
Abstract
A study of the interfacial properties of Ni-Al-V alloys is presented further, together with the interatomic potentials of the Ni-Al-V ternary phase system given in this paper, with the application of the Phase-field method and atomistic simulations, based on the second nearest-neighbor modified embedded-atom method (2NN MEAM) potential formalism. It is discovered that the alloy elements have different preferences of segregation or depletion at the (100) FCC/L12 interface (the γ-FCC-Ni(Al, V) and the L12-Ni3(Al, V)) of diffusion phases. Nickel atoms segregate first and then deplete, Vanadium atoms segregate while aluminum atoms deplete at the (100) FCC/L12 interfaces forming new diffusion phases. Both, vanadium segregation and aluminum depletion affect the alloy’s interfacial properties which in turn, will lower the interfacial energy and increase the work of separation. The results of the Phase-field method coincide with the results of the atomistic computation based on the 2NN MEAM method. Furthermore, the applicability of the atomistic approach to an elaborate alloy design of advanced Ni-based superalloys through the investigation of the effect of alloying elements at the interfacial properties is discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
In Ni-based superalloys, the application of the intermetallic compound Ni3Al (L12) in the series of high temperature structural materials is restricted due to severe brittleness problems. There are two reasons for this poor brittleness, one is the existing slip system, and another is the poor grain boundary cohesive force. A single crystal Ni3Al has good plasticity, while the poly-crystalline Ni3Al shows brittleness at both room and high temperatures, which suggests that its intrinsic brittleness of the interface, determines the brittleness of the Ni3Al crystal. Many researchers have studied that the addition of the element B, which is partially scattered at the interfaces of the Ni3Al crystal, can improve the alloy’s tensile plasticity significantly.[1,2] Some other alloy elements can also improve the intrinsic brittleness of the Ni3Al crystal plane interface by adsorbing the alloying elements which are segregated at the interface, and therefore improving the alloy plasticity.[3] As a result, the occupied behavior of alloy elements in the Ni3Al alloy[4,5] and the segregation of the alloy elements on the Ni3Al crystal’s plane interface[6-8] have been widely studied. What should be further pointed out is that the segregation of atoms can change the anisotropy in the interfacial energy and the shape of the γ′ precipitates. It is believed that the material’s grain size distribution can also be changed by alloying and, consequently change the lattice mismatch or the misfit strain energy at the interface of the crystal due to the atoms segregation.[9-11] Therefore, information on the interfaces in Ni-based superalloys, particularly regarding the structure, energy, migration and segregation dynamics behavior, is highly required to get a better understanding of their strengthening effect.
The experimental measurement of interfacial energy, migration and solute segregation is very difficult to conduct. Alternatively, the forehand parameters can be calculated rather easily using the atomistic simulations that combine molecular dynamics (MD) or Monte Carlo (MC) methods with the Phase-field method. These atomistic simulations have been successfully applied in both binary and ternary systems, whereas for exploring various interfacial properties of solids. Particularly, the interface structure and the interfacial energy as well, with simultaneous use for searching of candidate alloying elements that modify the interfacial energy, misfit the strain or the change in the volume fraction of precipitates for an improvement of the alloys’ mechanical properties.[12-15]
First, the interfacial migration and the elements segregation will be studied by the Phase-field method. Then, the alloying elements that modify the interfacial energy and the work of separation will be studied by the use of both MD and MC simulations.
