
Abstract
The aim of this study was to investigate the effect of pre-oxidation treatment at 400 °C for 1000 h on the corrosion behavior of 316L stainless steel in a high-temperature water environment. The oxide film was characterized via x-ray diffraction, x-ray photoelectron spectroscopy, scanning electron microscopy, and transmission electron microscopy. The weight gain of the pre-oxidized samples was lower than that of samples without pre-oxidation treatment. An (Ni,Fe)Fe2O4 outer layer with a spinel structure and an inner layer consisting of fine spinel oxides (FexCryNizO4) were formed on the surface of the untreated sample. In contrast, the pre-oxidized samples showed the formation of a triple layer: a hydroxide outer layer, a Cr-rich middle layer, and an Fe-rich inner layer. Moreover, the oxide film thickness was thinner than that of the sample without treatment. The results clearly indicate that the selected pre-oxidation process improved the corrosion resistance of 316L stainless steel in a high-temperature water environment.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
As very important structural materials, austenitic stainless steels of the 316 and 304 series are widely used as different components in nuclear power plants (NPPs) due to their excellent mechanical properties and corrosion resistance at high temperatures (Ref 1). The mechanical properties and corrosion resistance of 316L stainless steel (316LSS) are further improved by adding 2-3% Mo compared with 304 stainless steel (Ref 2), so 316LSS has been intensively applied in pressurized water reactors (PWRs).
However, there are still a great number of problems to be resolved for the use of 316LSS in NPPs (Ref 3,4,5). One main problem is to seek ways to resolve or reduce stress corrosion cracking (SCC) behavior because this behavior can cause a sudden rupture of 316LSS, which will then threaten the safety of NPPs (Ref 6). The critical issue to reduce or solve the problem of SCC behavior in high-temperature water is to establish the relationship between cracks and oxide films (Ref 7). It is well known that surface conditions determine the deposition and dissolution of oxides on out-of-core surfaces of structural materials in NPPs (Ref 8). A large amount of oxide deposition or dissolution on the surface will accelerate the SCC behavior if the surface condition is less resistant to corrosion (Ref 8). Therefore, a great number of surface modification methods have been proposed to improve the corrosion resistance of the materials (Ref 9,10,11,12,13,14,15). Han et al. investigated the effect of electropolishing treatment on the corrosion behavior of 316LSS and determined that it could improve the oxidation resistance of 316LSS in simulated PWR primary water, especially at the early stages of oxidation, since there is a compact oxide film monolayer (Ref 9). Seo et al.’s results indicated that the corrosion rate of nickel-based alloy 690TT in PWR primary water decreased by approximately 64% as the roughness values decreased from 710 to 150 nm, whereas no significant difference in the corrosion rate was observed for the roughness variation in the range of 25-150 nm (Ref 10). Wang et al.’s results revealed that the machined surface state of 316SS affected the thickness and elemental distribution of oxide films in a simulated PWR environment (Ref 11). Although the corrosion resistance of materials in high-temperature water is improved using these methods, it takes a long time to apply most of the methods to actual industrial production because of the special required equipment and treatments.
Currently, pre-oxidation treatment is gaining attention due to its practical application in industrial production. Moreover, previous studies have demonstrated that the pre-oxidation treatment significantly enhances the oxidation resistance of 316LSS in high temperature environments (Ref 16). Although a lot of research has been performed on the corrosion behavior of 316LSS used in NPPs over the past few years (Ref 9, 11, 17,18,19,20), no studies have examined the corrosion behavior of pre-oxidized 316LSS in water conditions with both high temperature and high pressure. Additionally, previous investigations on pre-oxidation treatment only focused on high temperatures over short time periods (Ref 16, 21), whereas there is limited understanding of whether long- or short-term exposure at a relatively lower temperature achieves the same effect as that at high temperatures. Therefore, to promote the application of pre-oxidation for industries, it is necessary to investigate the corrosion behavior and mechanism of 316LSS in high-temperature water after short- or long-term pre-oxidation at a relatively lower temperature.
To obtain the preliminary corrosion behavior and mechanism of pre-oxidized 316LSS in high-temperature water, the samples were oxidized in 400 °C air for 1000 h and subsequently exposed to high-temperature water for different periods. This paper mainly investigates the effects of exposure periods on the corrosion behavior and mechanism of long-term pre-oxidated (marked as LP) 316LSS in high-temperature water.
2 Experimental Materials and Methods
2.1 Materials
The material used in the present study is 316LSS, and its chemical composition (in wt.%) is listed in Table 1. A solid round bar of 316LSS, which was purchased from the Taiyuan Iron and Steel Co., Ltd., with a 50 mm diameter and a 1.5 m length was used to prepare the samples. The solid solution treatment was conducted at 1050 °C for 60 min. Specimens of approximately 20×10×3 mm3 were polished on SiC paper up to a 2000 polishing grade, washed with acetone, and finally dried in a drying box. The optical microstructure of 316LSS is shown in Fig. 1, where austenite phases with different grain sizes and a substantial number of twins along the grain boundaries appear.

