
Abstract
All-atom molecular-level computations are carried out to infer the dynamic response and material microstructure/topology changes of fused silica subjected to ballistic impact by a hard projectile. The analysis was focused on the investigation of specific aspects of the dynamic response and of the microstructural changes such as the deformation of highly sheared and densified regions and the conversion of amorphous fused silica to SiO2 crystalline allotropic modifications (in particular, α-quartz and stishovite). The microstructural changes in question were determined by carrying out a post-processing atom-coordination procedure. This procedure suggested the formation of stishovite (and perhaps α-quartz) within fused silica during ballistic impact. To rationalize the findings obtained, the all-atom molecular-level computational analysis is complemented by a series of quantum-mechanics density functional theory (DFT) computations. The latter computations enable determination of the relative potential energies of the fused silica, α-quartz, and stishovite under ambient pressure (i.e., under their natural densities) as well as under imposed (as high as 50 GPa) pressures (i.e., under higher densities) and shear strains. In addition, the transition states associated with various fused-silica devitrification processes were identified. The results obtained are found to be in good agreement with their respective experimental counterparts.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The problem addressed in the present work involves the formation of crystalline phases (specifically α-quartz and stishovite) within a fused-silica (ceramic glass containing a high-purity SiO2) target during a ballistic impact. Hence, the main aspects of the present work include (a) short- and intermediate-range order microstructure of fused silica, (b) (crystalline) allotropic modifications of SiO2, and (c) devitrification (i.e., crystallization of glass taking place under the ballistic-impact loading conditions). A brief overview of these aspects of the problem at hand is provided in the remainder of this section.
Short- and Intermediate-Range Order Microstructure of Fused Silica
Oxide-based glasses (like fused silica) are amorphous materials. The molecular-level microstructure of these materials involves entities such as a random network of covalently bonded atoms, atomic free volumes, (network-former) bridging and non-bridging oxygen atoms, and cations of glass-modifier species. Despite the absence of a crystalline structure, however, the microstructure of ceramic glasses is not completely random, but rather involves different extents of short- and intermediate-range order spanning over a range of length scales (from the quantum mechanical to the continuum level). Various experimental techniques (e.g., neutron diffraction, nuclear magnetic resonance, small-angle x-ray scattering (SAXS), etc.) have been used to capture the structure of ceramic glasses. Typically, this structure is described using the so-called “random network model” (Ref 1), which represents an amorphous material as a three-dimensional linked network of polyhedra. In this model, the character (number of facets) of the polyhedra is controlled by the species-specific coordination of the central (glass-forming) atom. In the case of silicate-based glasses such as fused silica and soda-lime glass, the polyhedron-center atoms are all silicon, with each silicon atom being surrounded by four oxygen atoms (while each oxygen atom is connected to, or bridges, two silicon atoms) forming an SiO4 4− tetrahedron. Other types of polyhedra may exist in glasses having different formulations. Since silicon has a tendency to form a continuous network with (bridging) oxygen atoms, SiO2 is commonly referred to as a “network former.” Other potential network formers in glass are boron and germanium oxides.
Numerous oxides and other additives are used to modify the basic silica tetrahedra network of silicate-based glasses in order to tailor their properties to specific applications. When alkali (or alkaline earth) oxides are added to a pure silicate-based glass, in order to accommodate the introduction of oxygen anions due to the alkali (or alkaline earth) oxide dissociation, the continuity of the silica tetrahedra network becomes disrupted. The resulting glass structure contains additional non-bridging (connected to only one silicon atom) oxygen atoms. Charge transfer from the alkali (or alkaline earth) metal atoms converts the non-bridging oxygen atoms into singly charged anions. The alkali (or alkaline earth) metallic cations formed in this process tend to hover around the non-bridging oxygen ions to provide local charge neutrality. Since alkali (or alkaline earth)-based oxides cause a disruption in the continuous glass network, they are typically referred to as “network modifiers.” As mentioned earlier, fused silica investigated in the present work is chemically pure SiO2 (i.e., free of glass modifiers) and, hence, contains a continuous network of Si-O bonds (i.e., it is free of non-bridging oxygen ions and alkali (or alkaline earth) metallic cations).
