNA, a mismatch distribution was constructed (Rogers and Harpending 1992) using the software Arlequin. Time of expansion was calculated using the formula T = τ/2U, where U represents the mutation rate over the total length of the sequence used in the mismatch distribution (calculated by multiplying the calibrated mutation rate μ by 918 bp). The mutation rate μ was calculated based on the biogeographical method used in Moura et al. (2013b), using the software IMa (Hey and Nielsen 2007). We compared the estimated time of expansion obtained from two sources of mutation rate variation: different values for the closing of the Bosporus Strait (the biogeographical calibration used in Moura et al. (2013b); and inference derived from calculating mutation rate using the whole mtDNA as in Moura et al. (2013b), and using the control region only. This was done to provide an idea of the error introduced in our interpretations, resulting from using an inappropriate mutation rate, a well described source of phylogeographic bias (Ho et al. 2008).
Historical variation in effective population size was reconstructed using the Bayesian skyline method implemented in the software BEAST (Drummond et al. 2012), using the mutation rate estimated from cetacean whole mtDNA by independent studies (Ho and Lanfear 2010; Moura et al. 2013b), and an alternative mutation rate devised for control region only using the same method as in Moura et al. (2013b).
Results
Measures of Genetic Diversity
The test for duplicates yielded two pairs of samples with matching microsatellite genotypes, but the mtDNA haplotype sequences were different for both pairs and were therefore kept. Probabilities of identities across microsatellites loci were low in all populations, which suggests enough power to differentiate between individuals. For microsatellites, diversity levels were similar between the different basins analysed. Observed heterozygosity was usually lower than expected, but differences were not significant and samples from all basins did not significantly deviate from HWE (Table 1). For mitochondrial DNA, pairwise identity (π) was similar between all basins (0.010 ± 0.003), except for Levantine where it was lowest (0.003). Conversely, haplotype diversity (H) was lowest in the Tyrrhenian Sea (0.714), but high overall (Table 1).
Microsatellite Genetic Structure and Gene Flow Estimates
A significant level of genetic differentiation was detected among all basins in the Eastern Mediterranean Sea. AMOVA revealed significant divergence among populations (FST = 0.071; P > 0.001), although most genetic variation occurred within rather than among populations (VWI = 75 %, VAI = 18 %, WAP = 6 %). No evidence of spatial autocorrelation (r) was found among samples from all basins (Table 1). Pairwise F ST comparisons were also applied to the Adriatic basin, both as a uniform basin and as a subdivided basin, to check for potential sub-structure within the Adriatic, given its environmental heterogeneity. Between basins all were significant, except for comparisons involving the Aegean Sea (Table S2). Within the Adriatic Sea, the significant F ST differences were found between samples from the Gulf of Trieste (GT) and the rest of the Adriatic, and between the East and West coasts, both keeping or excluding the GT area (respectively: F ST = 0.016, P = 0.000; F ST = 0.024, P = 0.000). Furthermore, the comparison between the two coasts was also significant when only the North Adriatic was considered, both keeping and excluding the GT area (respectively: F ST = 0.026, P = 0.002; F ST = 0.034, P = 0.000).
Bayesian clustering analyses collectively suggest genetic differentiation between all main basins, although most clusters include individuals from multiple regions (Figs. S1, S2). From the Structure analysis, the highest posterior probability was obtained for K = 15 (Fig. S3), although at K = 15 no further resolution was detectable relative to K = 9 (Fig. S1). ObStruct analyses for K = 9 resulted in an average R2 = 0.31, with pairwise values ranging from 0.03 (between Adriatic and Aegean) to 0.31 (between Adriatic and Levantine). PCA plot revealed some degree of overlap between the different basins cluster assignment, but different basins generally correspond well to genetic clusters. Exceptions are the Aegean and Ionian Seas, which exhibit a high degree of overlap with samples from most basins. Individual ancestry plots for K = 9 clearly separate the main oceanographic basins analysed (Fig. S1), with further subdivision found within the Adriatic and the Levantine regions (Fig. S1). Samples from the GT separate clearly from other Adriatic Sea samples, but cluster together with samples from the Aegean Sea (Fig. S1). Within the Adriatic Sea, different patterns can be seen between North/Central/South regions of the basin (Fig. S1), but the pattern is not clear. In the Levantine basin, three well separated clusters can be identified, which roughly segregate between North/Central/South of the basin (Fig. S1). However, spatial resolution is low due to all samples resulting from strandings, which limits inference (Bilgmann et al. 2011).
TESS results were consistent with Structure (Fig. S2) in separating the GT from the rest of the Adriatic, however this cluster is shared with individuals from North Adriatic, South Adriatic and Aegean Seas. No further structure was identified within the Levantine. The Ionian Sea shared clusters with all other basins. Both Structure and TESS individual ancestry plots organized by sample origin are included in the supplementary material (Figs. S1, S2).
BayesAss showed generally low recent migration rates between basins. Exceptions are migration rates from the Ionian into Tyrrhenian/Adriatic/Aegean, which are all above 0.10, and from Adriatic into Ionian/Aegean, also all above 0.10 (Table 2). Sex biased dispersal suggests females are the dispersing gender, with negative AIc for females and positive AIc for males (Fig. S4). However, Mann–Whitney U test was not significant (P = 0.7).
mtDNA Genetic Structure
Analyses of molecular variance (AMOVA) revealed significant divergence among populations (FST = 0.285; P > 0.001), although most genetic variation occurred within rather than among populations (VWP = 71.47 %; WAP = 28.53 %).
The median-joining network showed a main torso composed of well differentiated and equally represented haplotypes (mean 5.4 mutations) with no clear geographic correspondence, and two terminal star shaped sections. In both these sections, the central haplotype is found in multiple basins (Tyrrhenian/Adriatic for one, and Levantine/Aegean/Adriatic for the other), but haplotypes branching from those central ones were generally private to either Tyrrhenian/Adriatic/Aegean in one case, or Levantine in the other (Fig. 3).