The light optical microscope image of as-received 316LSS
2.2 Pre-Oxidation Treatment
Oxidation experiments were performed in a high temperature muffle furnace, with the maximum temperature of 1200 °C. The samples were placed in quartz crucibles during the high temperature oxidation process to reduce the error of the weight gain data. The oxidation temperature was 400 °C in air, held for 1000 h, and subsequently cooled to room temperature at a rate of 40 °C/min.
2.3 Corrosion Experiments
Samples with and without long-term pre-oxidation treatment (LPT) were exposed to a high-temperature water environment (320 ± 2 °C, 15 ± 1 MPa) in a static 0.5 L 316LSS autoclave. The test solution was simulated PWR primary water containing 1200 ppm B in H3BO3 and 2.2 ppm Li in LiOH. The dissolved oxygen concentration of the inlet ultrapure water was controlled by continuously bubbling with 99.999% high purity nitrogen gas for 24 h, and three discontinuous tests were performed within one hour before the experiments using a dissolved oxygen meter to ensure that the oxygen concentration in the solution was less than 25 ppb. Various of exposure periods of 100 to 1000 h were selected during the experiments. The LP samples after 100 and 1000 h of exposure are marked as LP-100 and LP-1000 in the following sections. Three samples were removed when each exposure period was finished and subsequently put into a vacuum oven for 10 min at 80 °C. Then, all samples were characterized using different equipment.
2.4 Characterization Methods
An FEI Nova Nano 450 scanning electron microscope (SEM) with energy-dispersive x-ray spectroscopy (EDS) was used to investigate the chemical composition and morphologies of the oxide films. D8 ADVANCE x-ray diffraction (XRD) with Cu Kα (λ= 0.154051 nm) was used to characterize the phase and analyze the crystal structure of the oxides. The diffracted x-ray signal was collected over an angle theta of 5-45. X-ray photoelectron spectroscopy (XPS) measurements were performed on a Thermo ESCALAB 250Xi x-ray photoelectron spectrometer using a monochromatic Al Kα source operating at 150 W with an initial photoenergy of 1486.6 eV. The C1s peak from carbon at 285 eV was used as a reference to correct the charging shifts. Depth profiling was performed over an area of 2.5×2.5 mm2 under 2 keV Ar-ion sputtering, and the spectra were collected within a 900 μm spot. The sputtering rate was set at 0.3 nm/s with reference to the Ta2O5 layer.
The cross section of the samples was characterized using transmission electron microscopy (TEM). A thin-foil sample for TEM observation was prepared using a Helios NanoLab 600i dual-beam focused ion beam (DB-FIB) with Ga ion sputtering after a protective Pt layer was deposited on the oxide scale. The large size of Fe-rich particles on the surface of the samples was avoided when we prepared our TEM sample. Microstructure and selected area electron diffraction (SAED) analyses were performed using JEM-2100F TEM equipment with EDS operating at 200 kV.
3 Results
The surface morphologies of 316LSS after 1000 h of exposure at 400 °C air are illustrated in Fig. 2. The chemical composition of oxide particle A is 72.90% for Fe, 19.46% for Cr, and 7.64% for O (at.%). The total atomic ratio of Fe, Cr, and O is approximately 1:1:3 after subtracting the concentration of these elements in the matrix, which is consistent with the atomic ratio of an oxide mixture of Cr2O3 and Fe2O3. The surface morphologies and EDS results show that Fe2O3 and Cr2O3 oxide particles are formed along the scratches, and a continuous Cr-rich oxide layer is identified on the surface of 316LSS. The XPS results further confirm that the surface oxide film is composed of Fe2O3 and Cr2O3 (Ref 22,23,24,25) and the oxide film under the surface is composed of FeCr2O4 and Cr2O3, as shown in Fig. 3.

Surface morphologies of 316LSS after 1000 h of exposure at 400 °C air

The XPS spectra of Cr 2p3/2, Fe 2p3/2, Ni 2p3/2, O1s in oxide films from the free surface to the deeper site of 316LSS after exposure at 400 °C for 1000 h
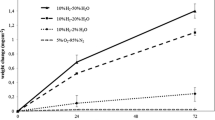
The weight gain of 316LSS with and without LPT after exposure to high-temperature water for different periods is illustrated in Fig. 4. The pre-oxidized samples had lower weight gain than of the samples without LPT, which implies that the pre-oxidation treatment can improve the corrosion resistance of 316LSS in high-temperature water.