2 Atomistic Modeling
2.1 Phase Field Model
The phase field equation, based on Onsager and Ginzburg-Landau theories,[16-19] describes the atomic configuration and precipitation pattern of the ordered phases by the occupation probability \(P(\vec{r},t)\) at the crystal lattice site \(\vec{r}\) and the random time t, with a proportional change rate to the corresponding variation of free energy. The atomic occupation probabilities of the ternary systems are \(P_{A} (\vec{r},t) + P_{B} (\vec{r},t) + P_{C} (\vec{r},t) = 1\), where the subscripts A, B and C designate three kinds of atoms (A, B and C designate Ni, Al and V respectively, in this paper). In this simulation, the phase field equation puts the simplification for crystalline defects, and just considers the integrated lattice to response atomic diffusion. Then the kinetic equation can be written as:
The free energy F for the ternary system is given by:
where the first sum represents the chemical energy, the second sum represents the thermal dynamic energy of the system, k B is the Boltzmann constant and \({\text{V}}_{\text{ab}}^{{}} (\vec{r} - \vec{r}^{\prime } )\) is the effective interactive energy. Details regarding Microscopic phase field modeling are referred.[16-19]
It is proven convenient and even computationally advantageous for the kinetic equations to be solved in the reciprocal space. First, the Euler method was used for solving the equation, whereas consequently the inverse Fourier transformation was executed; finally the relationship between the occupation probability and the random time were obtained.
2.2 MEAM Potential Formalism
During the MEAM, the total energy of a system is:
where F i is the embedding function for an atom i embedded in a background electron density \(\mathop {\rho_{i} }\limits^{\_}\), \(S_{ij}\) and \(\varphi_{ij} (R_{ij} )\) are the screening function and pair interaction between atoms i and j separated by a distance \(R_{ij}\). In order for energy calculations to be executed, the functional forms for F i and \(\varphi_{ij}\) should be utilized. The background electron density at each atomic site is computed with directionality of bonding consideration, such as by several partial electron density terms combinations for various angular contributions with the weight factors of t (h)(h = 1–3). Each partial electron density constitutes a function of both atomic configuration and atomic electron density. The atomic electron densities ρ a(h) (h = 0–4) are:
whereρ 0 (the atomic electron density scaling factor) and β (h) (the decay lengths) are adjustable parameters, and r e is the nearest-neighbor distance in the equilibrium reference structure. A specific form is given to the embedding function F i , but not to the pair interaction \(\varphi_{ij}\). Instead, a reference structure where individual atoms are on the exact lattice points was defined and the total energy per atom of the reference structure was estimated from the zero-temperature universal equation of state of metals by Rose et al.[20] Following, the value of the pair interaction was estimated from the known values of total energy per atom and the embedding energy as a function of the nearest-neighbor distance. In the original MEAM,[21] only first-nearest-neighbor interactions were considered. Neglect of second- and more distant nearest-neighbor interaction was accomplished by a strong, screening and many-body effects function.[22] The second-nearest-neighbor interactions during the modified formalism were considered by an adjustment in the screening parameters C min , in order for the screening and many-body effects to become less severe. In addition, a radial cutoff function[22] was applied to reduce calculation time. Details of the (2NN) MEAM formalism have been published.[21-26]
3 Results and Discussion
As mentioned forehand, the interfacial properties are crucial to the control of size distribution and the shape of precipitates. Therefore, it is necessary to know their migration formation and the effect of alloying elements on the interfaces in order to prepare the design of the Ni-Al-V-based superalloys. In this section, three methods of study are used. One method is the Microscopic Phase-field method, which leads to an easy study of the interfacial migration and the alloy elements performance. Additional methods are the MD and MC simulations based on the modified embedded-atom method, which leads to an easy calculation of the interfacial energy and separation.
The simulation based on the Microscopic phase field method was performed in a supercell consisting of 128×128 unit cells, in a periodical boundary condition imposed along both dimensions. The time increment Δt was 0.0002 and the thermal fluctuations were removed after the nucleation process, leading the system to choose the dynamic path automatically. The simulated pictures included here are two-dimensional projections along the [001] direction and depicted with different colors, if the occupation probability of aluminum is 1.0, consequently the site is green, therefore the L12-Ni3Al phase is green, whereas if the occupation probability of vanadium is 1.0, consequently the site is red, consequently the DO22-Ni3V phase is red, and if the occupation probability of nickel is 1.0, then the site is blue, so the matrix is almost blue. The precipitation sequences will be various; therefore the DO22 phase will be referred as a new phase or a second phase in this paper.