Within the random network model, the microstructure of glass is described using several network state parameters. Among these, the most frequently used are:
-
(a)
the so-called R parameter, defined as the average number of oxygen atoms per network-forming atom which is used to describe the overall connectivity of a given network. In the case of fused silica, in which there are two (bridging) oxygen atoms for every network-forming silicon atom, the R value is 2.0. In general, a larger value of the R parameter (brought about by the addition of network modifiers) implies a more open (less-connected) weaker glass network. The effect of network formers on the R parameter is more complicated and depends on the network-former coordination number and the strength of its bond with oxygen as well as its concentration;
-
(b)
the so-called X parameter which defines the average number of non-bridging (connected to only a single network-forming atom) oxygen atoms per network polyhedron and takes on a zero value in the case of fused silica; and
-
(c)
the so-called Y parameter which defines the average number of bridging (connected to two network-forming atoms) oxygen atoms per network polyhedron and takes on a value of 4.0 in the case of fused silica.
In addition to the three parameters mentioned above, the “seemingly” random microstructure of oxide-based glasses is also described using pair-wise correlation functions and bond length, bond angle, and Voronoi-cell volume distribution functions. A more detailed description of some of these microstructural parameters will be provided in section 4, as part of a discussion of the results pertaining to the structure of the as-received and as-impacted fused-silica ballistic targets.
Allotropic Modifications of SiO2
Previous investigations (e.g., Ref 2-12) established that under high-rate (shockwave or ballistic) loading conditions, fused-silica targets can, at least in the vicinity of the impact region, experience transformation of the amorphous structure into a crystalline one. To help rationalize potential transformations of fused silica into various (crystalline) allotropic modifications of SiO2, the corresponding temperature-pressure phase diagram is depicted in Fig. 1. Before the phase diagram is analyzed briefly, three points should be made: (a) the phase diagram is of an equilibrium character and, thus, shows the regions of existence of various crystallographic allotropic modifications of SiO2 after sufficient amount of time is given to the system to equilibrate (i.e., to acquire the state of minimal free energy); (b) there is no fused-silica region in the phase diagram since this SiO2 state is of a metastable rather than a stable/equilibrium character; and (c) as far as the silica melt is concerned, it is a high-temperature molten state of SiO2 and, despite its amorphous nature, it is structurally different than fused silica, i.e., it possesses a much lower level of short- and intermediate-range order. While, according to the phase diagram depicted in Fig. 1, there are six SiO2 allotropic modifications (α-quartz, β-quartz, coesite, stishovite, β-tridymite, and β-cristobalite), only three (α-quartz, coesite, and stishovite) appear at room temperature and a temperature slightly higher than room temperature. Consequently, during a ballistic impact into a fused-silica target, these three SiO2 allotropic modifications are the most likely to be formed (provided that such impact produces a local crystallization of the fused silica). Below, a brief description is provided of the main crystallographic features of these three SiO2 allomorphs:

Temperature-pressure phase diagram for SiO2
-
i.
α-quartz—Structure: Trigonal; Space Group: P3221 (No. 154); Lattice parameters: a = b = 4.9137 Å, c = 5.4047 Å, α = β = 90.0°, γ = 120.0°, and Z = 3 (where Z is the number of SiO2 units within the unit cell); Mass density: 2.66 g/cm3. The atomic arrangement within the non-primitive unit cell of α-quartz is depicted in Fig. 2(a).
Fig. 2 
The atomic arrangements within the non-primitive unit cells of (a) α-quartz, (b) stishovite, and (c) coesite
-
ii.
Coesite—Structure: Monoclinic; Space group: C2/c; Lattice parameters: a = 7.1356 Å, b = 12.3692 Å, c = 7.1736 Å; α = 90.0°, β = 120.34°, γ = 90.0°, and Z = 16; Mass density: 2.92 g/cm3. The atomic arrangement within the non-primitive unit cell of coesite is depicted in Fig. 2(b).
-
iii.
Stishovite—Structure: Tetragonal (ditetragonal dipyramidal); Space group: P 42/mnm (No. 136) P42/mnm {P42/m 21/n 2/m}; Lattice parameters: a = b = 4.179 Å, c = 2.6649 Å, and Z = 2; Mass density: 4.29 g/cm3. The atomic arrangement within the non-primitive unit cell of stishovite is depicted in Fig. 2(c).

Dynamic-Loading-Induced Crystallization of Glass
A review of the public-domain literature carried out as part of the present work revealed a number of experimental and computational analyses involving the mechanical response of fused silica to dynamic loading. Some of these studies revealed the formation of shear bands within otherwise amorphous glass, others established the formation of crystalline phases (α-quartz and stishovite, but not coesite), while still others demonstrated increased hardness of the material surrounding the impact region without establishing the microstructural cause for this property change.
Chakraborty et al. (Ref 13) performed a series of nano-indentation tests on soda-lime silica glass. Their results showed that, as the loading rate increases, the extent of shear band formation in the region surrounding the indentation decreases, while the hardness value increases. No crystal-structure analysis was carried out to determine potential formation of any of the crystalline phases as a result of loading or to provide a rationale for the observed effect of the loading rate.