The weight gain of 316LSS with and without LPT after exposure to high-temperature water for different exposure periods
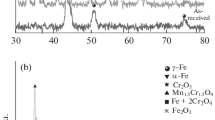
Figure 5 shows the XRD patterns of 316LSS without pre-oxidation exposed to high-temperature water for 100, 500, and 1000 h at 320 °C/15 MPa. The diffraction peaks are consistent with the peaks of FeCr2O4 (JCPDS 24-0511) and Fe3O4 (JCPDS 28-0491). However, it is not possible to distinguish the oxides of magnetite (Fe3O4) and spinel (FeCr2O4) because of the overlap of their diffraction peaks, which are located at 35.764°, 43.472° and 74.760° two-theta (Ref 26, 27). In addition to FeCr2O4/Fe3O4, hematite Fe2O3 (JCPDS 16-0653) and chromic oxide Cr2O3 (JCPDS 01-1294) are formed on the surface, and the signal intensity of hematite Fe2O3 increases with increasing exposure time. These results indicate that the main corrosion phase formed on the surface of untreated samples is magnetite or spinel, which is consistent with previous results for 316LSS exposed to high-temperature water (Ref 28, 29).

XRD patterns of 316LSS LPT after exposure to high-temperature water for different periods
Figure 6 displays the XRD patterns of LP 316LSS after exposure to high-temperature water for 100, 500, and 1000 h. There is a slight difference in corrosion phases compared to untreated samples, as shown in Fig. 5, which suggests that the pre-oxidation treatment does not significant affect the formation of oxides.

XRD patterns of LP 316LSS after exposure to high-temperature water for different periods
Figure 7 shows the SEM morphologies of the oxide films formed on 316LSS without LPT after exposure to high-temperature water for different lengths of time. The surfaces of all the exposed samples are covered with faceted oxide particles of different sizes. The distribution of these particles is inhomogeneous, where the larger Fe-rich oxide particles disperse on the surface and small oxide particles appear in the subsurface layer, as shown in Fig. 7(d), (e) and (f). Moreover, the density and size of these oxide particles gradually increase with increasing exposure time, which can be found elsewhere (Ref 9, 30). The SEM images of the oxide films formed on the surface of LP samples after different exposure periods in high-temperature water are shown in Fig. 8. As observed, there is a difference in corrosion morphologies of pre-oxidized samples compared with that of samples without LPT. A few Fe-rich faceted oxide particles with a large size are dispersed on the surface, and many oxide particles with nanometer sizes are present in a subsurface layer, as shown in Fig. 8(d), (e) and (f). Moreover, the size and density of the oxide particles significantly decrease for specimens with LPT.

SEM morphologies of the oxide films formed on the surface of samples without LPT after exposure to high-temperature water for different periods: (a) and (d) for 100 h; (b) and (e) for 500 h; (c) and (f) for 1000 h

SEM images of oxide films formed on the surface of LP samples after exposure to high-temperature water for different periods: (a) and (d) for 100 h; (b) and (e) for 500 h; (c) and (f) for 1000 h
Figure 9 shows the cross-sectional morphology of the oxide film that formed on the surface of 316LSS without LPT after 1000 h of exposure. The oxide film consists of a double-layer structure. The chemical composition of the outer layer (position I) obtained from EDS-TEM is 34.54% for Fe, 3.71% for Ni, and 1.03% for Cr (at.%). The electron diffraction pattern and chemical composition demonstrate that the outer layer is (Ni,Fe)Fe2O4 with a spinel structure. Meanwhile, the inner layer (position II) that consists of fine spinel oxides (FexCryNizO4) is also identified using the same methods. The thickness of the oxide film is approximately 372.2 nm, as determined by the EDS line scanning profile and mapping images, as shown in Fig. 10.

TEM image and corresponding electron diffraction patterns of oxide film formed on untreated 316LSS after exposure to high-temperature water for 1000 h at 320 °C

TEM-EDS mapping images and EDS line scanning profile of oxide film formed on untreated 316LSS after exposure to high-temperature water for 1000 h
Figure 11 illustrates the XPS spectra of Cr 2p3/2, Fe 2p3/2, Ni 2p3/2, and O 1s in oxide films from the free surface to the deeper site of the LP-100 sample. Possible species in oxide films are assessed based on the characterized standards from references (Ref 9, 24, 31,32,33,34,35), as shown in Table 2. For the Cr 2p3/2 peaks, two components at 575.9 and 576.9 eV are identified on the sample surface, while they are systematically decomposed into up to three components with an increasing sputter depth: one at a binding energy (BE) of 574.3 eV, and the two others at BEs of 576.1 and 577.3 eV, which indicates that the appearance of different valences for Cr appears beneath the surface layer. The peak at the lowest BE of 574.3 eV is assigned to Cr0, the peak at 576.1 eV is assigned to Cr3+ in Cr2O3/MCr2O4 (Ref 9, 36), and the peak at 577.3 eV is assigned to Cr3+ in Cr(OH)3 (Ref 33). By increasing the sputtering time, the signal intensity of Cr(OH)3 tends to decrease, while the signal intensity first increases and subsequently decreases for Cr2O3/MCr2O4. When the sputtering time further increases, the intensity of Cr0 becomes predominant and the intensity of Cr(OH)3 gradually disappears, which suggests that Cr(OH)3 and Cr2O3/MCr2O4 appear at the outer and inner layers of the oxide films, respectively.

