
Abstract
The time-dependent experiment was performed to investigate the corrosion behavior of low-temperature liquid nitrocarburized (LNC) 304 austenitic stainless steel in wet H2S environments. Characteristics of H2S corrosion products, as well as localized corrosion behavior, were investigated using X-ray diffraction (XRD), scanning electron microscopy, X-ray photoelectron spectroscopy (XPS), and optical profilometry. The results revealed that the untreated steels, of which the H2S corrosion product layer on the untreated surface thickened but displayed a layered defect structure as the corrosion proceeded, had a higher weight loss than the LNC. Energy-dispersive spectroscopy (EDS) served to reveal the relationship between corrosion behavior and the content of sulfur, chromium, and other elements, and the valences of these elements were illustrated by XPS. The surface morphology after removing corrosion products showed that the presence of nitrocarburized S phase could prevent general corrosion and inhibit pit propagation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
To meet the requirements for energy, a large amount of methane and other hydrocarbons is conveyed from acidic oil and gas fields. Such oil and gas fields generally contain various corrosive media, such as H2S and Cl−, which are harsh on the environment and can easily cause severe corrosion of the pipelines. The high concentrations of H2S promote ionized hydrogen into pipeline steel, thus increasing the possibility of failure related to sulfide stress cracking and hydrogen embrittlement (HE).[1,2,3] Austenitic stainless steel has been expected to be the structural material used in modern oil-gas fields due to its good corrosion resistance and formability. During the transportation of oil and gas, pipeline steel is susceptible to erosion-corrosion owing to the existence of various phases of the gas phase, liquid phase, and solid phase (sand).[4] The impact of solid particles included in the oil and gas on the pipe wall can accelerate the mass-transfer process and promote the depolarization agent (such as oxygen) to reach the surface of the material.[5,6,7] This process scrapes off the passive film, causing the H2S corrosion product film to detach from the steel, thereby accelerating the erosion process. Austenitic stainless steel, however, has lower hardness and poor friction properties, thus limiting its application in the oil and gas extraction industry.
To overcome the preceding limitations of austenitic stainless steel and broaden its application range, as well as to respond to the needs of the petrochemical industry, some surface treatment processes have emerged.[8,9] Low-temperature gas carburizing[10] and nitriding[11] are well-established processes for surface hardening. Direct current plasma carburizing has been indicated to be beneficial to prevent surface plastic deformation in the dry rolling-sliding wear tests[12] on the basis of ensuring primitive corrosion resistance.[13] However, electrochemical as well as wear-corrosion testing showed that the corrosion resistance of high-temperature ion nitriding stainless steel was significantly decreased.[14] The high-temperature ion nitriding treatment merely enhanced the corrosion resistance of the active region, but the self-corrosion potential was not influenced.[15] As for erosion-corrosion behavior, low-temperature nitriding increased the corrosion resistance of austenitic stainless steel by more than 80 pct.[16] Hydrogen-induced cracking of stainless steel often occurs in the oil and natural gas exploitation environment; that is, H2S decomposes into atomic hydrogen on the metal surface. Hydrogen atoms enter the metal and play a decisive role in the fracture.[17] Wei et al. found that 304 stainless steel treated by ion carburizing could effectively reduce the tendency of HE.[18] In the case of liquid nitriding, up to 15 at. pct carbon atoms and 20 at. pct nitrogen atoms enter and remain in the austenite lattice,[10] which is about 105 times that of the interstitial atoms in the solid solution equilibrium state, therefore forming expanded austenite (also called the S phase). Xu et al. observed masses of stacking faults and twins in low-temperature plasma nitrocarburized 304 austenitic stainless steel.[19] In our previous studies,[20] a large number of entangled dislocations, stacking faults, and twins existed in austenitic stainless steel after liquid nitriding for 8 hours at 703 K (430 °C). These defects are most likely caused by stress resulting from supersaturated nitrogen and carbon atoms. The entanglement of immovable dislocations is a favorable trap for capturing hydrogen atoms,[21] which can provide a large number of locations for the distribution of hydrogen atoms, helping to avoid high hydrogen atom enrichment in localized regions and microregional HE.
Nonetheless, the effect of nitridation on the corrosion resistance of stainless steel in the H2S-containing environment has not been researched in depth. Low-temperature liquid nitrocarburizing is a valid technique employed to improve tribological properties as well as guarantee the original corrosion resistance of austenitic stainless steel,[22] duplex stainless steel,[23] precipitation hardening stainless steel,[22] and die steel.[24] The oxide layer formed outside of the nitrocarburized layer can reduce the diffusion coefficient of hydrogen in austenitic steel to prevent HE at 25 °C in the H2S environment.[25]
The corrosion behavior of LNC materials in saturated H2S environments at different reaction times, however, has not been revealed, which is related to the operational difficulties caused by the highly toxic H2S gas. Besides, the invalid mechanisms of the nitrocarburized composite layer and their respective roles in the anticorrosion of H2S are still not well studied. In the present work, the corrosion phenomena of untreated and LNC 304 austenitic stainless steel corrosion in saturated H2S solution were studied to reveal the failure process after immersion for 4, 24, 72, 168, 480, and 1080 hours. Surface characterization techniques were used to analyze the formation and evolution mechanisms of H2S corrosion products formed on austenitic stainless steel. Moreover, the failure mechanism of the nitrogen-rich layer and the carbon-rich layer was discussed.