Tschauner et al. (Ref 14) investigated the formation of stishovite in soda-lime glass during 57 GPa shock loading experiments and the reversion of this phase during subsequent release/unloading. They demonstrated that upon loading, high-density non-fully crystallized SiO2 phase was present in the “shocked” fused silica. Upon static loading to only 13 GPa, this phase was fully converted into the crystalline stishovite, suggesting that the shock loading was able to devitrify fused silica and form crystalline stishovite.
Salleo et al. (Ref 15) demonstrated the formation of a defective form of stishovite in the surface region of fused-silica wafers through irradiation with a high-powered laser. The formation of such a phase and its continuous growth has been found to be the main cause of failure in optics used for high-power photonics.
Mantisi et al. (Ref 16) carried out a comprehensive atomic-scale simulation of fused silica under combined pressure/shear loading conditions. The results obtained established permanent/irreversible densification of the fused silica test sample and the change of the silicon and oxygen coordination relative to that present in the as-received fused silica. However, these microstructural changes could not be considered to be the result of glass devitrification/crystallization processes, i.e., the glass remained amorphous. One of the potential reasons for this observation was inadequacy of the interatomic potentials used in the simulation of the fused-silica mechanical behavior.
Kubota et al. (Ref 17) used molecular dynamics simulations to infer the atomic-scale structural changes in fused silica induced by shock-compression loading. The results obtained revealed that shock-compressive loading involving stress levels exceeding the Hugoniot Elastic Limit gives rise to dramatic changes in the structure and topology of the fused-silica network and densifications in excess of 20%. Coordination analysis of the as-shocked fused silica revealed the formation of under- and over-coordinated Si atoms. While under-coordinated Si atom regions could be interpreted as shock-induced fused-silica flaws, over-coordinated Si atom regions showed some resemblance to the crystalline stishovite. In addition to the coordination changes just described, changes in glass topology (such as increases in the number of the threefold, fourfold, sevenfold, and larger rings) were observed.
Main Objectives
The main objective of the present work is to carry out a series of all-atom molecular-level computational investigations of the ballistic impact by a hard projectile onto a fused-silica target plate and to characterize the target-plate material response to such impact. In addition, in order to help rationalize some of the findings regarding the microstructure and topology of the fused silica following the impact, a series of quantum-mechanical density functional theory (DFT) analyses will be carried out. These analyses will help reveal the relative potential energies of the SiO2 amorphous state and two SiO2 crystalline allotropic modifications, i.e., α-quartz and stishovite, and the changes in these energies as a function of the extent of material compression and shear.
Paper Organization
Details regarding the all-atom molecular-level computational procedure used to simulate the ballistic impact by a hard projectile onto a fused-silica target plate are presented in section 2. The quantum-mechanical DFT procedure used to determine the relative potential energies of the SiO2 amorphous state and the two SiO2 crystalline allotropic modifications, as well as the associated transition states (to be defined later), is provided in section 3. Key results obtained in the present work are presented and discussed in section 4, while a summary of the main findings and conclusions is provided in section 5.
Molecular-Level Analysis of Ballistic Impact
As mentioned earlier, one of the objectives of the present work is to carry out a series of all-atom molecular-level computational analyses of the problem of a ballistic impact by a hard projectile onto a fused-silica target plate. Within the all-atom molecular-level computational methods and tools, every atom and bond is explicitly accounted for and molecular mechanics and dynamics algorithms are used to quantify the state and behavior of the material under investigation. All-atom molecular-level simulation problems typically require the specification of the following: (a) a molecular-level computational model consisting of atoms, ions, functional groups, and/or molecules; (b) a set of force-field functions (mathematical expressions describing bonding and non-bonding interactions between the model constituents, e.g., atoms, ions, etc.); and (c) computational method(s) to be used in the simulation. More details of these three aspects of the molecular-level modeling and simulation of fused silica are provided below.
Computational Model
The computational model used in this portion of the work consists of two distinct sub-domains: (a) the projectile sub-domain and (b) the target-plate sub-domain. The two sub-domains are shown and labeled in Fig. 3(a).