Microstructure evolution of the Ni75Al19V6 alloy under the temperature of 1000K is shown in Fig. 1. At the t = 2.0 × 103 time step as shown in Fig. 1(a), there are some small ordered structures called non-stoichiometric ordered phases that randomly appear in the matrix, since their degree of order is low, leading to an uneasy observation of the corresponding structure. Subsequently, at the t = 2.0 × 104 time step as shown in Fig. 1(b), a large number of ordered L12 precipitates appear (L12 phase is green) and there are many interfaces between these ordered L12 phases. While time increases, at the t = 1.0 × 105 time step shown in Fig. 1(c), the L12 phases increase in size gradually, and the interfaces reduce and migrate (certain interfaces disappear), and the new DO22 phase crystals (DO22 phase is red) were encircled by the L12 phases. The region within the white arrow squares A/B is enlarged and shown in the boxes located on the lower left side, whereas the new DO22 phases increased in size and the L12 phases were coarser as shown in Fig. 1(d). Conclusively from Fig. 1, the interfaces between L12 phases are observed with the existence of DO22 phases formed on these interfaces.

Ni75Al19V6 alloy simulated atomistic microstructure temporal evolution pictures at 1000K, (a) t = 2.0 × 103, (b) t = 2.0 × 104, (c) t = 1.0 × 105, (d) t = 5.0 × 105, arrow squares A/B indicate the interface
For further clarification of the effect of the alloying elements performance on the new DO22 phase formed on the interfaces, the distribution and evolution of the three alloying elements across the ordered domain interfaces in different steps according to Fig. 1 arrow square A are consequently analyzed as shown in Fig. 2 of the Ni75Al19V6 alloy, precipitated at the temperature of 1000K. The definition of distance (lattice plane) of the arrow square A interface in Fig. 1(d) is presented in Fig. 2(f). In Fig. 2(a), (b), (c) the variation of the Ni, Al, V concentrations at different steps on the arrow square A interface is respectively shown. In these figures, segregation is declared as the raised above concentration and depletion is declared as the pushed below concentration, of the curves. In the early stage of precipitation at the time step of t=2.0×103, the concentration curves of Ni, Al, V have little effect, according to Fig. 1(a), in the matrix. As time increases to a 1.0 × 104time step, the concentration curves of Ni, is rising above (segregation) and the Al curves are pushing below (depletion) but the V curves change little and according to the Fig. 1(b) the interface is formed. At the 1.0 × 105 and 2.0 × 105 time steps, the concentration curves of Ni, Al, and V have a similar trend and the distribution of the three elements at the 2.0×105 time step as shown in Fig. 1(d), in which the Al is pushing below the curves and the Ni elements are pushing less below the curves and the V curves are raising above (the Al pushing below the curve in a nearly equivalent manner to the V curve raising above) according to the new DO22 phase formed in the arrow square A ordered at the interfaces, being in accordance with Fig. 1(c, d). Furthermore, the volume fraction of both L12 and DO22 phases as presented in Fig. 2(e) was calculated. It is concluded that the DO22 phase increased and the L12 phase decreased gradually from the 1.0 × 105 step, correlating with forehand results that the new DO22 phase was formed among L12 phases at the corresponding interfaces as presented in Fig. 1 and Fig. 2(a-d).