2 Experimental
2.1 Materials and Specimen Preparation
The materials were provided by PetroChina Southwest Oil and Gas Field Company, and the chemical compositions of 304 austenitic stainless steel were as follows (in wt pct): C, 0.035; Cr, 18.640; Ni, 8.010; Mn, 1.100; Si, 0.436; P, 0.013; S, 0.020; and Fe balance. Each specimen was machined with a size of 7.24 cm × 1.15 cm × 0.2 cm. Each specimen was wet ground with 600-, 1000-, 1500-, and 2000-grit silicon carbide sandpaper in sequence. Afterward, these specimens were cleaned first with distilled water and then with absolute ethyl alcohol. After rapid drying by air, each specimen was weighed and recorded using an FR-300MKII analytical balance.
2.2 Low-Temperature Liquid Nitrocarburizing and H2S Corrosion Tests
The raw materials used in the low-temperature liquid nitrocarburizing process were cyanate, chloride, and carbonate.[8] Their specific compositions were as follows (wt pct): 15 to 50 potassium cyanate, 10 to 40 sodium cyanate, 10 to 20 potassium carbonate, 10 to 20 sodium carbonate, 5 to 10 lithium carbonate, 3 to 25 sodium chloride, and 3 to 25 potassium chloride. All reagents were of analytical grade. For gas nitriding/carburizing, the typical oxide film formed on austenitic stainless steel alloys was quite resistant to nitrogen/carbon permeation. As for LNC treatment, there was no need to add processes to remove the oxide film of the 304 stainless steel because the nitrocarburizing salt with strong reducibility can dissolve it.[20] Stainless steel was immersed in molten salt for liquid nitrocarburization at 430 °C for 8 hours. Then, the specimens were taken out and cooled to room temperature. Finally, the specimens were ultrasonically cleaned with absolute ethyl alcohol for 20 minutes. When the heating temperature reaches the melting point, molten salt reacts and decomposes to produce carbon and nitrogen atoms, resulting in a higher atomic concentration on the surface of stainless steel. Under the action of the concentration gradient, the carbon/nitrogen atoms enter the stainless steel, thus forming expanded austenite (S phase). The details of the LNC treatment were presented in a previous report.[26]
The NACE A solution used in the immersion corrosion experiment was composed of 5 wt pct sodium chloride, 0.5 wt pct acetic acid, and deionized water. N2 or H2S gas passed through the order from left to right at a specific rate. After passing through the antipump bottle, it entered the container where the specimens were fixed. The H2S gas then entered the absorption bottle and was absorbed by the 10 wt pct NaOH solution. After sealing the device, the prepared NACE A solution was slowly poured into a sealed test vessel through a funnel to soak the specimen in the NACE A solution completely. Then, the solution was deoxidized with high-purity nitrogen gas for 3 hours. After that, pure H2S gas was bubbled into the solution for 60 minutes to ensure that H2S in the NACE A solution has been saturated. During the immersion corrosion process, pure H2S gas was continuously introduced into the container. All immersion corrosion tests were carried out at 60 °C for 4, 24, 72, 168, 480, and 1080 hours, respectively. A temperature of 60 °C is common during oil and gas field equipment operation,[27,28] and a large amount of H2S corrosion research has been carried out at this temperature.[1,2] After the corrosion test, the specimens were taken out from the vessel, cleaned successively with deionized water and ethanol, and then dried by air. The schematic diagram of the entire experimental process is illustrated in Figure 1.

Schematic illustration showing the flow graph of liquid nitrocarburizing and the H2S corrosion test
2.3 Characterization Methods
The cross-sectional morphology of the liquid nitrocarburized as well as untreated specimens was shown after etching by a mixture of 50 vol pct hydrochloric acid, 25 vol pct nitric acid, and 25 vol pct deionized water. The surface and cross-sectional morphologies were observed under a scanning electron microscope (JSM-5900LV, Japan) with the following parameters: 15 kV, 500 to 2000 times magnification. The X-ray diffraction (XRD) instrument used for phase analysis of H2S corrosion products was a Philips X’Pert 1 diffractometer (Netherlands) with Co target; the scan angle was from 40 to 110 deg at a scan step size of 0.026 deg and time per step of 13.52 seconds. The X-ray photoelectron spectroscopy (XPS) instrument used was XSAM 800, and Al Kα was used as an X-ray source with an acceleration voltage of 15 kV and an emission current of 10 mA.
According to the ASTM G1-03 standard,[29] a chemical cleaning solution composed of 500 mL hydrochloric acid, 500 mL distilled water, and 3.5 g C6H12N4 was prepared to remove corrosion products. H2S corroded austenitic stainless steels were immersed in the solution at room temperature for 10 minutes. Afterward, the specimen was cleaned with deionized water and ethanol, dried by air, and then weighed by analytical balance to calculate the corrosion rate. Three parallel specimens were set for each group under the same conditions. The calculation formula for the corrosion rate is as follows:
where v is the corrosion rate of the material in the H2S environment (mm/a), m0 is the mass of the specimen before corrosion (g), mt is the mass of the specimen after removing the corrosion products (g), S is the superficial area (cm2), ρ is the density of stainless steel (generally 7.9 g/cm3), and t is the H2S corrosion time (hours).
The surface profile after removal of the corrosion product was carried out using a ContourGT-K 3D optical microscope to obtain the relationship between the localized corrosion behavior and the immersion time. The pitting behavior of the material was reflected by averaging the three pits with the most serious pitting corrosion from observing the specimen surface.