(a) The computational model used in the molecular-level simulations, employed in this portion of the work, consisting of two distinct sub-domains for the projectile and the target plate; and (b) close-up of the resulting fused-silica molecular-level random network microstructure. Larger ball sizes are used in this figure to highlight a pair of SiO4 4− tetrahedra sharing a common oxygen atom
The projectile sub-domain is of a right circular solid cylindrical geometry (height-over-diameter ratio = 1.0, axis aligned with the z-direction). While, at least, the core of ballistic projectiles is typically made of hard and heavy metallic materials (e.g., tungsten), metallic materials could not be used in the construction of the projectile in the present work. The reason for this is the absence of metallic force-field functions (in the pure metallic environment) within the force-field function database used in the present work. Consequently, the projectile was made of another hard material, diamond. Typically, the projectile sub-domain contained 150 covalently bonded carbon atoms forming a perfect single-crystalline diamond structure.
As far as the target-plate sub-domain is concerned, it is of a rectangular parallelepiped (plate-like) shape and is made of fused silica. At the molecular level, fused silica is modeled as a discrete-particle-based material consisting of silicon (Si) and oxygen (O) atoms mutually bonded via a single covalent bond and forming a connected, non-structured/amorphous network of silica (SiO4 4−) tetrahedra. While fused silica is an amorphous material and does not possess any long-range regularity in its atomic/molecular structure, modeling of bulk behavior of fused silica is typically done at the molecular level by assuming the existence of a larger (amorphous) unit cell. Repetition of this cell in the three orthogonal directions (the process also known as application of the “periodic boundary conditions”) results in the formation of an infinitely large bulk-type material. This procedure was adopted in the present work. The parallelepiped-shaped target-plate computational sub-domain used in the present (ballistic-impact) analysis contained 9600 particles (3200 Si atoms and 6400 O atoms). The edge lengths of the computational cell were initially set to a = b ~ 7.9 nm, c ~ 2.9 nm (approximately), yielding a fused-silica initial nominal density of 2.19 g/cm3. The three edges (a, b, and c) of the cell were aligned, respectively, with the three coordinate axes (x, y, and z) (with the target-plate thickness aligned with the z-direction).
To create the ambient temperature/pressure “equilibrium” atomic configuration within the computational cell, the following procedure was implemented within the Visualizer (Ref 18) program from Accelrys:
-
(a)
A starting computational cell was first constructed by stacking the appropriate number of α-quartz unit cells in the three orthogonal directions. This was followed by the affine distortion of the computational cell in order to obtain the correct mass density of the fused-silica amorphous state (obtained using the following procedure);
-
(b)
To convert the crystalline material into an amorphous one, a stochastic bond-switching algorithm was then implemented (Ref 19) using a Monte Carlo computational procedure. Within this algorithm, two neighboring Si-O pairs were randomly selected from the computational cell and the potential energy change ΔE resulting from the Si-O bond switching computed. In the ΔE < 0 case, the bond switching in question was accepted without any additional conditions. On the contrary, in the ΔE > 0 case, a Boltzmann probability factor \(P_{\text{B}} = \exp \left[ { - {{\Delta E} \mathord{\left/ {\vphantom {{\left( {{{3NkT} \mathord{\left/ {\vphantom {{3NkT} 2}} \right. \kern-0pt} 2}} \right)}}} \right. \kern-0pt} {\left( {{{3NkT} \mathord{\left/ {\vphantom {{3NkT} 2}} \right. \kern-0pt} 2}} \right)}}} \right]\) (where N is the number of atoms within the computational cell, k is the Boltzmann constant, and T is the absolute temperature) was first calculated and compared with a random number RN drawn from a (0,1) uniform distribution function. The bond switching in question was then adopted only if P B > RN;
-
(c)
The resulting structure was then subjected to a carefully devised set of NVT (where N (=9600) is the (fixed) number of atoms within the computational cell, V, the computational cell volume (also fixed), and T is a fixed temperature) molecular dynamics simulations. Specifically, the NVT simulations were started at a temperature of 5300 K and carried out in such a way that the temperature was controlled using the following “simulated-annealing” scheme: (i) a particle-velocity scaling algorithm was applied at every time step for the first 6000 steps. This enforced strict control of the temperature but produced particle velocities which were inconsistent with the target-temperature Maxwell-Boltzmann distribution function; (ii) within the next 6000 NVT simulation steps, the frequency of particle-velocity scaling was decreased every 40 time steps, while a Nosé-Hoover (Ref 20) temperature-control algorithm (“thermostat”) was applied between the particle-velocity scaling steps. A brief description of the Nosé-Hoover thermostat could be found in our prior work (Ref 21); and (iii) during the final 8000 steps, the temperature was controlled using only the Nosé-Hoover thermostat. This procedure ensured an efficient temperature control while yielding an equilibrium state of the material (i.e., a particle-velocity distribution consistent with the target-temperature Maxwell-Boltzmann distribution function).