Phylogenetic network of mtDNA from Mediterranean Tursiops truncatus, obtained using the software Network. Each circle represent a unique haplotype, with size being proportional to the number of samples carrying it. Links represent 1 point mutation between haplotypes, with longer links represented with 1 dark vertical bar for each mutation. Black circles represent haplotypes that were inferred by the software, but not found in the population. Numbered haplotypes (15, 17, 20, 22, 25, 26, 48) are all similar to North Atlantic Pelagic ecotype haplotypes (Fig. S4)
The network for North Atlantic Tursiops is characterized by several equally differentiated haplotypes at the centre of star shaped phylogenies, but there is no clear correspondence between network lineages and geographic origin. Haplotypes found in the Ionian Sea (which is considerably deeper than other basins) were generally closely related to haplotypes from the Western North Atlantic Pelagic (WNAP) ecotype (as defined in Hoelzel et al. 1998), though none was shared (Fig. S5). In contrast, haplotypes from Mediterranean basins with depth profiles similar to coastal regions (Tyrrhenian, Adriatic, Aegean and Levantine), were often shared with samples obtained from oceanic locations (Azores and Madeira), as well as open water coastal locations (mainland Portugal, Bay of Biscay, Gulf of Cadiz, Iroise Sea) and the English Channel. Interestingly, only two haplotypes are shared between WNAP and other oceanic locations (Azores and Madeira), although they tend to be separated by a small number of mutational steps. Haplotypes that are shared between multiple locations are generally found at the centre of star shaped phylogenies, while terminal haplotypes are usually private to specific locations, including both Mediterranean coastal basins and Atlantic open water coastal regions.
Historical Demography
Summary statistics suggested a recent expansion accompanied by low inbreeding. Tajima’s D and Fu’s F were both generally negative, consistent with demographic expansion for most basins, except for the Aegean where a positive value suggests contraction. Consistently, F IS values were positive for all basins except the Aegean Sea where it was negative, with only the Tyrrhenian and Levantine being significantly different from zero (Table 1).
Mismatch distribution showed a bimodal profile that did not significantly differ from the expected under a spatial expansion model (P = 0.162; Fig. 4), but it did significantly deviate from expectations under a demographic expansion model (P = 0.02). Estimates of expansion time suggest this has occurred recently, likely after the Last Glacial Maximum (LGM; Table 3). Using a mutation rate calibrated for the whole mitogenome, the estimated time of spatial expansion centres around the Eemian interglacial roughly 155 kya (Table 3; note that the most likely time for the opening of the Bosporus strait is around 5–7 kya). However, using the mutation rate calibrated for control region only (the fragment used in this study), this time moves forward to after the LGM.

Mismatch distribution of pairwise differences between all Mediterranean Tursiops truncatus mtDNA haplotypes, calculated using the software Arlequin. Solid line represents the expected distribution under a spatial expansion model, which does not significantly differ from the observed data (see “Results” section)
Bayesian skyline plots also retrieve an increase in effective population size close to the LGM. Using the mutation rate calibrated for the entire mitogenome, the increase in population size starts roughly around 35 kya (Fig. S6a), and it shifts to around 5 kya using the mutation rate calibrated for control region only (Fig. S6b). Note that the error associated with this calculation is likely to be large, due to the fact that only modern samples were used. Consistently, calculating the time of expansion using τ = 1 for the first modal peak (corresponding to differences between the star-shaped regions of the network), the time of demographic expansion is also after the LGM for both mutation rates (Table 3).
Discussion
Population Structure Within the Mediterranean Sea
The results of our study collectively suggest that the bottlenose dolphin in the Mediterranean Sea exhibits fine scale population structure. Geographical distribution of the main population groups appears to correspond well to the main Mediterranean basins, namely the Tyrrhenian, Ionian, Adriatic, Aegean and Levantine seas (Figs. 2, S1). The R2 statistic was not as high as observed in populations structured strongly according to geographic region, but it was still distinctively higher than expected if structure is not organized geographically (Gayevskiy et al. 2014). The Obstruct visual plot (Fig. 2) is also consistent with this by showing strong levels of correlation between geographic origin of samples and inferred genetic cluster, although it appears weaker for some geographic regions (i.e. Ionian; see below for more details). Furthermore, we found evidence of fine scale population division within the Adriatic and the Levantine. However, this pattern is likely confounded by patterns of migration and phylogeographic history (see below for details). In the Adriatic, samples from the GT clearly differentiate from other Adriatic samples, and within the Adriatic Sea our results indicate division both between North/Central/South basins, consistent with previous preliminary results (Gaspari et al. 2013).
Patterns of population structure across such small distances are commonly described for bottlenose dolphins around the world (e.g. Rosel et al. 2009; Ansmann et al. 2012; Kiszka et al. 2012), and our results of fine scale population structure within the Adriatic Sea are consistent with local reports of strong site fidelity (Bearzi et al. 1997; Genov et al. 2008; Pleslić et al. 2013). However, this fine scale structure is likely to have been established recently, and it is not clear how stable it will be in the long term.
The separation between the GT and the remaining Adriatic is similar to that observed elsewhere in European waters, such as the Moray Firth (Scotland), the Shannon estuary (Ireland), and the Sado estuary (Portugal). In all locations, small populations show strong site-fidelity to semi-enclosed bays, with limited interaction with populations outside the bays (Ingram and Rogan 2002; Augusto et al. 2011; Cheney et al. 2013). Genetically they are differentiated from the closest populations, but are often similar to those found further apart (Parsons et al. 2002; Fernández et al. 2011; Mirimin et al. 2011), just as observed in our study. In our case, the similarity between GT and the Aegean Sea is likely due to the stochastic distribution of genetic variation during a recent colonization of the Mediterranean (see below for details).
Phylogeographic History and Mediterranean Invasion
Our study suggests that population structure within the Mediterranean largely results from stochastic distribution of genetic variation, through a series of founder events (either sequential or concurrent) during a recent invasion of the Mediterranean Sea. An Atlantic origin of Mediterranean populations has been proposed earlier (Natoli et al. 2005; Moura et al. 2013b), and our study confirms this by showing that haplotypes private to individual basins occur in low frequencies and branch off from star shaped phylogenies, whose central haplotypes are shared across the Mediterranean/Atlantic. Our study further estimates a timing for this colonization, which likely occurred after the LGM, particularly if a faster mutation rate is used.