3 Results and Discussion
3.1 Weight Loss After H2S Corrosion
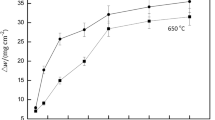
The relationship between the weight loss of austenitic stainless steel caused by H2S corrosion and the immersion time is shown in Figure 2(a). The weight loss of both the untreated and LNC materials increased over time: The growth tendency was evident when the soaking time was less than 72 hours, and then it decreased. It is worth noting that compared to the state at 480 hours, the weight loss due to immersion corrosion at 1080 hours did not show a significant increase; this was due to the inhibition of the corrosion course by H2S corrosion product film in the later stage.[30,31] For the LNC 304 stainless steel, the weight loss during each corrosion period was significantly lower than that of the untreated material under the same conditions.

(a) Weight loss and (b) corrosion rate of untreated and liquid nitrocarburized specimens after immersion in NACE solution A at 60 °C
Figure 2(b) illustrates the corrosion rate of untreated and liquid nitrocarburized 304 austenitic stainless steel after corrosion in H2S environments. Within 0 to 1080 hours of wet H2S immersion, the corrosion rate of the material basically decreased. At the initial stage of H2S corrosion reaction, the corrosion rate of the material was relatively high. The iron ions dissolved rapidly because the exposed stainless steel substrate was in the state of activated corrosion and the corrosion rate was larger. The iron sulfide formed in the later corrosion stage deposited on the stainless steel surface, which has a positive protective effect on the below substrate and made the corrosion rate gradually decrease.
3.2 XRD Analysis
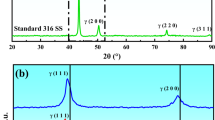
Figure 3 indicates the XRD pattern of untreated stainless steel after immersing in soaking H2S environments for different times. The austenite peaks only were detected after immersing in saturated H2S solution for both 4 and 24 hours and no sulfide peak was detected. After a 72-hour reaction, the characteristic peaks of marcasite (FeS2, 2θ = 43.51, 63.78, and 54.55 deg, PDF no. 37-0475) were detected on the surface of the specimen. The intensity of the peaks increased with a further extension of the immersion time. After the corrosion time was extended to 1080 hours, two diffraction peaks of Mackinawite (FeS, 2θ = 59.31 and 90.64 deg, PDF no. 15-0037) appeared.

XRD pattern of untreated specimens (a) before and (b) after immersing in NACE solution A at 60 °C for different times
The products formed on the untreated stainless steel in H2S environment are mainly composed of polymorphic iron-sulfide. Currently, seven kinds of sulfur-iron solid phases have been found, most of which exist in H2S environments.[32] The type of H2S corrosion product formed is dependent upon the environment to which the material is exposed. It is generally considered that mackinawite FeS is the primary product on the steel in saturated H2S solution,[33] while the most common H2S product in the working condition is amorphous ferrous sulfide, which is named for the fact that XRD cannot obtain the structural information of the material. Recently, some researchers have shown that amorphous FeS is essentially a nanocrystal of the mackinawite mineral. It was mistakenly defined as amorphous FeS because the nanocrystal of the mackinawite mineral is too small to detect the crystal structure by XRD.[34] This also explains the fact that no characteristic peak of sulfide is detected on the surface after immersion for both 4 and 24 hours in the solution, as shown in Figure 4.

XRD pattern of liquid nitrocarburized specimens after corrosion in NACE solution A at 60 °C for different times
The S phase is a supersaturated solid solution in which nitrogen atoms were introduced into the austenite lattice.[35] The (111) and (200) peaks of austenite shifted to lower angles with broader peak width due to the existence of interstitial atoms. An oxide film was introduced on the surface, which consisted of Fe3O4 (2θ = 41.36, 43.29, 67.19, and 74.08 deg, PDF no. 72-2303). The diffraction peaks of Fe3O4 maintained high intensity even after 1080 hours of immersion. This may be due to the fact that once the complete Fe3O4 layer with high corrosion resistance is broken, the corrosion continues inward through the breakage and, thus, no longer consumes Fe3O4.