Upon establishing the thermodynamic equilibrium at 5300 K, the target temperature was reduced by 500 K and then this procedure was re-applied at progressively (by 500 K) lower temperatures until the final temperature of 300 K was reached. The total system equilibration procedure typically involved simulation times on the order of 500 ps resulting in an average cooling rate of ~10 K/ps. A close-up of the resulting fused-silica molecular-level random network is displayed in Fig. 3(b). Larger ball sizes are used in this figure to highlight a pair of SiO4 4− tetrahedra sharing a common oxygen atom.
Force Fields
The behavior of a material system at the molecular level is governed by the appropriate force fields which describe, in an approximate manner, the various interactions taking place between the constituent particles, atoms, ions, charge groups, etc. In other words, the knowledge of force fields enables determination of the potential energy of a system in a given configuration. In addition, gradients of the force-field functions quantify the net forces experienced by the particles, the information that is needed in the molecular dynamics simulations.
In general, the potential energy of a system of interacting particles can be expressed as a sum of the valence (or bond), E valence, cross-term, E cross-term, and non-bond, E non-bond, interaction energies as follows:
The valence energy generally accounts for the contribution of valence electrons bonding and contains the following components: (a) a bond length/stretching term; (b) a two-bond angle term; (c) a three-bond dihedral/torsion angle term; (d) an inversion (or a four-atom out-of-plane interaction) term; and (e) the so-called “three-atom Urey-Bradley term” (i.e., the interaction of two atoms which are bonded to a common atom).
The cross-term interacting energy accounts for the cross-interactions between the aforementioned valence-energy components and includes terms like (a) stretch-stretch interactions between two adjacent bonds; (b) stretch-bend interactions between a two-bond angle and one of its bonds; (c) bend-bend interactions between two valence angles associated with a common vertex atom; (d) stretch-torsion interactions between a dihedral angle and one of its end bonds; (e) stretch-torsion interactions between a dihedral angle and its middle bond; (f) bend-torsion interactions between a dihedral angle and one of its valence angles; and (g) bend-bend-torsion interactions between a dihedral angle and its two valence angles.
The non-bond interaction term accounts for the interactions between non-bonded atoms and includes the van der Waals energy and the Coulomb electrostatic energy.
In the present work, the so-called “COMPASS” (Condensed-phase Optimized Molecular Potentials for Atomistic Simulation Studies) force field is used (Ref 22, 23). This highly accurate force field is of an ab initio type since most of its parameters were determined by matching the predictions made by the ab initio quantum-mechanics calculations to the condensed-matter experimental data. A summary of the COMPASS force-field functions can be found in our previous work (Ref 24).
Computational Method(s)
All the all-atom molecular-level calculations were carried out within the present work using Discover (Ref 25) (an atomic simulation program from Accelrys). Both equilibrium and non-equilibrium molecular dynamics (MD) analyses were employed in the present work. Within the molecular dynamics method, negative gradient of the potential energy evaluated at the location of each atom is first used to compute forces acting on each atom. Then, the associated Newton’s equations of motion (three equations for each atom) are integrated numerically over a femtosecond-long time interval. This procedure is repeated over a pico-to-nanosecond-long simulation time in order to determine the temporal evolution of the material molecular-level configuration. Equilibrium MD was used in the aforementioned Monte Carlo-based bond-switching procedure to generate amorphous state from the crystalline state in SiO2. Non-equilibrium MD was used during the simulation of the ballistic impact by a hard projectile onto a fused-silica target plate.
Within the equilibrium MD method, the system under consideration is coupled to an (external) environment (e.g., constant-pressure piston, constant-temperature reservoir, etc.) which ensures that the system remains in equilibrium (i.e., the system is not subjected to any thermodynamic fluxes). In the present work, NVT, NPT (P is pressure), and NVE (E is the total energy) equilibrium MD simulations were used. Equilibrium MD calculations enable determination of the (equilibrium) thermodynamic properties of a material system through the use of time averages of the state variables sampled along the calculated system trajectories.
Within non-equilibrium MD, the system is subjected to large mechanical and/or thermal perturbations (momentum transfer from the moving projectile to the initially stationary target plate, in the present case). As a consequence, the system experiences large fluxes of its thermodynamic quantities (mass, momentum, and energy, in the present case). Since Discover was initially designed to carry out equilibrium MD simulations, a procedure had to be devised to deactivate “equilibration” portions of this algorithm so that non-equilibrium MD calculations can be carried out. This procedure was implemented using a Discover input file (Ref 25). This file is written using the Basic Tool Command Language (BTCL) which enabled the use of a scripting engine that provides very precise control of simulation tasks (e.g., specification of the projectile constant incident velocity, deactivation of the thermal-equilibration algorithm, etc.)