Several other marine organisms in the Mediterranean exhibit star shaped networks (Natoli et al. 2005; Carreras et al. 2006; Charrier et al. 2006; Perez-Losada et al. 2007; Sušnik et al. 2007; Perez-Portela and Turon 2008; Zulliger et al. 2009) similar to those found in our study. In most cases, this expansion has been dated to around the Eemian interglacial (consistent with our older age estimate of ~155 kya), and never before the Pleistocene (Patarnello et al. 2007; Perez-Losada et al. 2007; Sušnik et al. 2007; Pujolar et al. 2010). However, mutation rates used in these earlier studies were not calibrated using biogeographical events, and are thus likely underestimated (Ho et al. 2005; Patarnello et al. 2007; Ho et al. 2008; Calvo et al. 2009). Indeed, recent studies on sand smelt (Pujolar et al. 2012) and green crab (Marino et al. 2011) using mutation rates derived from biogeographical calibrations events, also time the Mediterranean colonization to after the LGM. In our case, due to the control-region being a known mutational hotspot (Stoneking 2000), the rate calculated for the whole mitogenome (Moura et al. 2013b) is likely inappropriately slow for our study.
Geological data indicates that during the LGM, water exchange through the Strait of Gibraltar was much reduced (Mikolajewicz 2011). This likely led to lower temperatures and oxygen levels, with corresponding increases in salinity compared to present day, particularly in the Eastern Mediterranean basin (Mikolajewicz 2011). Because the Mediterranean has a net deficit of water, and limited connectivity with the Atlantic, this further led to changes in the sea level, with a likely drying of the Adriatic Sea, and physical separation between the Eastern and Western Mediterranean basins (Thiede 1978; Hayes et al. 2005).
High salinity during the LGM could have been a limiting factor for the survival of fish species in the Eastern Mediterranean, especially in combination with high water temperatures (Morris 1960; Gonzalez 2011). Dolphins are large predators with high resting metabolic rates (Williams et al. 2001), which require a high energy diet to survive (Spitz et al. 2012). Therefore, even if some fish species managed to survive the harsh environmental conditions in the LGM Mediterranean, they might not have been present in sufficient numbers to support a viable bottlenose dolphin population.
The differentiation of the population occupying the Levantine basin, together with known morphological differences (Sharir et al. 2011), could reflect longer term survival of local refugial population that diversified in isolation. However, the number of unique haplotypes and private alleles was comparable to those found in the Adriatic, where a refugial population was very unlikely. Nevertheless, if present genetic structure patterns reflect two independent colonisations of the Mediterranean, still most of its diversity results from a recent expansion from the Atlantic, as haplotypes from the Adriatic and Aegean seas are more closely related to Ionian/Atlantic haplotypes. Our data thus support the interpretation that bottlenose dolphin colonization of the Mediterranean occurred after the LGM, and that current patterns of populations structure are the result of chance distribution of haplotypes due to founder events during the spatial expansion.
Research from other high dispersal animals is also consistent with a post-LGM expansion. Common dolphin (D. delphis) mtDNA also shows a pattern consistent with a recent expansion into the Mediterranean from a larger Atlantic population (Natoli et al. 2008), but exhibits weak population structure possibly due to a more fluid social structure and lower natal philopatry (Moura et al. 2013a). Recent studies on white-sided dolphins (L. acutus, Banguera-Hinestroza et al. 2014) and harbour porpoises (Phocoena phocoena, Fontaine et al. 2014) also found evidence for a post-LGM expansions in the North Atlantic, as well as in killer whales (O. orca; Moura et al. 2015) in association with a strong decline during the LGM (Moura et al. 2014). Similarly, the low diversity observed in loggerhead sea turtles in Eastern Mediterranean (Carreras et al. 2006), has been linked with excessively low temperature for successful hatching during the LGM (Bowen et al. 1993), which would imply that present loggerhead turtles are descendent from post-glacial colonizers. Recently, comparison with simulated datasets also suggested that differentiation of certain estuarine/bay populations of bottlenose in the North Atlantic, has been achieved post LGM (Louis et al. 2014b). This suggests that the LGM might have had profound effects not only in the Mediterranean marine fauna, but also more broadly in the North Atlantic.
Integration with North Atlantic Structure
Integration of our samples with mtDNA data from the North Atlantic available in the literature, showed significant haplotype sharing not only between Mediterranean and Atlantic, but also more broadly across the Atlantic. In spite of this, shallow water basins within the Mediterranean had a high number of private alleles as compared to the deeper Ionian Sea, with mtDNA also showing close resemblance with the WNAP ecotype (Hoelzel et al. 1998; Tezanos-Pinto et al. 2008). This suggests a pelagic versus coastal differentiation as described elsewhere in the world (Hoelzel et al. 1998; Segura et al. 2006; Tezanos-Pinto et al. 2008; Caballero et al. 2012) including the Eastern North Atlantic (Natoli et al. 2004; Moura et al. 2013b). This distinction is described here within the Mediterranean for the first time, and is consistent with reports of vagrant individuals being sighted only occasionally in regions where other groups show strong site fidelity (Bearzi et al. 2005; Genov et al. 2008; Pleslić et al. 2013). It is also ecologically consistent, as the Ionian Sea is the deepest of all basins analysed (Becker et al. 2009).
However, the sharing of haplotypes indicates a different dynamic to that found in the Western North Atlantic, where genetic differentiation is not only much stronger (Hoelzel et al. 1998; Moura et al. 2013b), but is also accompanied by nearly complete spatial segregation (Torres et al. 2003). Instead, vagrant individuals with a fluid social structure are commonly reported in the same sites as coastal populations with tighter social structure (Bearzi et al. 2005; Genov et al. 2008; Pleslić et al. 2013; Martinho et al. 2014), and gene flow is detected between the two habitats (Quérouil et al. 2007, this study). Population dynamics of bottlenose dolphin in the North Atlantic and Mediterranean Sea, are thus more typical of a metapopulation (Nichols et al. 2007; Oremus et al. 2007). In such cases, populations that exhibit strong site fidelity and are demographically isolated (e.g. the Tyrrhenian and the Adriatic populations), can appear to be part of a single large population in a mtDNA phylogenetic network (Oremus et al. 2007).