Distribution and evolution of alloying elements across ordered domain interfaces formed among L12 phases in the Ni75Al19V6 alloy at 1000K, (a) Ni, (b) Al, (c) V, (d) t = 2.0 × 105; (e) volume fraction of L12 and DO22 phases in the Ni75Al19V6 alloy at 1000K; (f) the definition of distance (lattice plane) of the arrow square A interface in Fig. 1(d)
Conclusively, the Ni, Al, V elements have a different distribution and evolution trend during the formation of the interface; specifically both the Al and Ni elements are depleted and V is segregated (Al depletion according to V segregation) to form the new DO22 phase; Ni is segregated first and then depleted, as the interfaces migrate; the Al depletion is larger and segregation of the V is simultaneously larger as the new DO22 phase is formed; the new DO22 phase was formed among L12 phases at the interfaces. This shows that the interfaces have local characteristics in the structure of the DO22 phase, upon which the V atoms easily precipitate and to be the new DO22 phase nucleation position.
The distribution and evolution of the alloying elements across the ordered domain plane interfaces, in the Ni75Al22V3 alloy at 1000K presented in Fig. 3, were studied further and the enlargement of the region within the white arrow square C is presented. In Fig. 3(a), all the phases are L12 and there is no DO22 phase appearance (no red color). Although the Al and Ni elements are depleted and V is segregated in the same trend as in Fig. 2(d), the V concentration resulted being significantly low and could not increase to the concentration of 0.25 (or 25%)to form the DO22(Ni3V) phase.

(a) Ni75Al22V3 alloy simulated atomistic microstructure temporal evolution pictures aging at 1000K for t = 2.0 × 105, arrow C indicating the interface, (b) distribution and evolution of alloying elements across ordered domain interfaces formed among L12 phases of the C interface
For the study of the effect of the distribution and evolution of the alloy elements addition on the interfacial properties, the interfacial energy and the effect of separation should be calculated as it couldn’t be executed with the use of the Microscopic Phase-field method. The calculations of the interfacial energy and the work of separation based on the modified embedded-atom method were performed by the computation of the total energy of a supercell that involved an interface between the two phases and the sum of the total energies of the individual phase samples of equal size.[13,14] When a random solid solution is used in an atomistic computation, it becomes necessary for attention to be paid in the probable effect of a solute distribution on the computation results because it could result in a statistical error due to the limited atomic size (number of atoms) of the samples. For the probable statistical error to be reduced, a relatively large sample with 4000~12000 atoms was used and ten independent calculations were performed for the individual interfaces. The interfacial energy was calculated by the following equation:
where, \(E_{\sup ercell}\) is the total energy of the supercell, \(E_{one\;phase}\) and \(E_{another\;phase}\) are the total energies of individual phase samples, A represents the area of the interface. The work of separation is the following equation:
where, \(E_{\sup ercell}\) is also the total energy of the supercell (the same as in the calculation of interfacial energy), \(E^{\prime }_{one\;phase}\) and \(E^{\prime }_{another\;phase}\) are the total energies of individual phase samples (the difference between the calculations of the interfacial energy was that, at the side of each sample composing the interface was left as a free surface, without the application of the periodic boundary condition and the same lattice parameters were maintained in both directions parallel to the interface).
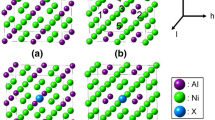
All samples with coherent interfaces of the γ-Ni(Al22, V3) and the L12-Ni3(Al22, V3) (100) interface were prepared by the combination of equal amounts of both γ and L12phase addition. The results are shown in Table 1 and Fig. 4. Before the Monte Carlo (MC) simulation, the interfacial energy of 0.0375 J/m2 and the work of separation of 3.90 J/m2were calculated. Following, the temperature was increased to 1000K and the MC simulation was performed in each situation to keep the interface stable. Moreover, the interfacial energy of −0.1418 J/m2 and the work of separation of 4.37 J/m2were also calculated. The results coincided with the Cr atoms lowering the interfacial energy when replacing the Al atoms on the interface.[27] It is known that a variety of segregations can cause three effects: a segregation that might enhance the interface; reversely, segregation might reduce the cohesion and the bonding strength of the interface; or the segregation might result in a new phase transformation.[9]The results showed that the interface was unstable as the interfacial energy was lower than zero and might form a new phase being in agreement with forehand results. In Fig. 4(a) the distribution and evolution of V across the interface is shown. It can be seen from the concentration of V that it segregated itself near the interface. Comparing Figures 4(c) and (d), the interface after the 200 steps MC simulation has a much higher concentration of vanadium atoms than measured before the MC simulation. The V concentration in Fig. 4(a) is approximately 0.08, nearly equal to the V concentration presented in Fig. 3(b) of being approximately 0.075. The MC simulation results shown in Table 1 and displayed in Fig. 4 resulted in vanadium atoms segregating on the γ/L12 interface; showing that the vanadium segregation was lower from the interfacial energy simultaneously increasing the work of separation; the MC method results matched the Microscopic Phase-field method results.