The XPS spectra of Cr 2p3/2, Fe 2p3/2, Ni 2p3/2, O1s in oxide films from the free surface to the deeper site of pre-oxidized 316LSS after exposure to high-temperature water for 100 h
For the Fe 2p3/2 peaks, two peaks were present on the sample surface, including a peak at 710.2 eV assigned to Fe3+ in Fe2O3 or Fe3O4 and a peak at 712. 3 eV assigned to Fe3+ in FeOOH (Ref 32, 34, 35). However, it is difficult to distinguish Fe2O3 or Fe3O4 because they have very similar peak positions and shapes (Ref 36), presenting a mixture of Fe2O3 and Fe3O4. The observation that the intensity of the FeOOH peak gradually weakens with increasing sputtering depth, which indicates that FeOOH is present in the outer layer of oxide films.
Regarding the XPS spectra of Ni 2p3/2, no significant peak is identified on the surface of the specimen, representing the absence of Ni-rich oxides in the surface oxide film. However, two peaks are detected with an increasing sputter depth: A peak at 852.8 eV assigned to Ni0, and a peak at 854.1 eV assigned to Ni2+ in NiO (Ref 35, 36), which reveals the formation of a Ni-rich oxide layer beneath the surface oxide layer (Ref 37).
The O 1s peaks in Fig. 11 show that an OH- peak at 531.5 eV and an O2- peak at 530.5 eV are identified on the sample surface (Ref 33, 36). The presence of OH- indicates the existence of Fe and Cr hydroxides, mostly in the outer layer of the oxide films. With a further increase in sputter depth, the signal intensity of O2- gradually strengthens, and OH- gradually weakens. This result implies that the surface of the pre-oxidized sample after exposure to high-temperature water is dominated by hydroxides, while the oxide film under surface oxides consists of a mixture of different oxides.
Table 2 summarizes the binding energies of XPS-peaks of Cr 2p3/2, Fe 2p3/2, Ni 2p3/2, and O 1s in oxide films from the free surface to the deeper site of the LP-1000 sample. No notable difference is observed in the chemical valences of Cr, Ni, and O compared with those of the LP-100 sample. Regarding the Fe peaks, a single peak at 711. 6 eV assigned to Fe3+ in FeOOH is observed, revealing that the outer oxide layer is dominated by hydroxide instead of a mixture of hydroxide and oxides.
The XPS depth profiles of the oxide films developed on LP-100 and LP-1000 h are shown in Fig. 12. A duplex structure is observed, which includes a Cr-rich outer oxide layer and an Fe-rich inner oxide layer. This duplex structure is quite different from the chemical composition of the oxide film formed on the untreated sample, as shown in Fig. 9. In addition, the results also reveal that the thickness of the oxide films developed on the LP-100 and LP-1000 sample are 34.58 and 162.9 nm, respectively, which is calculated by the oxygen atom concentration dropping to 50% of its maximum value (Ref 38). The oxide film on pre-oxidized 316LSS is significantly thinner than the sample without a pre-oxidation treatment. This result also shows that pre-oxidation treatment can inhibit the growth of oxide films and improve the corrosion resistance.

The XPS depth profiles of oxide layers of samples with LPT after 100 and 1000 h exposure tests
Schematic diagrams of the oxide film structure developed on LP 316LSS are shown in Fig. 13. Cr-rich and Fe-rich oxide particles on a Cr-rich continuous layer are formed after the pre-oxidation treatment in 400 °C air for 1000 h, as shown in Fig. 13(b). The pre-existing oxides will be partially dissolved at the initial stage, whereas the remaining oxides will gradually transform to Fe3O4, Cr(OH)3, and FeOOH through a series of chemical reactions (Ref 33), which forms a hydroxide layer on the surface of the samples, as shown in Fig. 13(c). Moreover, the metal cations from the matrix, including Fe2+, Fe3+, Cr3+, and Ni2+, will diffuse to the interface of water and oxide films to form oxides via an interstitial diffusion mechanism, and therefore, Fe-rich oxides (FeCr2O4) will develop on the surface first, as shown in Fig. 13(d).