Recent expansion into unoccupied habitats contributes to the sharing of haplotypes across distant regions, but metapopulations are usually characterized by specific source-sink dynamics, for which there is evidence in our study. Microsatellite data suggest that gene flow is stronger from the Ionian pelagic basin to the other coastal basins than in the opposite direction, and also low between different coastal basins. This implies that gene flow between shallow water basins (e.g. Tyrrhenian, Adriatic and Aegean) might be mediated by the pelagic ecotype, and explains how coastal populations exhibiting strong site fidelity (Bearzi et al. 2005; Genov et al. 2008; Pleslić et al. 2013) can still be genetically similar (e.g. Tyrrhenian and Adriatic seas). Gene flow appears to be female mediated, which would explain why lineage sorting between pelagic and Mediterranean haplotypes is incomplete (Natoli et al. 2005; Moura et al. 2013b) in spite of the good differentiation seen here in nuclear DNA.
A metapopulation dynamics would account for the weak genetic differentiation found between oceanic and coastal open water samples in the Atlantic (Quérouil et al. 2007; Louis et al. 2014a). Regardless, our North Atlantic network shows that open water coastal areas still exhibit private alleles at the tips of star shaped phylogenies, suggesting that these regions too are characterized by local specializations following expansion. Populations inhabiting estuarine/bay habitats will differentiate faster due to a low carrying capacity of those environments, meaning large populations of a top predator cannot be supported and new individuals (either migrants or born locally) not readily accepted. Consistently, coastal populations found to have strong genetic differentiation, typically inhabit enclosed or semi-enclosed habitats and have low census and effective population sizes (Parsons et al. 2002; Fernández et al. 2011; Mirimin et al. 2011; Louis et al. 2014a). Therefore, population differentiation in North Atlantic/Mediterranean bottlenose dolphins is likely the result of the low carrying capacity of coastal environments, which makes them unable to act as sink populations for the larger pelagic source population.
Conservation Implications
From our genetic data, no evidence was found for recent local population contractions in the Mediterranean, except for the Aegean Sea. This inference controverts previous concerns of up to 50 % decline of bottlenose dolphins in the Adriatic Sea since the 1950s (Bearzi and Fortuna 2006). However, bottleneck tests often lack power if pre-bottleneck effective population size was low, and if the test is carried out a small number of generations after the bottleneck (Peery et al. 2012). A recent study in the Adriatic found evidence for an increase in abundance for one local population (Pleslić et al. 2013), suggesting that natural local fluctuations could be interpreted as reductions if quantitative data is limited to a short time period or sparse over a long period. For the Aegean Sea, genetic data support a recent bottleneck (although sample size is low), but no abundance estimates are currently available to corroborate the inference from genetic data. Our results highlight the need for accurate abundance estimates in the region, as it is possible that local declines are occurring undetected.
A metapopulation dynamics has important conservation implications. Although coastal populations could be replaced through migration from the source population, local declines likely reflect an inability of the environment to support a viable population, and thus emphasise the need for conservation measures. Similarly, pelagic source populations should be seen as a valuable reservoir of individuals and genetic variation, and be the target of conservation measures well before its numbers appear to be depleted.
Concluding Remarks
Our study suggests that present bottlenose dolphin genetic structure patterns in the Mediterranean Sea, largely result from the stochastic distribution of Atlantic genetic diversity during a recent post-glacial expansion. Furthermore, North Atlantic and Mediterranean populations likely constitute a single metapopulation, with pelagic populations acting as genetic source for coastal ones. Current population differentiation appears to be the result of the combined effects of past climatic variations, local carrying capacity associated with differences in social structure and site fidelity, and potentially ecological differences, particularly between the pelagic and coastal populations. Adaptation might further contribute to differentiation, but this will likely not be possible to address using only neutral genetic markers.
Our results have important implication for the understanding of Mediterranean biodiversity. Previous studies have suggested that Mediterranean biodiversity was the result of endemism from glacial refugia. Our study further suggests that the Mediterranean might have also been a sink for many Atlantic species post-LGM. This could explain why some of the Mediterranean oceanographic boundaries do not appear to constitute genetic boundaries for all marine species (e.g. Patarnello et al. 2007). Patterns of population structure will likely results from a combination of pre-expansion diversity, dispersal and colonization mechanisms, as well as specific limiting factors and behavioural characteristics in post-colonization environments.
References
Amaral, A. R., Beheregaray, L. B., Bilgmann, K., Boutov, D., Freitas, L., Robertson, K. M., et al. (2012). Seascape genetics of a globally distributed, highly mobile marine mammal: The short-beaked common dolphin (genus Delphinus). PLoS One, 7, e31482.
Almada, V. C., Oliveira, R. F., Goncalves, E. J., Almeida, A. J., Santos, R. S., & Wirtz, P. (2001). Patterns of diversity of the north-eastern Atlantic blenniid fish fauna (Pisces: Blenniidae). Global Ecology and Biogeography, 10, 411–422.
Ansmann, I. C., Parra, G. J., Lanyon, J. M., & Seddon, J. M. (2012). Fine-scale genetic population structure in a mobile marine mammal: Inshore bottlenose dolphins in Moreton Bay, Australia. Molecular Ecology, 21, 4472–4485.
Augusto, J. F., Rachinas-Lopes, P., & dos Santos, M. E. (2011). Social structure of the declining resident community of common bottlenose dolphins in the Sado Estuary, Portugal. Journal of Marine Biology Association of United Kingdom, 92, 1773–1782.
Bahri-Sfar, L., Lemaire, C., Hassine, O. K. B., & Bonhomme, F. (2000). Fragmentation of sea bass populations in the western and Eastern Mediterranean as revealed by microsatellite polymorphism. Proceeding of the Royal Society of London B, 267, 929–935.
Bandelt, H.-J., Forster, P., & Röhl, A. (1999). Median-joining networks for inferring intraspecific phylogenies. Molecular Biology and Evolution, 16, 37–48.
Banguera-Hinestroza, E., Bjørge, A., Reid, R. J., Jepson, P., & Hoelzel, A. R. (2010). The influence of glacial epochs and habitat dependence on the diversity and phylogeography of a coastal dolphin species: Lagenorhynchus albirostris. Conservation Genetics, 11, 1823–1836.