γ-Ni(Al22, V3) and L12-Ni3(Al22, V3) (100) interface at 1000 K. (a) V concentration, (b) the supercell before MC simulation, (c) the dark profile layer before MC simulation, (d) the same layer after MC simulation. Yellow, gray, and red circles denote Ni, Al, and V atoms, respectively (Color figure online)
With the combination of the Microscopic Phase-field method and the modified embedded-atom method results, the conclusion that the alloying elements segregation or depletion can affect the interfacial properties and form a new phase on the interfaces if the concentration of the segregated elements was adequate, could be reached.
Elements segregation in alloys on the interface is related to a new phase formation and the grain stability in the alloys, also being closely connected with the materials’ mechanical properties. Many researches showed that interface segregation is affected by the materials properties and directly related to the alloying elements, varying in the concentration of the alloying elements and the position of the segregation elements: Elements such as H, O, S, or P causing interface segregation could reduce the material plasticity to be detrimental to the material as a brittle fracture; and the elements Re, Co, Mo, B, and C, in interface segregation could enhance the performance of materials by strengthening the interfacial cohesion; the fundamental physical reason was that the interface segregation was caused by the electronic structure on the interface.[1-8,28,29,30,31,32] Therefore, an important field in the interface segregation study is that it is used in microstructure and alloy design. A difficult challenge in these researches is the structure of the interface and the chemistry of the interfacial properties at an atomic scale. Historically, studies showed that each method, such as experimenting, ab initio, MD, MC and phase field simulation, has different disadvantages. Preferably, a new simplifying method or combination of several methods for better results to be reached should be developed. More research to improve the materials’ mechanical properties is proven to be needed.
4 Conclusions
The main conclusions are:
-
1.
The results showed that the Ni, Al, V elements have different preferences of segregation or depletion during the interface forming in the Ni75Al19V6 alloy; both the Al and Ni elements were depleted and V segregated (Al depletion percentage nearly equal to V segregation) to form the new DO22 phase; Ni first segregated and then was depleted according to the interface migration.
-
2.
Also the Al and Ni elements were depleted and the V was segregated in the Ni75Al22V3 alloy in the same trend as in the Ni75Al19V6 alloy, but the V element concentration was not enough to form a new DO22 phase.
-
3.
The results of the 2NN MEAM method matched the results based on the Phase-field method. Vanadium segregation and aluminum depletion both affect the interfacial properties which in turn, both lower the interfacial energy and increase the work of separation.