Problem Formulation
The problem addressed in the present work involves all-atom molecular-level computational modeling of the ballistic impact by a hard projectile onto a fused-silica target plate and the coordination/topology analysis of the as-impacted fused silica. The projectile is of a solid right circular cylindrical shape and impacts the target plate normally (i.e., at a 0° obliquity angle through-the-thickness direction). The projectile is driven at a constant velocity and the target plate is confined only along its bottom rim.
Quantum-Mechanical Analysis of Glass Devitrification
In order to rationalize the molecular-level computational results pertaining to the ballistic impact of a projectile onto the fused-silica target plate, and the potentially accompanying changes in the glass microstructure, a quantum-mechanical DFT analysis of the fused-silica devitrification process is carried out in the present work. The main purpose of this analysis was the establishment of (a) the energy difference between the as-received glass material state and the crystalline α-quartz, stishovite, and coesite SiO2 states; (b) the determination of the transition-state energy barriers associated with the three (glass → α-quartz, glass → stishovite, or glass → coesite) fused-silica devitrification processes. The transition state is the material state along the devitrification pathway which is associated with the maximum potential energy; and (c) the effect of high pressure and shear strains on the energetics of the initial, transition, and final states.
Computational Models
As mentioned earlier, all three room-temperature SiO2 polymorphs (α-quartz, stishovite, and coesite) are investigated in this portion of the work. The respective computational cells are shown in Fig. 2(a)-(c). Since fused silica possesses an amorphous structure, without long-range order, a substantially larger computational cell had to be used for this (initial) state of SiO2, in order to more accurately determine its average potential energy. An example of such a computational cell is given in Fig. 3(a), the target-plate computational sub-domain.
In the portion of the analysis dealing with determination of the transition state, the initial and final states of the material have to contain the same number of atoms and species. For that reason, fused-silica computational cells containing six Si and 12 O atoms (the numbers match those seen in Fig. 2a) are used to investigate fused-silica → α-quartz transition state. Likewise, fused-silica computational cells containing two Si and four O atoms (the numbers match those seen in Fig. 2c) are used to investigate fused-silica → stishovite transition state. In order to account for the statistical effects associated with the extraction of such small fused-silica computational cells from a larger computational cell, ten small fused-silica computational cells (not shown for brevity) are generated for each of the two transition state analyses and the results obtained are averaged out.
Computational Method
All calculations in the present work are carried out using the ab initio density functional theory code DMol3 developed by Accelrys Inc. (Ref 26). In this code, each electronic wave function is expanded in a localized atom-centered basis set with each basis function defined numerically on a dense radial grid. No pseudopotential approximation is used for the near-core electrons. Instead, all-electron calculations are performed with a double numerical polarized (DNP) basis set, the most complete basis set available in the DMol3 code. This basis set is equivalent to the commonly used analytical 6-31g** basis set, a split-valence basis set with polarization functions p to H and d to C, and the halogens (Ref 27).
To improve the computational speed, the local-density approximation (LDA) (Ref 28) for the exchange-correlation potential is often used within the DFT formulation, which assumes that the electron-charge density varies slowly on the atomic length scale. However, the LDA method is found not to have the correct asymptotic behavior and generally overestimates the magnitude of the chemical-bond energy. To overcome this overbinding phenomenon, one of the existing density gradient expansion schemes (also referred to as Generalized Gradient Approximations, GGA), which includes the effect of charge-density inhomogeneity, needs to be utilized. Following Grujicic et al. (Ref 29), the Perdew-Burke-Ernzerhof (PBE) gradient-corrected functional (Ref 30) is used in the present work. A standard value of 5.5 Å is assigned to the finite basis-set cutoff radius.
Determination of the Transition States
In general, the transition state lies on a Minimum Energy Pathway connecting the initial and final states’ potential energy minima, where the pathway and the two minima all reside on the associated potential energy hypersurface. The defining feature of the transition state is that it is associated with a minimum potential energy in all the directions but one (in which the potential energy experiences a maximum). In the case of a system with two degrees of freedom, the transition state corresponds to a saddle point, as depicted in Fig. 4. Moving the system under consideration from the transition state point (in either direction) along the steepest descent path leads to the initial/final states.

The initial-to-final state transition in a two-degree-of-freedom system and identification of the corresponding transition state (the saddle point)
There are several algorithms which are commonly used for determination of the transition state. The three most frequently used include (a) linear synchronous transit (LST) (Ref 31), used in the present work; (b) quadratic synchronous transit (QST) (Ref 31); and (c) nudged elastic band (Ref 32).