In the first 24 hours of H2S corrosion, no characteristic peaks of sulfide were detected on the LNC surface. After 72 hours, the presence of pyrrhotite (Fe1–xS, 2θ = 43.88 and 62.63 deg, PDF no. 20-0534) was detected at the position of 2θ = 62.63 deg. The other diffraction peak of Fe1–xS almost overlaps with one of the characteristic peaks of Fe3O4. Zhang et al. reported that the corrosion product film formed by Fe1–xS has a relatively dense structure, which can effectively prevent the diffusion of hydrogen atoms into the matrix, thereby restraining HE.[36] At the same time, the film with Fe1–xS corrosion product has high electric resistance and prevents uniform corrosion of steel.[37] Previous results have shown that Fe1–xS was also produced on the nitriding stainless steel after 720 hours of corrosion in the NACE A solution at 25 °C.[8]
3.3 Surface Topography and Cross-Sectional Analysis
Figure 5 presents the corrosion products of untreated and liquid nitrocarburized materials after corrosion in saturated H2S environments at different times before removing corrosion products. The grain boundaries and corrosion pits in tens of microns can be observed on the untreated surface after immersion for 4 hours for untreated 304 materials. From the energy-dispersive spectroscopy (EDS) results in Figure 6, it was found that sulfur content reached 4.76 at. pct, which was related to sulfides formed on the untreated surface. It is known that sulfide existed in the form of nanocrystalline mackinawite FeS in the early stage of H2S corrosion.[34] As the immersion time extended to 24 hours, the corrosion pits grew in size in hundreds of microns, accompanied by sulfur content up to 5.42 at. pct on the entire surface. Meanwhile, corrosion developed inside the substrate, with granular austenite crystals lying below the loose corrosion product layer. As the immersion time increased to 72 hours, H2S corrosion spread continuously, which caused the entire surface to be covered with loose and cracked corrosion products. In the meantime, the S content was 19.01 at. pct. Local corrosion products fell off and then covered the stainless steel again after immersion for 168 and 480 hours, respectively. The brighter and small granular corrosion products were also embedded in the corrosion product layer. The corrosion products began to delaminate when the immersion time was further increased to 1080 hours. The inner layer was the cracked corrosion product layer, while the outer layer was clusterlike corrosion products developed from the previous granular corrosion products. Table I shows the elemental composition of H2S corrosion products of points A and B in Figure 5(k). Iron, chromium, and nickel are the substrate components of austenite stainless steel, while S exists in the sulfur-iron compound. The outer corrosion layer has a higher S content, but the inner corrosion layer has a higher Fe content. Combined with the results in Figure 3, it is evident that the weight loss increased rapidly over immersion time when the untreated material was immersed in a saturated H2S solution for a relatively short time (< 72 hours). When soaking for a relatively long time (> 72 hours), the rate of weight loss decreased. Bai investigated the polarization behavior of low-carbon steel immersed in H2S solution at the different times.[38] The research demonstrated that potential increased and polarization current decreased as the corrosion time extended, indicating that thicker product films help to inhibit further corrosion.

Surface morphology of untreated and liquid nitrocarburized specimens after immersion in NACE solution A at 60 °C for different times: (a) untreated, 4 h; (b) LNC, 4 h; (c) untreated, 24 h; (d) LNC, 24 h; (e) untreated, 72 h; (f) LNC, 72 h; (g) untreated, 168 h; (h) LNC, 168 h; (i) untreated, 480 h; (j) LNC, 480 h; (k) untreated, 1080 h; and (l) LNC, 1080 h. Images are acquired before removal of the corrosion product layer

Curves showing changes in Cr, S, and N elements on the surface of liquid nitrocarburized and untreated specimens with immersion time
In the case of LNC specimens, there was no significant change on the surface after immersion in saturated H2S solution for 24 hours. The significant intergranular corrosion occurred on the surface of the LNC specimen after 72 hours. The surface partially fell off, and the corrosion was intensified after 1080 hours of immersion.
The content of sulfur, chromium, and nitrogen of both the untreated and LNC specimens changed with immersion time. Figure 6 sketches these relationships by EDS. After 4 hours of immersion, the sulfur content of the untreated specimen surface reached 4.76 at. pct and further increased to 25.96 at. pct for 1080 hours. The total sulfur content of the LNC material was low, below 6 at. pct at each stage during H2S immersion corrosion, indicating that the content of surface corrosion products of the LNC treatment was low. After H2S immersion, the chromium content of the untreated specimen surface was less than 3 at. pct, indicating that the corrosion products contained almost no chromium. The surface chromium content of the LNC specimen was above 14 at. pct due to the enrichment of chromium in the S phase layer induced by low-temperature liquid nitrocarburizing. Chromium can increase the electrode potential of the material, thereby strengthening the corrosion resistance of stainless steel in corrosive media. The nitrogen content of the LNC specimen decreased continuously with the immersion time. After 1080 hours of exposure, the nitrogen content on the surface of the specimen decreased to 1.8 at. pct.
Figure 7 shows the cross-sectional microstructure of untreated and nitrocarburized specimens after immersing in H2S environments for different times. The cross section was etched after polishing for better observation of structure. After immersing for a short time, local pitting occurred on the untreated surface. The corrosion pitting spread to a larger size as the immersion time increased. For the LNC specimens, the overall nitrided S-phase layer was relatively complete at the initial 24 hours of H2S immersion corrosion. Corrosion pits appeared at the interface between the nitrogen-rich layer and the corrosion products after 168 hours. Obvious cracks appeared in the nitrogen-rich layer at the 480-hour period, which penetrated through the entire nitrogen-rich layer, while the carbon-rich layer was unbroken. When the immersion time reached 1080 hours, the cracking in the nitrogen-rich layer was more severe and breakage occurred locally. Zhang et al. studied the stress corrosion behavior of nitrocarburized stainless steel in the H2S solution.[8] Once the stress reaches 205 MPa and higher (246 MPa), pitting following cracking will occur in the S-phase layer when the applied tensile stress reaches the yield strength of the material.