Schematic diagrams of the structure of the oxide films developed on LP 316LSS
4 Discussion
It is generally accepted that a double-layered oxide film is formed on the surface of 316LSS after immersion in a simulated PWRs primary water environment, whereby the outer oxide layer constitutes Fe-rich magnetite grains and the inner oxide layer is composed of small crystallites enriched in Cr (Ref 34, 39). Moreover, Ni is enriched in a substrate metal adjacent to the inner oxide layer (Ref 29, 30, 37). The current findings are in line with a previous investigation on the corrosion behavior of 316LSS without LPT in a simulated PWR environment (Ref 9). A comparison of the oxide film composition of samples with and without LPT indicates an absence of an Fe-rich outer layer formed on the surface of samples with pre-oxidation treatment, which is consistent with SEM images, as shown in Fig. 7 and 8. Moreover, the pre-oxidized samples have lower oxide film thickness and weight gain than the samples without pre-oxidation, as shown in Fig. 4, 9, and 12. All of these results indicate that LPT improves the corrosion resistance of 316LSS by hindering the growth of the outer oxide layer. Our previous investigation on the oxidation behavior of 316LSS exposed to 400 °C air for 1000 h displayed that a continuous Cr-rich oxide layer was formed on the surface of the sample, and that this film is thicker than the untreated sample (Ref 40). A pre-existing oxide film with a high Cr concentration will effectively suppress the diffusion of anions and cations across oxide films (Ref 40, 41) and then enhance the corrosion resistance of 316LSS in high-temperature water.
In addition to hindering the diffusion of anions and cations, the appearance of an oxide film will alter the electrochemical characteristics of 316LSS in high-temperature water and subsequently improve the corrosion resistance (Ref 42). It can be inferred that the icorr, icrit, and ipass of the pre-oxidized 316LSS will be reduced compared with those of samples without pre-oxidation because the existence of a Cr-rich oxide film will prevent the combination and migration of interstitial atoms and vacancies, according to the point defect model (Ref 43, 44). The reduction in the electrochemical parameters enhances the corrosion resistance of pre-oxidized 316LSS in high-temperature water.
The growth mechanism of the oxide films on 316LSS in high-temperature water is believed to include metal dissolution/oxide precipitation for the outer oxide layer and solid-state growth for the inner oxide layer (Ref 36, 45). For samples without surface treatment, each element in 316LSS forms corresponding oxides at the initial stage, e.g., Fe for Fe2O3 or Fe3O4, Cr for Cr2O3, and Ni for NiO (Ref 28). After the nucleation and growth of these oxides, a double-oxide layer that contained an Fe-rich outer layer and a Cr-rich inner layer is formed via elemental diffusion, i.e., the anions gradually diffuse inward and metal cations gradually diffuse outward (Ref 37). However, for the pre-oxidized samples, the continuous Cr-rich oxide film on the surface reduces the rate of diffusion for both cations and anions, restricting the formation of Fe-rich oxides. Therefore, only a few Fe-rich oxide particles with larger sizes are formed for pre-oxidized 316LSS after 100 h of exposure. With an increasing exposure period, the number of cations that diffuse to the interface between the water and oxide film gradually increases, especially for Fe2+/Fe3+, leading to an increase in the density and size of Fe-rich oxides. However, the total number of diffused ions will be significantly lower than that of the samples without pre-oxidation treatment due to the hindering effect of the Cr-rich oxide layer, which will reduce the size and number of Fe-rich oxides on the surface of the pre-oxidized samples. Thus, oxide particles with small sizes are observed on the pre-oxidized samples even if exposure periods extend to 1000 h, as shown in Fig. 8(c) and (f). In addition, the increase in number of Fe ions that diffuse to the interface will make Cr and Ni elements accumulate beneath the Cr-rich oxide layer. However, notably, most of the Cr elements on the surface have been consumed to form the Cr-rich oxide film during the pre-oxidation process, which leaves an Fe-rich oxide layer under this oxide film. The Cr-rich oxide film reduces the diffusion rate of Fe ions, resulting in the accumulation of Fe, as shown in Fig. 13(d). These two reasons cause the formation of an Fe-rich oxide layer under the Cr-rich oxide film. This oxide film gradually thickens with an increasing exposure period, as shown in Fig. 12 and 13.