Banguera-Hinestroza, E., Evans, P. G. H., Mirimin, L., Reid, R. J., Mikkelsen, B., Couperus, A. S., et al. (2014). Phylogeography and population dynamics of the white-sided dolphin (Lagenorhynchus acutus) in the North Atlantic. Conservation Genetics,. doi:10.1007/s10592-014-0578-z.
Bearzi, G., & Fortuna, C. M. (2006). Common bottlenose dolphin Tursiops truncatus (Mediterranean subpopulation). In R. R. Reeves & G. Notarbartolo di Sciara (Eds.), The status and distribution of cetaceans in the Black Sea and Mediterranean Sea (pp. 64–73). Malaga: IUCN Centre for Mediterranean Cooperation.
Bearzi, G., Fortuna, C. M., & Reeves, R. R. (2009). Ecology and conservation of common bottlenose dolphins (Tursiops truncatus) in the Mediterranean Sea. Mammal Review, 39, 92–123.
Bearzi, G., Notarbartolo di Sciara, G., & Politi, E. (1997). Social ecology of bottlenose dolphins in the Kvarneric (northern Adriatic Sea). Marine Mammal Science, 13, 650–668.
Bearzi, G., Politi, E., Agazzi, S., Bruno, S., Costa, M., & Bonizzoni, S. (2005). Occurrence and present status of coastal dolphins (Delphinus delphis and Tursiops truncatus) in the eastern Ionian sea. Aquatic Conservation: Marine Freshwater Ecosystem, 15, 243–357.
Becker, J. J., Sandwell, D. T., Smith, W. H. F., Braud, J., Binder, B., Depner, J., et al. (2009). Global bathymetry and elevation data at 30 arc seconds resolution: STRM30_PLUS. Marine Geodesy, 32, 355–371.
Bérubé, M., & Palsbøll, P. (1996). Identification of sex in cetaceans by multiplexing with three ZFX and ZFY specific primers. Molecular Ecology, 5, 283–287.
Bierne, N., Bonhomme, F., & David, P. (2003). Habitat preference and the marine-speciation paradox. Proceeding of the Royal Society of London B, 270, 1399–1406.
Bilgmann, K., Möller, L. M., Harcourt, R. G., Kemper, C. M., & Beheregaray, L. B. (2011). The use of carcasses for the analysis of cetacean population genetic structure: A Comparative study in two dolphin species. PLoS One, 6(5), e20103.
Borsa, P., Blanquer, A., & Berrebi, P. (1997). Genetic structure of the flounders Platichthys flesus and P. stellatus at different geographic scales. Marine Biology, 129, 233–246.
Bowen, B., Avise, J. C., Richardson, J. I., Meylan, A. B., Margaritoulis, D., & Hopkins-Murphy, S. R. (1993). Population structure of loggerhead turtles (Caretta caretta) in the northwestern Atlantic Ocean and Mediterranean Sea. Conservation Biology, 7, 834–844.
Caballero, S., Islas-Villanueva, V., Tezanos-Pinto, G., Duchene, S., Delgado-Estrella, A., Sanchez-Okrucky, R., et al. (2012). Phylogeography, genetic diversity and population structure of common bottlenose dolphins in the wider Caribbean inferred from analyses of mitochondrial DNA control region sequences and microsatellite loci: Conservation and management implications. Animal Conservation, 15, 95–112.
Caldwell, M., Gaines, M. S., & Hughes, C. R. (2002). Eight polymorphic microsatellite loci for bottlenose dolphin and other cetacean species. Molecular Ecolology Notes, 2, 393–395.
Calvo, M., Templado, J., Oliverio, M., & Machordom, A. (2009). Hidden Mediterranean biodiversity: Molecular evidence for a cryptic species complex within the reef building vermetid gastropod Dendropoma petraeum (Mollusca: Caenogastropoda). Biological Journal of Linnean Society, 96, 898–912.
Carreras, C., Pascual, M., Cardona, L., Aguilar, A., Margaritoulis, D., Rees, A., et al. (2006). The genetic structure of the loggerhead sea turtle (Caretta caretta) in the Mediterranean as revealed by nuclear and mitochondrial DNA and its conservation implications. Conservation Genetics, 8, 761–775.
Carreras-Carbonell, J., Macpherson, E., & Pascual, M. (2006). Population structure within and between subspecies of the Mediterranean triplefin fish Tripterygion delaisi revealed by highly polymorphic microsatellite loci. Molecular Ecology, 15, 3527–3539.
Casado-Amezúa, P., Goffredo, S., Templado, J., & Machordom, A. (2012). Genetic assessment of population structure and connectivity in the threatened Mediterranean coral Astroides calycularis (Scleractinia, Dendrophylliidae) at different spatial scales. Molecular Ecology, 21, 3671–3685.
Charrier, G., Chenel, T., Durand, J. D., Girard, M., Quiniou, L., & Laroche, J. (2006). Discrepancies in phylogeographical patterns of two European anglerfishes (Lophius budegassa and Lophius piscatorius). Molecular Phylogenetics and Evolution, 38, 742–754.
Cheney, B., Thompson, P. M., Ingram, S. N., Hammond, P. S., Stevick, P. T., Durban, J. W., et al. (2013). Integrating multiple data sources to assess the distribution and abundance of bottlenose dolphins Tursiops truncatus in Scottish waters. Mammal Review, 43, 71–88.
Coll, M., Piroddi, C., Steenbeek, J., Kaschner, K., Lasram, F. B. R., Aguzzi J., et al. (2010). The biodiversity of the Mediterranean Sea: Estimates, patterns, and threats. PLoS One, 5, e11842.
Cornuet, J. M., & Luikart, G. (1996). Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics, 144, 2001–2014.
Domingues, V. S., Bucciarelli, G., Almada, V. C., & Bernardi, G. (2005). Historical colonization and demography of the Mediterranean damselfish, Chromis chromis. Molecular Ecology, 14, 4051–4063.
Domingues, V. S., Santos, R. S., Brito, A., Alexandrou, M., & Almada, V. C. (2007). Mitochondrial and nuclear markers reveal isolation by distance and effects of pleistocene glaciations in the northeastern Atlantic and Mediterranean populations of the white seabream (Diplodus sargus, l.). Journal of Experimental Marine Biology and Ecology, 346, 102–113.