Cross-sectional morphology of untreated and liquid nitrocarburized specimens after immersion in NACE solution A at 60 °C for different times: (a) untreated, 24 h; (b) LNC, 24 h; (c) untreated, 168 h; (d) LNC, 168 h; (e) untreated, 480 h; (f) LNC, 480 h; (g) untreated, 1080 h; and (h) LNC, 1080 h
3.4 XPS Analysis
Chromium is considered to be a key element in the formation of passive films. Figures 8(a) and (b) reveal the high-resolution XPS spectrum of Cr 2p3/2 obtained on the liquid nitrocarburized and the untreated stainless steel, both of which were immersed for 4, 24, and 72 hours in saturated H2S solution. In the case of the LNC specimens (Figure 8(a)), the chromium (Cr 2p3/2) contribution in the passive film can be represented by chromium oxides (Cr2O3) at 576.4 eV and chromium hydroxides (Cr(OH)3) at 577.3 eV. In the case of the untreated specimens, additional metallic Cr peaks were also detected on the surface in the early stages of H2S corrosion. The content of metallic Cr decreases gradually as the corrosion time is prolonged, which may be related to the increase of sulfide content on the untreated surface. The presence of metallic Cr indicates that the passive film is thinner and more susceptible to damage.[39]

Typical high-resolution Cr 2p3/2, S 2p3/2, Ni 2p3/2, and N 1s XPS spectra after immersion in NACE solution A at 60 °C for 4, 24, and 72 h: (a) LNC, Cr 2p3/2; (b) untreated, Cr 2p3/2; (c) LNC, S 2p3/2; (d) untreated, S 2p3/2; (e) LNC, Ni 2p3/2; (f) untreated, Ni 2p3/2; and (g) LNC, N 1s
In the point defect model,[40] it was pointed out that H2O molecules directly participate in the formation of passive films, which makes it possible to keep the stainless steel in passivation without O2. Since the H2S molecule has a chemical structure similar to that of H2O, HS– has a higher polarization ability than OH–, which makes it easily adsorbed and detected on the passive film. Sulfur peaks can be separated into three main contributions, as shown in Figures 8(c) and (d), the photoelectrons of which correspond to S2– and S22–. Some researchers have theoretically summarized the electrode reaction. In response to the anion-anode reaction of H2S in the corrosion process, they proposed the corresponding reaction mechanism. The anode reaction process mainly included chemical adsorption of gas, ion or dipole compound, desorption, oxidation, and hydrolysis reactions.[41,42]
The FeS generated by the preceding reaction has a strong binding force between the surface and substrate, and the corrosion of the material can be slowed because of its dense structure. If the FeS produced by the preceding reaction has a compact structure, it produces a strong binding force to the substrate; the corrosion of the material can be alleviated. If the formed FeS structure is loose and can easily fall off, as shown in Figure 7, an occlusion cell is built to accelerate local corrosion of the substrate. Figures 8(c) and (d) show the valence of sulfur in the passive film after H2S corrosion. For both untreated and LNC specimens, sulfur presents in the form of S22– (161.6 eV) and S2– (162.5 eV). Since the detection depth of XPS analysis is in the surface nanometer range, this indicates that there are many sulfides on the outer surface of the two materials in the first 72 hours of corrosion. It is worth noting that the S22– content in the untreated specimen is higher than that in the LNC specimen. King et al. studied the effects of different corrosion products on steel corrosion by using corrosion weight loss.[43] The results showed that FeS2 caused the most severe corrosion for steel, while Fe1–xS had little effect on the deterioration of materials because there is a difference in sulfur content between the preceding two corrosion products.
It is generally considered that the passive film has a small content of nickel.[44] The nickel is mainly concentrated at the bottom of the passive film and can avoid reduction in the oxide film, thereby improving the stability of the film. Some scholars[39] have detected the existence of NiO and Ni(OH)2 in stainless steel passive films after H2S corrosion. Laszczyńska et al. found that Ni(OH)2 was the major form of Ni in the passivation film by studying the passivation behavior of Ni-Mo alloy coating in 0.5 mol/L sodium chloride solution.[45] Recorded for the untreated and LNC materials subjected to saturated H2S environment, the valences coming from NiO (853.8 eV) and Ni(OH)2 (856.2 eV) were shown in Figures 8(e) and (f). With the prolongation of H2S soaking time, the content of NiO decreased slightly, while the content of Ni(OH)2 increased correspondingly.
Nitrogen peaks can be separated into three main contributions, as shown in Figure 8(g), and their photoelectrons correspond to NH3 and CrN. The reactive nitrogen within the nitrocarburized layer can raise the pH inside the corrosion pit and promote the repassivation of stainless steel. The reaction is as follows: [N] + 4H+ + 3e– → NH4+. CrN can also form a passive film by the following reaction: 2CrN + 3H2O → Cr2O3 + 2NH3 (ligands). It can be seen from Table II that the content of NH3 gradually decreases as the corrosion time increases.
3.5 Localized Corrosion Behavior
In addition to accentuating the formation of corrosion products in a saturated H2S solution, the presence of S phase changes the surface corrosion morphology by influencing H2S corrosion behavior. Figure 9 illustrates the three-dimensional surface profiles of untreated and LNC materials after removing H2S corrosion products. The micropits appear on the steel surface and their depths reached 9.2 μm after 4 hours of reaction (Figure 9(a)) for the untreated 304 materials. After 24 hours, the corrosion pit continued to expand to the extent of more than 10 μm. This process is considered to be the first stage of corrosion reaction. Uniform corrosion appeared after 72 hours (Figure 9(e)). Moreover, in the subsequent H2S corrosion, the surface of the untreated steel presented the mixed corrosion morphology of uniform corrosion and local corrosion. More bottomless small corrosion pits are located in the large corrosion pit (Figure 9(i)). These corrosion pits, which are 100-μm wide and 31.1-μm deep, are interconnected (Figure 9(k)). Corrosion pits with surface widths greater than depth show the characteristics of mesa corrosion.[53] With the further progress of corrosion, the pitting would transform into mesa corrosion. Mesa corrosion is a kind of severe corrosion frequently occurring in oil and gas fields,[54] which also leads to higher pipeline penetration rates. For liquid nitrocarburized materials, the size of the corrosion pit is small after reaction for a short time. After 480 hours of reaction (Figure 8(j)), the depth of the corrosion pits reached 10.8 μm and cracks in the nitrogen-rich layer lay under the corrosion pits (Figure 7(f)). After 1080 hours, the depth and surface size of the corrosion pits increased continuously (Figure 9(l)), while obvious uniform corrosion did not appear. To summarize, the existence of the highly protective S-phase layer influences the topography significantly and completely prevents general corrosion and also minimizes the extent of pit propagation.