5 Conclusions
The corrosion behavior of LP 316LSS in 320 °C and 15 MPa high-temperature water was investigated by using different characterization methods. The following conclusions are reached:
-
(1)
The weight gain of pre-oxidized samples is lower than that of samples without LPT. A double-layered oxide film, including an outer oxide particle layer consisting of (Ni,Fe)Fe2O4 with a spinel structure and an inner oxide layer consisting of fine spinel oxides (FexCryNizO4), was developed on samples without PLT after 1000 h of exposure in high-temperature water. The thickness of the oxide film was approximately 372.2 nm.
-
(2)
A few Fe-rich faceted oxide particles with a large size on the surface and many oxide particles with nanometer sizes in the subsurface layer were developed on the LP samples after exposure to high-temperature water. The density and size of these oxide particles increased with increasing exposure periods.
-
(3)
A triple layer, including a hydroxide outer layer, a Cr-rich middle layer, and an Fe-rich inner layer, was developed on pre-oxidized samples after 100 and 1000 h of exposure. The thickness of the oxide film after 100 h of exposure is 34.58 nm, whereas it is 162.9 nm after 1000 h. In the comparison of the thickness of the oxide films and weight gain, it can be inferred that a LPT can improve the corrosion resistance of 316LSS in high-temperature water by inhibiting the formation of an outer oxide layer.
References
S.J. Zinkle and J.T. Busby, Structural Materials for Fission & Fusion Energy, Mater. Today, 2009, 12(11), p 12–19.
N. Karimi, F. Riffard, F. Rabaste, S. Perrier, R. Cueff, C. Issartel, and H. Buscail, Characterization of the Oxides Formed at 1000°C on the AISI 304 Stainless Steel by X-Ray Diffraction and Infrared Spectroscopy, Appl. Surf. Sci., 2008, 254(8), p 2292–2299.
S. Bush, Structural Materials for Nuclear Power Plants, J. Test. Eval., 1974, 2(6), p 435–462.
T. Allen, J. Busby, M. Meyer, and D. Petti, Materials Challenges for Nuclear Systems, Mater. Today, 2010, 13(12), p 14–23.
Y. Behnamian, A. Mostafaei, A. Kohandehghan, B.S. Amirkhiz, J. Li, W. Zheng, D. Guzonas, M. Chmielus, W. Chen, and J.L. Luo, Characterization of Oxide Layer and Micro-Crack Initiation in Alloy 316L Stainless Steel after 20,000 h Exposure to Supercritical Water at 500 °C, Mater. Charact., 2017, 131, p 532–543.
N.A. Tiner and C.B. Gilpin, Microprocesses in Stress Corrosion of Martensitic Steels, Corrosion, 1966, 22(10), p 271–279.
L. Chang, M.G. Burke, and F. Scenini, Stress Corrosion Crack Initiation in Machined Type 316L Austenitic Stainless Steel in Simulated Pressurized Water Reactor Primary Water, Corros. Sci., 2018, 138, p 54–65.
W.-I. Choi, G.D. Song, S.-H. Jeon, S.J. Kim, and D.H. Hur, Magnetite-Accelerated Stress Corrosion Cracking of Alloy 600 in Water Containing 100 Ppm Lead Oxide at 315 °C, J. Nucl. Mater., 2019, 522, p 54–63.
G. Han, Z. Lu, X. Ru, J. Chen, Q. Xiao, and Y. Tian, Improving the Oxidation Resistance of 316L Stainless Steel in Simulated Pressurized Water Reactor Primary Water by Electropolishing Treatment, J. Nucl. Mater., 2015, 467, p 194–204.
M.J. Seo, H.-S. Shim, K.M. Kim, S.-I. Hong, and D.H. Hur, Influence of Surface Roughness on the Corrosion Behavior of Alloy 690TT in PWR Primary Water, Nucl. Eng. Des., 2014, 280, p 62–68.
S. Wang, Y. Hu, K. Fang, W. Zhang, and X. Wang, Effect of Surface Machining on the Corrosion Behaviour of 316 Austenitic Stainless Steel in Simulated PWR Water, Corros. Sci., 2017, 126, p 104–120.
W. Schulz, M. Nofz, M. Feigl, I. Dörfel, R. Saliwan Neumann, and A. Kranzmann, Corrosion of Uncoated and Alumina Coated Steel X20CrMoV12-1 in H2O–CO2–O2 and Air at 600°C, Corros. Sci., 2013, 68, p 44–50.
A. Radhamani, H.C. Lau, and S. Ramakrishna, Nanocomposite Coatings on Steel for Enhancing the Corrosion Resistance: a Review, J. Compos. Mater., 2020, 54(5), p 681–701.
B.N. Mordyuk, S.M. Voloshko, V.I. Zakiev, A.P. Burmak, and V.V. Mohylko, Enhanced Resistance of Ti6Al4V Alloy to High-Temperature Oxidation and Corrosion by Forming Alumina Composite Coating, J. Mater. Eng. Perform., 2021, 30(3), p 1780–1795.