Drummond, A. J., Suchard, M. A., Xie, D., & Rambaut, A. (2012). Bayesian phylogenetics with BEAUti and the BEAST 1.7. Molecular Biology and Evolution, 29, 1969–1973.
Earl, D. A., & vonHoldt, B. M. (2012). Structure harvester: A website and program for visualizing structure output and implementing the Evanno method. Conservation Genetics Resources, 4, 359–361.
Excoffier, L., Laval, G., & Schneider, S. (2005). Arlequin ver. 3.0: An integrated software package for population genetics data analysis. Evolutionary Bioinformatics Online, 1, 47–50.
Fernández, R., Santos, M. B., Pierce, G. J., Llavona, Á., López, A., Silva, M. A., et al. (2011). Fine-scale genetic structure of bottlenose dolphins, Tursiops truncatus, in Atlantic coastal waters of the Iberian Peninsula. Hydrobiologia, 670, 111–125.
Fontaine, M. C., Roland, K., Calves, I., Austerlitz, F., Palstra, F. P., Tolley, K. A., et al. (2014). Postglacial climate changes and rise of three ecotypes of harbor porpoises, Phocoena phocoena, in western palearctic waters. Molecular Ecology,. doi:10.1111/mec.12817.
Foote, A. D., Kaschner, K., Schultze, S. E., Garilao, C., Ho, S. Y. W., Post, K., et al. (2013). Ancient DNA reveals that bowhead whale lineages survived late pleistocene climate change and habitat shifts. Nature Communications, 4, 1677.
Galarza, J. A., Carreras-Carbonell, J., Macpherson, E., Pascual, M., Roques, S., Turner, G. F., et al. (2009). The influence of oceanographic fronts and early-life-history traits on connectivity among littoral fish species. Proceeding of the National Academy of Sciences USA, 106, 1473–1478.
Gaspari, S., Airoldi, S., & Hoelzel, A. R. (2007a). Risso’s dolphins (Grampus griseus) in UK waters are differentiated from a population in the Mediterranean Sea and genetically less diverse. Conservation Genetics, 8, 727–732.
Gaspari, S., Azzelino, A., Airoldi, S., & Hoelzel, A. R. (2007b). Social kin associations and genetic structuring of striped dolphin populations (Stenella coeruleoalba) in the Mediterranean Sea. Molecular Ecology, 16, 2922–2933.
Gaspari, S., Holcer, D., Mackelworth, P., Fortuna, C., Frantzis, A., Genov, T., et al. (2013). Population genetic structure of common bottlenose dolphins (Tursiops truncatus) in the Adriatic Sea and contiguous regions: Implications for international conservation. Aquatic Conservation: Marine Freshwater Ecosystem. doi:10.1002/aqc.2415.
Gayevskiy, V., Klaere, S., Knight, S., & Goddard, M. R. (2014). ObStruct: A method to objectively analyse factors driving population structure using Bayesian ancestry profiles. PLoS One, 9, e85196.
Genov, T., Kotnjek, P., Lesjak, J., Hace, A., & Fortuna, C. M. (2008). Bottlenose dolphins (Tursiops truncatus) in Slovenian and adjacent waters (northern Adriatic Sea). Annales Series Historia Naturalis, 18(2), 227–244.
Gonzalez, R. J. (2011). The physiology of hyper-salinity tolerance in teleost fish: A review. Journal of Compatrative Physiology B, 182, 321–329.
Goudet, J. (2001). FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3). http://www.unil.ch/izea/softwares/fstat.html
Hayes, A., Kucera, M., Kallel, N., Sbaffi, L., & Rohling, E. J. (2005). Glacial Mediterranean Sea surface temperatures based on planktonic foraminiferal assemblages. Quaternary Science Review, 24, 999–1016.
Hey, J., & Nielsen, R. (2007). Integration within the Felsenstein equation for improved Markov Chain Monte Carlo methods in population genetics. Proceeding of the national academy of sciences USA, 104, 2785–2790.
Ho, S. Y., & Lanfear, R. (2010). Improved characterisation of among-lineage rate variation in cetacean mitogenomes using codon-partitioned relaxed clocks. Mitochondrial DNA, 21, 138–146.
Ho, S. Y., Phillips, M. J., Cooper, A., & Drummond, A. J. (2005). Time dependency of molecular rate estimates and systematic overestimation of recent divergence times. Molecular Biology and Evolution, 22, 1561–1568.
Ho, S. Y., Saarma, U., Barnett, R., Haile, J., & Shapiro, B. (2008). The effect of inappropriate calibration: Three case studies in molecular ecology. PLoS One, 3, e1615.
Hoelzel, A. R., Natoli, A., Dahlheim, M. E., Olavarria, C., Baird, R. W., & Black, N. A. (2002). Low worldwide genetic diversity in the killer whale (Orcinus orca): Implications for demographic history. Proceedingof the Royal Society of London B, 269, 1467–1473.
Hoelzel, A. R., Potter, C. W., & Best, P. B. (1998). Genetic differentiation between parapatric `nearshore’ and `offshore’ populations of the bottlenose dolphin. Proceedingof the Royal Society of London B, 265, 1177–1183.
Ingram, S. N., & Rogan, E. (2002). Identifying critical areas and habitat preferences of bottlenose dolphins Tursiops truncatus. Marine Ecology Progress Series, 244, 247–255.
Kiszka, J., Simon-Bouhet, B., Gastebois, C., Pusineri, C., & Ridoux, V. (2012). Habitat partitioning and fine scale population structure among insular bottlenose dolphins (Tursiops aduncus) in a tropical lagoon. Journal of Experimental Marine Biology and Ecology, 416–417, 176–184.
Louis, M., Fontaine, M. C., Spitz, J., Schlund, E., Dabin, W., Deaville, R., et al. (2014a). Ecological opportunities and specializations shaped genetic divergence in a highly mobile marine top predator. Proceedingof the Royal Society of London B, 281, 20141558.
Louis, M., Viricel, A., Lucas, T., Peltier, H., Alfonsi, E., Berrow, S., et al. (2014b). Habitat-driven population structure of bottlenose dolphins, Tursiops truncatus, in the North-East Atlantic. Molecular Ecology, 23, 857–874.