The LST method constructs the initial-state → final-state transition pathway by connecting each atom in its initial and final states using a straight-line pathway and by constructing the intermediate configurations by linearly and synchronously interpolating the atomic positions along the pathway of each atom. In other words, the structure of the intermediate states is a rule of mixtures of the initial structure and the final structure, with a weighting factor for the product state \(f = \left( {0,1} \right)\). The potential energy is next determined for all the intermediate states, and the one associated with the largest potential energy is taken as a first approximation of the system transition state. The location of the transition state is further improved by carrying out a constrained optimization of its first approximation.
Results and Discussion
Validation of the As-Received Fused-Silica Material State
In this section, an attempt is made to validate the fused-silica room-temperature/ambient-pressure structure obtained through application of the previously described bond-switching and simulated-annealing computational procedures. In particular, the material mass density, the partial radial distribution functions for the three (Si-Si, Si-O, and O-O) atomic pairs, and distributions of the Si-atom and O-atom coordination numbers are calculated and compared with their experimental counterparts.
To compute the material mass density, the partial radial distribution functions, and atomic coordination combinations, NPT equilibrium molecular dynamics simulations were run at the room temperature and the ambient pressure. Simulations were carried out for over 10,000 (0.1 fs-long) time steps while maintaining the temperature using the Nosé-Hoover thermostat and scaling the particle velocities at every ten time steps. Pressure was controlled using a Berendsen barostat (Ref 33). A brief overview of this barostat is provided in our prior work (Ref 34).
Mass Density
The average mass density of fused silica computed from the computational cell average volume and the cell mass has been found to be about 1.0% greater than its commonly cited experimental counterpart of 2.19 g/cm3. This computation/experiment agreement has been deemed to be reasonably good.
Radial Distribution Functions
The partial radial distribution (often also referred to as the partial pair correlation) function provides a measure of the probability that, given the presence of an atom of type α at the origin of an arbitrary reference frame, there will be an atom of type β within a spherical shell of infinitesimal thickness dr at a distance r from the reference atom. Alternatively, this function can be considered as a function which defines a ratio of the probability of finding an α-β atomic pair with the separation distance r, and the average probability of finding an α-β atomic pair at the same distance. In amorphous materials like fused silica, the partial pair correlation functions are quite important since they (a) provide an insight into the short-range order of the system, (b) can be used in the assessment of continuum-level thermodynamic material properties, and (c) provide a way of validating the molecular-level calculations since these quantities can also be determined experimentally using x-ray diffraction.
The computed partial radial distribution functions for the as-received/initial state of fused silica, Fig. 5(a), are compared with their counterparts based on the shell model molecular dynamics calculations (Ref 35), Fig. 5(b). This comparison reveals that the present computational results are qualitatively similar to the ones reported in Ref 35. As far as the quantitative agreement between the two sets of results is concerned, it could be characterized as being fair to good.

A comparison between the fused-silica Si-Si, O-O, and Si-O partial radial distribution functions computed in (a) the as-received/initial state, determined in the present work, and (b) the equilibrium initial state as reported in Tilocca et al. (Ref 35)
Si- and O-Atom Coordinations
Since fused silica does not contain any network modifiers and Si is the only network-forming element, each Si atom is expected to be bonded to four O atoms, while each O atom is expected to be bonded to two Si atoms. This is confirmed through post-processing of the molecular dynamics results, as shown in Fig. 6(a)-(b) (the data labeled “Initial State”). The results shown in Fig. 6(a) pertain to the Si-atom coordination, while those displayed in Fig. 6(b) refer to the O-atom coordination.

Fractional distribution of the (a) Si-atom and (b) O-atom coordination numbers in the initial and the as-impacted states of fused silica
Analysis of the Ballistic Impact
Temporal evolution of the computational domain at four (0.5, 1.5, 2.5, and 3.5 ps) times following the initial contact between the diamond solid right circular cylindrical projectile moving at a high velocity and the fused-silica target plate is depicted in Fig. 7(a)-(d), respectively. For improved clarity, the symbols for the target-plate atoms made smaller. Examination of the results displayed in Fig. 7(a)-(d) reveals that the target-plate material in the close vicinity of the projectile experiences major changes. However, the nature of these changes, i.e., the accompanying alterations in the material microstructure and topology, is difficult to infer from the results displayed in Fig. 7(a)-(d). To overcome this problem, a few selected results revealing the as-impacted fused-silica local microstructure in the region adjacent to the projectile are presented and discussed below.