U. Trdan, M. Hočevar, and P. Gregorčič, Transition from Superhydrophilic to Superhydrophobic State of Laser Textured Stainless Steel Surface and Its Effect on Corrosion Resistance, Corros. Sci., 2017, 123, p 21–26.
H. Buscail, S. El Messki, F. Riffard, S. Perrier, and C. Issartel, Effect of Pre-Oxidation at 800 °C on the Pitting Corrosion Resistance of the AISI 316L Stainless Steel, Oxid. Met., 2011, 75(1–2), p 27–39.
M. Sun, X. Wu, Z. Zhang, and E.-H. Han, Oxidation of 316 Stainless Steel in Supercritical Water, Corros. Sci., 2009, 51(5), p 1069–1072.
Z.R. Xu, W. Zhu, and S.H. Htar, Partial Oxidative Gasification of Municipal Sludge in Subcritical and Supercritical Water, Environ. Technol., 2012, 33(11), p 1217–1223.
Y. Behnamian, A. Mostafaei, A. Kohandehghan, B.S. Amirkhiz, D. Serate, Y. Sun, S. Liu, E. Aghaie, Y. Zeng, M. Chmielus, W. Zheng, D. Guzonas, W. Chen, and J.L. Luo, A Comparative Study of Oxide Scales Grown on Stainless Steel and Nickel-Based Superalloys in Ultra-High Temperature Supercritical Water at 800 °C, Corros. Sci., 2016, 106, p 188–207.
Y. Behnamian, A. Mostafaei, A. Kohandehghan, B. Zahiri, W. Zheng, D. Guzonas, M. Chmielus, W. Chen, and J.L. Luo, Corrosion Behavior of Alloy 316L Stainless Steel after Exposure to Supercritical Water at 500 °C for 20,000 h, J. Supercrit. Fluids, 2017, 127, p 191–199.
Y.-C. Liu, S.-M. Chen, F.-Y. Ouyang, and J.-J. Kai, Corrosion Behavior of Pre-Oxidized HR-224 Superalloy in Supercritical Water Environment at 700 °C, J. Nucl. Mater., 2018, 505, p 7–14.
Z. Wang, E.-M. Paschalidou, A. Seyeux, S. Zanna, V. Maurice, and P. Marcus, Mechanisms of Cr and Mo Enrichments in the Passive Oxide Film on 316L Austenitic Stainless Steel, Front. Mater., 2019, 6, p 232.
L. Ma, F. Wiame, V. Maurice, and P. Marcus, New Insight on Early Oxidation Stages of Austenitic Stainless Steel from in Situ XPS Analysis on Single-Crystalline Fe–18Cr–13Ni, Corros. Sci., 2018, 140, p 205–216.
T. Yamashita and P. Hayes, Analysis of XPS Spectra of Fe2+ and Fe3+ Ions in Oxide Materials, Appl. Surf. Sci., 2008, 254(8), p 2441–2449.
M.C. Biesinger, B.P. Payne, A.P. Grosvenor, L.W.M. Lau, A.R. Gerson, and R.S.C. Smart, Resolving Surface Chemical States in XPS Analysis of First Row Transition Metals, Oxides and Hydroxides: Cr, Mn, Fe, Co and Ni, Appl. Surf. Sci., 2011, 257(7), p 2717–2730.
D. Laverde, T. Gómez-Acebo, and F. Castro, Continuous and Cyclic Oxidation of T91 Ferritic Steel under Steam, Corros. Sci., 2004, 46(3), p 613–631.
K. Yin, S. Qiu, R. Tang, Q. Zhang, and L. Zhang, Corrosion Behavior of Ferritic/Martensitic Steel P92 in Supercritical Water, J. Supercrit. Fluids, 2009, 50(3), p 235–239.
T. Terachi, K. Fujii, and K. Arioka, Microstructural Characterization of SCC Crack Tip and Oxide Film for SUS 316 Stainless Steel in Simulated PWR Primary Water at 320°C, J. Nucl. Sci. Technol., 2005, 42(2), p 225–232.
T. Terachi, T. Yamada, T. Miyamoto, K. Arioka, and K. Fukuya, Corrosion Behavior of Stainless Steels in Simulated PWR Primary Water—Effect of Chromium Content in Alloys and Dissolved Hydrogen—, J. Nucl. Sci. Technol., 2008, 45(10), p 975–984.
M. da Cunha Belo, M. Walls, N.E. Hakiki, J. Corset, E. Picquenard, G. Sagon, and D. Noël, Composition, Structure and Properties of the Oxide Films Formed on the Stainless Steel 316L in a Primary Type PWR Environment, Corros. Sci., 1998, 40(2–3), p 447–463.