Three-dimensional profiles showing untreated and LNC steel surfaces after immersion in NACE solution A at 60 °C for 4, 24, 72, 168, 480, and 1080 h: (a) untreated, 4 h; (b) LNC, 4 h; (c) untreated, 24 h; (d) LNC, 24 h; (e) untreated, 72 h; (f) LNC, 72 h; (g) untreated, 168 h; (h) LNC, 168 h; (i) untreated, 480 h; (j) LNC, 480 h; (k) untreated, 1080 h; and (l) LNC, 1080 h. Images are acquired after removal of the corrosion product layer
In the process of oil and gas production, pipeline perforation often occurs. The wall thickness at the maximum pitting depth is low, with poor mechanical properties and stress concentration, which makes the perforation more likely to happen in this position.[55] Figure 10 demonstrates the maximum pitting depth as a function of H2S corrosion time, which is roughly the same as the corrosion weight loss tendency in Figure 2. For the untreated materials, pitting depth increased rapidly when the corrosion time was less than 720 hours and increased slowly in the later stage of corrosion. For LNC materials, the initial increase in pitting depth was slow because the top dense oxide film formed by nitrocarburizing was resistant to H2S corrosion.[25] Once the oxide film failed, the pitting depth increased rapidly. After 480 hours of corrosion, the pitting depth expansion rate decreased again because the complete carbon-rich layer hindered its development, as illustrated in Figures 7(f) and (h). The relationship between pitting depth changes and corrosion time satisfies the Sigmoidal (Boltzmann) formula[56] as follows:

Maximum pitting depth of stainless steel after immersion in NACE solution A at 60 °C for different corrosion times
where h is the maximum pitting depth at different corrosion times (μm), h0 and hmax correspond to the initial and final pitting depth (μm), t0 is the time value corresponding to the median of the maximum pitting depth (hours), and ht is constant. The pitting depth in Figure 10 was fitted to obtain a function equation, as shown subsequently.
For the untreated material,
For the LNC material,
The correlation indices of the untreated specimen and the LNC specimen are R2 = 0.995 and R2 = 0.993, respectively, indicating that the fitting result is credible. It can be seen from the preceding equation that as the corrosion time increases, the pitting depth of both specimens increases slowly, while the pitting depth of LNC specimens increases at a lower trend, demonstrating that nitrocarburizing treatment can equip steel with remarkable anti-H2S corrosion in long-term immersion.
Based on the preceding analyses, a model is proposed to clarify the degradation and corrosion product formation on stainless steel in a wet H2S environment, as revealed in Figure 11. First, when an untreated specimen is exposed to H2S solution, the H+/HS–/S2–species preferentially migrate onto the passive film/solution interface, as exhibited in Figure 11(a). This action suppresses the hydration of Cr and, consequently, impedes the formation of the passive film. Furthermore, even if the passive film can form on the surface, the H+/HS–/S2– species, especially by their collaborative action with Cl–, can still lead to the failure of the passive film, as demonstrated by the breakdown mechanism of the passive film.[57,58] The reduction in the surface Cr content also makes repassivation of the stainless steel difficult (Figure 6). After 4 hours of corrosion, the substrate begins to dissolve and the resulting Fe ions combine with the sulfur ions to form a sulfur-iron compound and deposit on the metal surface. Pitting was observed on the surface simultaneously. As the corrosion proceeds, the new layer of corrosion products is continuously produced and deposited on the stainless steel surface until the entire surface is coated with FeS corrosion products. As H2S products thicken, it becomes more difficult for the metal cation of the substrate to pass through and sulfur ions in the solution penetrate the corrosion products layer. Finally, the corrosion products begin to delaminate, with Fe-depleted phases on the outer layer and sulfur-depleted phases in the cracked inner layer.[59] The corresponding element content is shown in Table II. The corrosion characteristics of the substrate also change from pitting to a mixture of general corrosion and pitting, as demonstrated in Figure 9(k).

Schematic model for the degradation and corrosion product formation on stainless steel in wet H2S environment: (a) untreated materials and (b) LNC materials
The dense oxide film on the surface is resistant to H2S corrosion within a specific time. After 72 hours of reaction, corrosion occurs along the grain boundaries (Figure 5(f)). Once the oxide film is broken, H2S enters the S phase through the grain boundary to make the corrosion proceed further. After 168 hours, pits can be observed at the interface of the corrosion products layer and the nitrogen-rich layer. As immersion time is further extended, the obvious crack and local breakage occur in the nitrogen-rich layer. The pitting corrosion is a prerequisite for crack formation, and stress-induced hydrogen permeation is also an important cause.[60] Besides, this phenomenon can be attributed to the fact that the consumption of reactive nitrogen atoms not only reduces the corrosion resistance of the material but also leads to a reduction in residual compressive stress. Under the barrier of the outer nitrogen-rich layer, the inner carbon-rich layer is relatively complete. Therefore, the substrate can still be completely protected.
4 Conclusions
-
1.