Marino, I. A. M., Pujolar, J. M., & Zane, L. (2011). Reconciling deep calibration and demographic history: Bayesian inference of post glacial colonization patterns in Carcinus aestuarii (nardo, 1847) and C. Maenas (Linnaeus, 1758). PLoS One, 6, e28567.
Martinho, F., Pereira, A., Brito, C., Gaspar, R., & Carvalho, I. (2014). Structure and abundance of bottlenose dolphins (Tursiops truncatus) in coastal Setúbal Bay, Portugal. Marine Biology Research. doi:10.1080/17451000.2014.894244.
Mendez, M., Subramaniam, A., Collins, T., Minton, G., Baldwin, R., Berggren, P., et al. (2011). Molecular ecology meets remote sensing: Environmental drivers to population structure of humpback dolphins in the Western Indian Ocean. Heredity, 107, 349–361.
Mikolajewicz, U. (2011). Modeling Mediterranean ocean climate of the Last Glacial Maximum. Climate of the Past, 7, 161–180.
Mirimin, L., Miller, R., Dillane, E., Berrow, S. D., Ingram, S., Cross, T. F., et al. (2011). Fine-scale population genetic structuring of bottlenose dolphins in Irish coastal waters. Animal Conservation, 14, 342–353.
Morris, R. W. (1960). Temperature, salinity, and southern limits of three species of Pacific cottid fishes. Limnology Oceanography, 5, 175–179.
Mossman, C. A., & Waser, P. M. (1999). Genetic detection of sex-biased dispersal. Molecular Ecology, 8, 1063–1067.
Moura, A. E., Janse van Rensburg, C., Pilot, M., Tehrani, A., Best, P. B., Thornton, M., et al. (2014). Killer whale nuclear genome and mtDNA reveal widespread population bottleneck during the last glacial maximum. Molecular Biology and Evolution, 31, 1121–1131.
Moura, A. E., Kenny, J. G., Chaudhuri, R. R., Hughes, M. A., Reisinger, R. R., de Bruyn, P. J. N., et al. (2015). Phylogenomics of the killer whale indicates ecotype divergence in sympatry. Heredity, 114, 48–55.
Moura, A. E., Natoli, A., Rogan, E., & Hoelzel, A. R. (2013a). Atypical panmixia in a European dolphin species (Delphinus delphis): Implications for the evolution of diversity across oceanic boundaries. Journal of Evolutionary Biology, 26, 63–75.
Moura, A. E., Nielsen, S. C. A., Vilstrup, J. T., Moreno-Mayar, J. V., Gilbert, M. T. P., Gray, H., et al. (2013b). Recent diversification of a marine genus (Tursiops spp.) tracks habitat preference and environmental change. Systematic Biology, 62, 865–877.
Naciri, M., Lemaire, C., Borsa, P., & Bonhomme, F. (1999). Genetic study of the Atlantic/Mediterranean transition in sea bass (Dicentrarchus labrax). Journal of Heredity, 90, 591–596.
Natoli, A., Birkun, A., Aguilar, A., Lopez, A., & Hoelzel, A. R. (2005). Habitat structure and the dispersal of male and female bottlenose dolphins (Tursiops truncatus). Proceeding of the Royal Society of London B, 272, 1217–1226.
Natoli, A., Cañadas, A., Vaquero, C., Politi, E., Fernandez-Navarro, P., & Hoelzel, A. R. (2008). Conservation genetics of the short-beaked common dolphin (Delphinus delphis) in the Mediterranean Sea and in the Eastern North Atlantic Ocean. Conservation Genetics, 9, 1479–1487.
Natoli, A., Peddemors, V. M., & Hoelzel, A. R. (2004). Population structure and speciation in the genus Tursiops based on microsatellite and mitochondrial DNA analyses. Journal of Evolutionary Biology, 17, 363–375.
Nichols, C., Herman, J., Gaggiotti, O. E., Dobney, K. M., Parsons, K., & Hoelzel, A. R. (2007). Genetic isolation of a now extinct population of bottlenose dolphins (Tursiops truncatus). Proceedingof the Royal Society of London B, 274, 1611–1616.
Norris, R. D. (2000). Pelagic species diversity, biogeography, and evolution. Paleobiology, 26, 236–258.
Oremus, M., Poole, M. M., Steel, D., & Baker, C. S. (2007). Isolation and interchange among insular spinner dolphin communities in the South Pacific revealed by individual identification and genetic diversity. Marine Ecology Progress Seriess, 336, 275–289.
Park, S. D. E. (2008). Excel microsatellite toolkit. Computer program and documentation distributed by the author. http://animalgenomics.ucd.ie/sdepark/ms-toolkit/. Accessed June 2013.
Parsons, K. M., Noble, L. R., Reid, R. J., & Thompson, P. M. (2002). Mitochondrial genetic diversity and population structuring of UK bottlenose dolphins (Tursiops truncatus): Is the NE Scotland population demographically and geographically isolated? Biology Conservation, 108, 175–182.
Patarnello, T., Volckaert, F. A., & Castilho, R. (2007). Pillars of Hercules: Is the Atlantic-Mediterranean transition a phylogeographical break? Molecular Ecology, 16, 4426–4444.
Peakall, R. O. D., & Smouse, P. E. (2006). Genalex 6: Genetic analysis in Excel. Population genetic software for teaching and research. Molecular Ecology Notes, 6, 288–295.
Peery, M. Z., Kirby, R., Reid, B. N., Stoelting, R., Doucet-Bëer, E., Robinson, S., et al. (2012). Reliability of genetic bottleneck tests for detecting recent population declines. Molecular Ecology, 21, 3403–3418.
Perez-Losada, M., Nolte, M. J., Crandall, K. A., & Shaw, P. W. (2007). Testing hypotheses of population structuring in the Northeast Atlantic Ocean and Mediterranean Sea using the common cuttlefish Sepia officinalis. Molecular Ecology, 16, 2667–2679.
Perez-Portela, R., & Turon, X. (2008). Cryptic divergence and strong population structure in the colonial invertebrate Pycnoclavella communis (Ascidiacea) inferred from molecular data. Zoology, 111, 163–178.