F. Ning, X. Wu, and J. Tan, Crevice Corrosion Behavior of Alloy 690 in High-Temperature Water, J. Nucl. Mater., 2019, 515, p 326–337.
A. Machet, A. Galtayries, P. Marcus, P. Combrade, P. Jolivet, and P. Scott, XPS Study of Oxides Formed on Nickel-Base Alloys in High-Temperature and High-Pressure Water, Surf. Interface Anal., 2002, 34(1), p 197–200.
Z. Duan, F. Arjmand, L. Zhang, and H. Abe, Investigation of the Corrosion Behavior of 304L and 316L Stainless Steels at High-Temperature Borated and Lithiated Water, J. Nucl. Sci. Technol., 2016, 53(9), p 1435–1446.
Y. Han, J. Mei, Q. Peng, E.-H. Han, and W. Ke, Effect of Electropolishing on Corrosion of Nuclear Grade 316L Stainless Steel in Deaerated High Temperature Water, Corros. Sci., 2016, 112, p 625–634.
L. Marchetti, F. Miserque, S. Perrin, and M. Pijolat, XPS Study of Ni-base Alloys Oxide Films Formed in Primary Conditions of Pressurized Water Reactor, Surf. Interface Anal., 2015, 47(5), p 632–642.
Y. Guo, E.-H. Han, and J. Wang, Effects of Forging and Heat Treatments on the Microstructure and Oxidation Behavior of 316LN Stainless Steel in High Temperature Water, J. Mater. Sci. Technol., 2015, 31(4), p 403–412.
R. Soulas, M. Cheynet, E. Rauch, T. Neisius, L. Legras, C. Domain, and Y. Brechet, TEM Investigations of the Oxide Layers Formed on a 316L Alloy in Simulated PWR Environment, J. Mater. Sci., 2013, 48(7), p 2861–2871.
Y. Gui, X.B. Meng, Z.J. Zheng, and Y. Gao, Critical Temperature Determination of Detectable Cr Diffusion Enhancement by Nanostructure through Structural Evolution Analysis of the Oxide Films at 25–450 °C on 304 Stainless Steel, Appl. Surf. Sci., 2017, 419, p 512–521.
J. Chen, Q. Xiao, Z. Lu, X. Ru, H. Peng, Q. Xiong, and H. Li, Characterization of Interfacial Reactions and Oxide Films on 316L Stainless Steel in Various Simulated PWR Primary Water Environments, J. Nucl. Mater., 2017, 489, p 137–149.
X. Huang, K. Xiao, X. Fang, Z. Xiong, L. Wei, P. Zhu, and X. Li, Oxidation Behavior of 316L Austenitic Stainless Steel in High Temperature Air with Long-Term Exposure, Mater. Res. Express, 2020, 7(6), p 066517.
F. Abe, H. Kutsumi, H. Haruyama, and H. Okubo, Improvement of Oxidation Resistance of 9 Mass% Chromium Steel for Advanced-Ultra Supercritical Power Plant Boilers by Pre-Oxidation Treatment, Corros. Sci., 2017, 114, p 1–9.
K.S. Raja and T. Shoji, Effect of Pre-Oxidation and Corrosion Potential on Electronic Properties of Passive Films on Ni-Cr-Fe Alloys in Pure Water at 288 °C, J. Mater. Sci., 2004, 39(3), p 1033–1036.
C.Y. Chao, L.F. Lin, and D.D. Macdonald, A Point Defect Model for Anodic Passive Films: I. Film Growth Kinetics, J. Electrochem. Soc., 1981, 128(6), p 1187–1194.
L.F. Lin, C.Y. Chao, and D.D. Macdonald, A Point Defect Model for Anodic Passive Films: II. Chemical Breakdown and Pit Initiation, J. Electrochem. Soc., 1981, 128(6), p 1194–1198.
B. Stellwag, The Mechanism of Oxide Film Formation on Austenitic Stainless Steels in High Temperature Water, Corros. Sci., 1998, 40(2–3), p 337–370.
Acknowledgments
This investigation was supported by Jiangxi Provincial Natural Science (20202BABL211014), Foundation of Jiangxi Educational Committee (GJJ180401), Jiangxi Key Laboratory for Mass Spectrometry and Instrumentation (JXMS2017017), and Doctoral Scientific Research Foundation of East China University of Technology (DHBK2017127).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Huang, X., Li, X., Zhan, Z. et al. Effect of Long-Term Pre-oxidation on the Corrosion Rate of 316L Stainless Steel in a High-Temperature Water Environment. J. of Materi Eng and Perform 31, 7935–7944 (2022). https://doi.org/10.1007/s11665-022-06878-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11665-022-06878-2