References
J.T. Guo, L.Y. Sheng, Y. Xie, Z.X. Zhang, V.E. Ovcharenko, and H.Q. Ye, Microstructure and Mechanical Properties of Ni3Al and Ni3Al-1B Alloys Fabricated by SHS/HE, Intermetallics, 2011, 19, p 137-142
M. Khitouni and N. Njah, Microstructure Evolution After Thermal Treatments of Nanocrystalline Ni3Al and Ni3Al-B Produced by Filling, Phys. Status Solidi (c), 2004, 7, p 1981-1984
X.X. Wu and C.Y. Wang, Influence of Alloying Elements Upon the Theoretical Tensile Strength of Ni-Based Model Superalloy: γ-Ni/γ′-Ni3Al Multilayer, Comput. Mater. Sci., 2016, 119, p 120-129
S. Miura, S. Takizawa, T. Suzuki, Y. Mishima, and T. Mohri, Effect of Tetragonal Distortion Introduced by Anti-site Defect Configuration on Additional Hardening in Off-Stoichiometric Al-rich Ni3Al Alloys, Acta Mater., 2005, 53, p 5175-5181
X.F. Gong, G.X. Yang, Y.H. Fu, Y.Q. Xie, J. Zhuang, and X.J. Ning, First-Principles Study of Ni/Ni3Al Interface Strengthening by Alloying Elements, Comput. Mater. Sci., 2009, 47, p 320-325
Y.J. Wang and C.Y. Wang, Influence of the Alloying Element Re on the Ideal Tensile and Shear Strength of γ′-Ni3Al, Scr. Mater., 2009, 61, p 197-200
T. Kitashima, T. Yokokawa, A.C. Yeh, and H. Harada, Analysis of Element-Content Effects on Equilibrium Segregation at Gamma/Gamma’ Interface in Ni-Based Superalloys Using the Cluster Variation Method, Intermetallics, 2008, 16, p 779-784
Y. Amouyal, Z.G. Mao, and D.N. Seidman, Segregation of Tungsten at γ′(L12)/γ(f.c.c.) Interfaces in a Ni-Based Superalloy: An Atom-Probe Tomographic and First-Principles Study, Appl. Phys. Lett., 2008, 93, p 201905
D. Raabe, M. Herbig, S. Sandlöbes, Y. Li, D. Tytko, M. Kuzmina, D. Ponge, and P.P. Choi, Grain Boundary Segregation Engineering in Metallic Alloys: A Pathway to the Design of Interfaces, Curr. Opin. Solid State Mater. Sci., 2014, 18, p 253-261
R.I. Babicheva, S.V. Dmitriev, Y. Zhang, S.W. Kok, N. Srikanth, B. Liu, and K. Zhou, Effect of Grain Boundary Segregations of Fe Co, Cu, Ti, Mg and Pb on Small Plastic Deformation of Nanocrystalline Al, Comput. Mater. Sci., 2015, 98, p 410-4166
M.A. Gibson and C.A. Schuh, Segregation-Induced Changes in Grain Boundary Cohesion and Embrittlement in Binary Alloys, Acta Mater., 2015, 95, p 145-155
Y.S. Li, Y.X. Pang, W. Liu, X.C. Wu, and Z.Y. Hou, Effect of Diffusivity on the Pseudospinodal Decomposition of the γ′ Phase in a Ni-Al Alloy, J. Phase Equilib. Diffus., 2016, 37, p 261-268
W.P. Dong, H.K. Kim, W.S. Ko, B.M. Lee, and B.J. Lee, Atomistic Modeling of Pure Co and Co-Al System, CALPHAD, 2012, 38, p 7-16
W.P. Dong, B.J. Lee, and Z. Chen, Atomistic Modeling for Interfacial Properties of Ni-Al-V Ternary System, Met. Mater. Int., 2014, 20, p 423-429
M.Y. Zhang, G.Q. Yue, J.Z. Zhang, K. Yang, and Z. Chen, Microscopic Phase Field Study on the Kinetics of Order-Disorder Transition of APDBs Formed Between DO22 Phases During Stress Aging, Mater. Sci. Forum, 2014, 789, p 530-535
A.G. Khachaturyan, Theory of Structural Transformation in Solids, Wiley, New York, 1983, p 129
L.Q. Chen and A.G. Khachaturyan, Computer Simulation of Structural Transformations During Precipitation of an Ordered Intermetallic Phase, Acta Metall. Mater., 1991, 39, p 2533-2551