Pleslić, G., Rako, N., Mackelworth, P., Wiemann, A., Holcer, D., & Fortuna, C. (2013). The abundance of common bottlenose dolphins (Tursiops truncatus) in the former marine protected area of the Cres-Lošinj archipelago, Croatia. Aquatic Conservation: Marine Freshwater Ecosystem. doi:10.1002/aqc.2416.
Pritchard, J. K., Stephens, M., & Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics, 155, 945–959.
Pujolar, J. M., Marčeta, T., Saavedra, C., Bressan, M., & Zane, L. (2010). Inferring the demographic history of the Adriatic Flexopecten complex. Molecular Phylogenetics and Evolution, 57, 942–947.
Pujolar, J. M., Zane, L., & Congiu, L. (2012). Phylogenetic relationships and demographic histories of the Atherinidae in the Eastern Atlantic and Mediterranean Sea re-examined by Bayesian inference. Molecular Phylogenetics and Evolution, 63, 857–865.
Quérouil, S., Silva, M. A., Freitas, L., Prieto, R., Magalhães, S., Dinis, A., et al. (2007). High gene flow in oceanic bottlenose dolphins (Tursiops truncatus) of the North Atlantic. Conservation Genetics, 8, 1405–1419.
Rogers, A. R., & Harpending, H. (1992). Population growth makes waves in the distribution of pairwise genetic differences. Molecular Biology and Evolution, 9, 552–569.
Rolland, J. L., Bonhomme, F., Lagardère, F., Hassan, M., & Guinand, B. (2006). Population structure of the common sole (Solea solea) in the Northeastern Atlantic and the Mediterranean Sea: Revisiting the divide with epic markers. Marine Biology, 151, 327–341.
Rosel, P. E., Forgetta, V., & Dewar, K. (2005). Isolation and characterization of twelve polymorphic microsatellite markers in bottlenose dolphins (Tursiops truncatus). Molecular Ecology Notes, 5, 830–833.
Rosel, P. E., Hansen, L., & Hohn, A. A. (2009). Restricted dispersal in a continuously distributed marine species: Common bottlenose dolphins Tursiops truncatus in coastal waters of the western North Atlantic. Molecular Ecology, 18, 5030–5045.
Segura, I., Rocha-Olivares, A., Flores-Ramírez, S., & Rojas-Bracho, L. (2006). Conservation implications of the genetic and ecological distinction of Tursiops truncatus ecotypes in the Gulf of California. Biological Conservation, 133, 336–346.
Sharir, Y., Kerem, D., Goldin, P., & Spanier, E. (2011). Small size in the common bottlenose dolphin Tursiops truncatus in the Eastern Mediterranean: A possible case of Levantine nanism. Marine Ecology Progress Seriess, 438, 241–251.
Shinohara, M., Domingo-Roura, X., & Takenaka, O. (1997). Microsatellites in the bottlenose dolphin Tusiops truncatus. Molecular Ecology, 6, 695–696.
Spitz, J., Trites, A. W., Becquet, V., Brind’Amour, A., Cherel, Y., Galois, R., et al. (2012). Cost of living dictates what whales, dolphins and porpoises eat: The importance of prey quality on predator foraging strategies. PLoS One, 7, e50096.
Stoneking, M. (2000). Hypervariable sites in the mtDNA control region are mutational hotspots. The American Journal of Human Genetics, 67, 1029–1032.
Sušnik, S., Snoj, A., Wilson, I. F., Mrdak, D., & Weiss, S. (2007). Historical demography of brown trout (Salmo trutta) in the Adriatic drainage including the putative S. letnica endemic to lake Ohrid. Molecular Phylogenetics and Evolution, 44, 63–76.
Tezanos-Pinto, G., Baker, C. S., Russell, K., Martien, K., Baird, R. W., Hutt, A., et al. (2008). A worldwide perspective on the population structure and genetic diversity of bottlenose dolphins (Tursiops truncatus) in New Zealand. Journal of Heredity, 100, 11–24.
Thiede, J. (1978). A glacial Mediterranean. Nature, 276, 680–683.
Torres, L. G., Rosel, P. E., D’Agrosa, C., & Read, A. J. (2003). Improving management of overlapping bottlenose dolphin ecotypes through spatial analysis and genetics. Marine Mammal Science, 19, 502–514.
Valsecchi, E., & Amos, W. (1996). Microsatellite markers for the study of cetacean populations. Molecular Ecology, 5, 151–156.
Williams, T. M., Haun, J., Davis, R. W., Fuiman, L. A., & Kohin, S. (2001). A killer appetite: Metabolic consequences of carnivory in marine mammals. Comparative Biochemistry and Physiology Part A: Molecular & Integrative Physiology, 129, 785–796.
Wilson, G. A., & Rannala, B. (2003). Bayesian inference of recent migration rates using multilocus genotypes. Genetics, 163, 1177–1191.
Zulliger, D. E., Tanner, S., Ruch, M., & Ribi, G. (2009). Genetic structure of the high dispersal Atlanto-Mediterreanean sea star Astropecten aranciacus revealed by mitochondrial DNA sequences and microsatellite loci. Marine Biology, 156, 597–610.
Acknowledgments
This research was funded by the Italian DG Fishery within the research framework of the Italian obligations to the Council Regulation (EC) n. 812/2004 (BYCATCH programme). The authors acknowledge the people that have provided samples: Israeli Marine Mammal Research and Assistance Center (IMMRAC, Dan Karem); Blue World Institute, Croatia, Marine Mammals Tissue Bank, University of Padoa, Italy (Bruno Cozzi, Maristella Giurisato); Università degli Studi di Siena, Italy (Letizia Marsili); Morigenos, Slovenia A.R.C.H.E. Porto Garibaldi, Italy (Carola Vallini); Fondazione Cetacea, Italy (Marco Affronte); Tethys Research Institute, Italy (Ada Natoli); Capitaneria di Porto di Brindisi, Italy (Paola Pino d’Astore). We would also like to thank the researchers and volunteers of IMMRAC, Israel.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Gaspari, S., Scheinin, A., Holcer, D. et al. Drivers of Population Structure of the Bottlenose Dolphin (Tursiops truncatus) in the Eastern Mediterranean Sea. Evol Biol 42, 177–190 (2015). https://doi.org/10.1007/s11692-015-9309-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11692-015-9309-8