Low-temperature nitrocarburizing treatment can significantly reduce the weight loss of the material during the entire H2S corrosion process. Compared with the LNC material, the untreated specimen had a high sulfur content. After 1080 hours of corrosion, the corrosion products delaminated, with Fe-depleted phases on the outer layer and sulfur-depleted phases in the inner cracked layer. XRD analysis showed that the sulfide on the untreated surface was present in the form of marcasite, while the LNC surface was the more protective pyrrhotite.
-
2.
XPS analysis indicates that the original passive film was not stable under such corrosive conditions. The chromium content in the nitrocarburized surface was higher and existed in the main form of Cr2O3 and Cr(OH)3; this helped to improve the corrosion resistance of the material.
-
3.
During the time-dependent H2S corrosion experiments, the untreated specimen pitted, followed by general corrosion. The existence of the highly protective S-phase layer influenced the topography significantly and completely prevented general corrosion and also minimized the extent of pit propagation.
-
4.
Dense oxide film improved the resistance to H2S corrosion within 72 hours at 60 °C; then, corrosion occurred along the grain boundaries. Owing to the consumption of nitrogen atoms, the nitrogen-rich layer of the LNC specimen was cracked, while the intact carbon-rich layer could well protect the substrate.
References
Z. Wang, L. Zhang, X. Tang, Z. Zhang, and M. Lu: Appl. Surf. Sci., 2017, vol. 423, pp. 457–64.
J. Ding, L. Zhang, M. Lu, J. Wang, Z. Wen, and W. Hao: Appl. Surf. Sci., 2014, vol. 289, pp. 33–41.
P. Wang, J. Wang, S. Zheng, Y. Qi, M. Xiong, and Y. Zheng: Int. J. Hydr. Energy, 2015, vol. 40, pp. 11925–11930.
L. Zeng, X.P. Guo, and G.A. Zhang: J. Alloy Compd., 2017, vol. 724, pp. 827–40.
E. Heitz: Electrochim. Acta, 1996, vol. 41, pp. 503–09.
X. Hu and A. Neville: Wear, 2009, vol. 267, pp. 2027–32.
J. Aguirre, M. Walczak, and M. Rohwerder: Wear, 2019, vols. 438–439, 203053.
X. Zhang, J. Wang, H. Fan, J. Yan, L. Duan, and T. Gu: Metall. Mater. Trans. A, 2018, vol. 49A, pp. 1–12.
E. Araújo, R. Bandeira, M. Manfrinato, J. Moreto, R. Borges, S. Vales, P. Suzuki, and L. Rossino: J. Mater. Res. Technol., 2019, vol. 8, pp. 2180–91.
J. Wang, Z. Li, D. Wang, S. Qiu, and F. Ernst: Acta Mater., 2017, vol. 128, pp. 235–40.
D. Wu, H. Kahn, J.C. Dalton, G.M. Michal, F. Ernst, and A.H. Heuer: Acta Mater., 2014, vol. 79, pp. 339–50.
Y. Sun and T. Bell: Tribol. Lett., 2002, vol. 13, pp. 29–34.
Y. Peng, C. Chen, X. Li, J. Gong, Y. Jiang, and Z. Liu: Surf. Coat. Technol., 2017, vol. 328, pp. 420–27.
X. Li, W. Dou, L. Tian, and H. Dong: Lubricants, 2018. doi:10.3390/lubricants6040093
G. Pintaude, A. Rovani, J. Das, L. Lagoeiro, X. Li, and H. Dong: J. Mater. Eng. Perform., 2019, vol. 28, pp. 3673–82.
X. Zhang, J. Wang, H. Fan, and D. Pan: Appl. Surf. Sci., 2018, vol. 440, pp. 755–62.
P. Wang, Z. Lv, S. Zheng, Y. Qi, J. Wang, and Y. Zheng: Int. J. Hydr. Energy, 2015, vol. 40, pp. 11514–11521.
W. Li, X. Zhu, C. Wang, and X. Jin: Mater. Today, 2015, vol. 2, pp. S691–S695.
X. Xu, L. Wang, Z. Yu, J. Qiang, and Z. Hei: Metall. Mater. Trans. A, 2000, vol. 31A, pp. 1193–99.
J. Wang, Y. Lin, and Q. Zhang: Metall. Mater. Trans. A, 2014, vol. 45A, pp. 4525–34.
M. Zhao, Y. Shan, R. Fu, Y. Ke, and H. Yu: Mater. Lett., 2002, vol. 57, pp. 141–45.
J. Wang, Y. Lin, M. Li, H. Fan, D. Zeng, and J. Xiong: Metall. Mater. Trans. B, 2013, vol. 44B, pp. 1010–16.
R. Huang, J. Wang, S. Zhong, M. Li, J. Xiong, and H. Fan: Appl. Surf. Sci., 2013, vol. 271, pp. 93–97.
G. Chen, J. Wang, H. Fan, D. Wang, X. Li, and H. Dong: J. Alloy Compd., 2019, vol. 776, pp. 702–11.
L. Li, J. Wang, J. Yan, L. Duan, X. Li, and H. Dong: Metall. Mater. Trans. A, 2018, vol. 49A, pp. 6521–32.
J. Wang, Y. Lin, J. Yan, D. Zen, Q. Zhang, and R. Huang: Surf. Coat. Technol., 2012, vol. 206, pp. 3399–3404.
K. Deng, Y. Lin, H. Ning, W. Liu, A. Singh, and G. Zhang: Appl. Geochem., 2018, vol. 99, pp. 22–30.
A. Groysman: Corrosion Problems and Solutions in Oil Refining and Petrochemical Industry, 1st ed., Springer International Publishing AG, Cham, Switzerland, 2017.