Y. Wang, L.Q. Chen, and A.G. Khachaturyan, Kinetics of the Strain-Induced Morphological Transformation in Cubic Alloys with Miscibility Gap, Acta Metall. Mater., 1993, 41, p 279-296
R. Poduri and L.Q. Chen, Computer Simulation of the Kinetics of Order-Disorder and Phase Separation During Precipitation of δ’(Al3Li) in Al-Li Alloys, Acta Mater., 1998, 46, p 1719-1729
J.H. Rose, J.R. Smith, F. Guinea, and J. Ferrante, Universal Features of the Equation of State of Metals, Phys. Rev. B, 1984, 29, p 2963-2969
B.J. Lee and M.I. Baskes, Second Nearest-Neighbor Modified Embedded-Atom Method Potential, Phys. Rev. B, 2000, 62, p 8564-8567
M.I. Baskes, Determination of Modified Embedded Atom Method Parameters for Nickel, Mater. Chem. Phys., 1997, 50, p 152-158
B.J. Lee, M.I. Baskes, H. Kim, and Y.K. Cho, Second Nearest-Neighbor Modified Embedded Atom Method Potentials for BCC Transition Metals, Phys. Rev. B, 2001, 64, p 184102
M.I. Baskes, Modified Embedded-Atom Method Potentials for Cubic Materials and Impurities, Phys. Rev. B, 1992, 46, p 2727-2742
B.J. Lee, J.H. Shim, and M.I. Baskes, Semi-Empirical Atomic Potentials for the FCC Metals Cu, Ag, Au, Ni, Pd, Pt, Al and Pb Based on First and Second Nearest Neighbor Modified Embedded Atom Method, Phys. Rev. B, 2003, 68, p 144112
W.P. Dong, Z. Chen, and B.J. Lee, Modified Embedded-Atom Interatomic Potential for Co-W and Al-W Systems, Trans. Nonferrous Met. Soc. China, 2015, 25, p 907-914
T. Hong and A.J. Freeman, Effect of Antiphase Boundaries on the Electronic Structure and Bonding Character of Intermetallic Systems: NiAl, Phys. Rev. B, 1991, 43, p 6446
M. Yamaguchi, Y. Nishiyama, and H. Kaburaki, Decohesion of Iron Grain Boundaries by Sulfur or Phosphorous Segregation: First-Principles Calculations, Phys. Rev. B, 2007, 76, p 18
E. Wachowicz and A. Kiejna, Effect of Impurities on Grain Boundary Cohesion in BCC Iron, Comput. Mater. Sci., 2008, 43, p 736-743
N. Garimella, M. Ode, M. Ikeda, H. Murakami, and Y.H. Sohn, Interdiffusion in L12-Ni3Al Alloyed with Re, J. Phase Equilib. Diffus., 2009, 30, p 246-253
Y.J. Wang and C.Y. Wang, The Alloying Mechanisms of Re, Ru in the Quaternary Ni-Based Superalloys γ-γ′ Interface: A First Principles Calculation, Mater. Sci. Eng. A, 2008, 490, p 242-249
M. Všianská and M. Šob, The Effect of Segregated sp-Impurities on Grain-Boundary and Surface Structure, Magnetism and Embrittlement in Nickel, Prog. Mater. Sci., 2011, 56, p 817-840
Acknowledgment
The authors gratefully acknowledge the financial support of the National Natural Science Foundation of China (NO. 51501165), the Zhejiang Provincial Natural Science Foundation of China (NO. LQ14E010002) and the Special Program for Applied Research on Super Computation of the NSFC-Guangdong Joint Fund (the second phase).We would like to thank all the members of the CMSE Lab at POSTECH for their assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Dong, Wp., Wang, L. & Chen, Z. Effects of Vanadium Intergranular Segregation at FCC/L12 Interfaces in Ni-Al-V Alloys. J. Phase Equilib. Diffus. 38, 102–109 (2017). https://doi.org/10.1007/s11669-017-0520-4
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11669-017-0520-4