Standard Practice for Preparing, Cleaning, and Evaluating Corrosion Test Specimens, ASTM, West Conshohocken, PA, 2003.
P. Tewari, M. Bailey, and A. Campbell: Corros. Sci., 1979, vol. 19, pp. 573–85.
M. Alizadeh and S. Bordbar: Corros. Sci., 2013, vol. 70, pp. 170–09.
D. Rickard and G. Luther: ChemInform, 2007, vol. 38, pp. 514–62.
J. Sardisco, W. Wright, and E. Greco: Corrosion, 1963, vol. 19, pp. 354–59.
P. Tewari and A. Campbell: Can. J. Chem., 1979, vol. 57, pp. 188–96.
H. Dong: Int. Mater. Rev., 2010, vol. 55, pp. 65–98.
W. Hao, L. Zhang, and H. Li: Paper No. 11293, NACE International, Houston, TX, 2011.
L. Zhang, H. Li, and F. Shi: Int. J. Min. Met. Mater., 2017, vol. 24, pp. 401–09.
P. Bai, S. Zheng, and C. Chen: Mater. Chem. Phys., 2015, vols. 149–150, pp. 295–301.
Z. Wang, L. Zhang, Z. Zhang, and M. Lu: Appl. Surf. Sci., 2018, vol. 458, pp. 686–99.
D. Macdonald, S. Biaggio, and H. Song: J. Electrochem. Soc., 1992, vol. 139, pp. 170–76.
X. Liu, P. Okafor, and Y. Zheng: Corros. Sci., 2009, vol. 51, pp. 744–51.
Z. Iofa and V. Batrakov: Electrochim. Acta, 1964, vol. 9, pp. 1645–53.
R. King, J. Miller, and J. Smith: Br. Corros. J., 1973, vol. 8, pp. 137–41.
W. Fredriksson, S. Malmgren, T. Gustafsson, M. Gorgoi, and K. Edstr: Appl. Surf. Sci., 2012, vol. 258, pp. 5790–97.
A. Laszczyńska, W. Tylus, J. Winiarski, and I. Szczygieł: Surf. Coat. Technol., 2017, vol. 317, pp. 26–37.
G. Myburg, K. Varga, W.O. Barnard, P. Baradlai, L. Tomcsányi, J.H. Potgieter, C.W. Louw, and M.J. van Staden: Appl. Surf. Sci., 1998, vol. 136, pp. 29–35.
L. Zeng, X.P. Guo, G.A. Zhang, and H.X. Chen: Corros. Sci., 2018, vol. 144, pp. 258–65.
J.E. Castle: J. Electrochem. Soc., 1990, vol. 137, p. 2031.
H.W. Nesbitt, D. Legrand, and G.M. Bancroft: Phys. Chem. Miner., 2000, vol. 27, pp. 357–66.
H. Feng, Z. Jiang, H. Li, P. Lu, S. Zhang, H. Zhu, B. Zhang, T. Zhang, D. Xu, and Z. Chen: Corros. Sci., 2018, vol. 144, pp. 288–300.
S. Ningshen, U. KamachiMudali, V.K. Mittal, and H.S. Khatak: Corros. Sci., 2007, vol. 49, pp. 481–96.
M. Monnot, R.P. Nogueira, V. Roche, G. Berthomé, E. Chauveau, R. Estevez, and M. Mantel: Appl. Surf. Sci., 2017, vol. 394, pp. 132–41.
M.B. Kermani and A. Morshed: Corrosion, 2003, vol. 59, pp. 659–83.
G. Schmidt: Advance in CO2 Corrosion, vol. 1, NACE, Houston, TX, 1984, pp. 1–2.
A. Jarrah, M. Bigerelle, G. Guillemot, D. Najjar, A. Iost, and J.-M. Nianga: Corros. Sci., 2011, vol. 53, pp. 2453–67.
L. Organ, J.R. Scully, A.S. Mikhailov, and J.L. Hudson: Electrochim. Acta, 2005, vol. 51, pp. 225–41.
P. Marcus, V. Maurice, and H. Strehblow: Corros. Sci., 2008, vol. 50, pp. 2698–2704.
J. Soltis: Corros. Sci., 2015, vol. 90, pp. 5–22.
P. Bai, H. Zhao, S. Zheng, and C. Chen: Corros. Sci., 2015, vol. 93, pp. 109–19.
R. Wei, X. Chen, Z. Ai, and Y. Jin: Int. J. Hydrogen Energy, 2018, vol. 43, pp. 9059–67.
Acknowledgments
The authors are grateful for the grants provided by the National Natural Science Foundation of China (Grant Nos. 51471112 and 51611130204), the Science and Technology Planning Project of Sichuan (Grant No. 2016GZ0173), and the Royal Society, United Kingdom (Newton Mobility Grant No. IE151027).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Manuscript submitted December 13, 2019.
Rights and permissions
About this article

Cite this article
Li, L., Wang, J., Tang, Z. et al. Low-Temperature Nitrocarburizing of Austenitic Stainless Steel for Combat Corrosion in H2S Environments. Metall Mater Trans A 51, 4242–4256 (2020). https://doi.org/10.1007/s11661-020-05802-4
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11661-020-05802-4