Temporal evolution of the computational domain at four times: (a) 0.5 ps, (b) 1.5 ps, (c) 2.5 ps, and (d) 3.5 ps, following the initial contact between the diamond solid right circular cylindrical projectile moving at a high velocity and the fused-silica target plate
Figure 8 shows a thin slice of the fused-silica target plate, having the faces parallel to the x-z plane, centered on the hole created by the projectile after penetrating the target by about half of its thickness. Examination of the fused-silica microstructure within this slice revealed the presence of numerous sixfolded Si and threefolded O atoms (the Si and O coordination characteristic of stishovite and not of fused silica). In Fig. 8, two six-coordinated Si and two three-coordinated O atoms are highlighted by assigning a larger sphere radius to the atoms involved. In addition, sixfolded Si atoms and threefolded O atoms are tagged with circular symbols. In addition to the highlighted atomic configurations, many more instances of six-coordinated Si and three-coordinated O are found in the region surrounding the penetration hole. Since, as mentioned earlier, the initial state of fused silica only contained fourfolded Si and twofolded O atoms, this finding suggests that ballistic impact can lead, at least locally, to the conversion of amorphous SiO2 into the crystalline (but highly deformed) stishovite structure.

Local stishovite-like microstructure showing two sixfolded Si and two threefolded O atoms. For clarity, a larger sphere radius is assigned to the atoms involved. In addition, sixfolded Si atoms and threefolded O atoms are tagged with circular symbols
Additional changes observed in the as-impacted fused silica pertain to the distribution of the smallest Si-O rings. To quantify the size distribution of the smallest Si-O rings in both the initial and the as-impacted fused-silica states, a computational method was developed. This method solves the class of so-called “shortest path problems” and is a simple modification of the Dijkstra’s algorithm (Ref 36). The main modifications in this algorithm are associated with the fact that, in the present case, the starting point and the destination point of the path are identical. The smallest-ring size distribution results for the initial and as-impacted fused-silica states are displayed in Fig. 9(a)-(b), respectively. Examination of these results reveals that ballistic impact alters the ring structure within fused silica. Specifically, while no five-membered rings were present in the initial state of fused silica, such rings are found in the as-impacted state of the same material. Additional changes observed pertain to the topology of the (smallest, pre-existing) six-membered rings. That is, while in the initial state of fused silica these rings resemble the corresponding rings found in cristobalite (another allotropic modification of SiO2), Fig. 10(a), in the as-impacted fused silica the topology of the six-membered rings was found to resemble more that found in α-quartz, Fig. 10(b).

Size distribution function for the smallest Si-O rings in the fused silica: (a) initial state and (b) as-impacted state

Two views of the atomic structure of six-membered Si-O rings in (a) cristobalite and (b) quartz
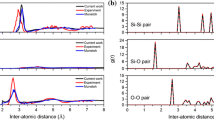
Three partial radial distribution functions for the fused-silica target plate after the diamond impactor has penetrated halfway through the target-plate thickness are depicted in Fig. 11(a). A comparison of these results with their as-received fused-silica counterparts, Fig. 5(a), reveals that the ballistic impact causes distinct changes in the short-range order and atomic coordination within this material. To help rationalize these changes, the same partial radial distribution functions are calculated for α-quartz, Fig. 11(b), and stishovite, Fig. 11(c). A comparison of the results displayed in Fig. 5(a) and 11(a)-(c) further confirms that the ballistic-impact-induced changes in the pair correlation functions are associated with the devitrification of fused silica and the formation of α-quartz and stishovite. In other words, differences in the partial pair correlation functions in the as-impacted fused silica relative to those in the as-received fused silica appear to be caused by the presence of Si-Si, Si-O, and O-O atomic pairs with an atomic environment similar to those found in stishovite (and α-quartz).

Three partial radial distribution functions for (a) the fused-silica region adjacent to the projectile, after the diamond impactor has penetrated approximately halfway through the target-plate thickness, (b) α-quartz, and (c) stishovite
To further demonstrate the conversion of fused-silica to stishovite as a result of the ballistic impact, Si- and O-atom coordinations in the fused-silica region surrounding the penetration hole are determined. The results of this procedure, labeled “As-impacted State,” are shown in Fig. 6(a)-(b). Examination of the results displayed in these figures reveals the presence of six-coordinated Si and three-coordinated O atoms, the atomic coordination which characterizes the stishovite crystal structure.
It should be noted that the results depicted in Fig. 6(a)-(b) and 11(a)-(c) represent average results obtained for ten different (but statistically identical) starting configurations of the fused-silica target plate. A statistical analysis of the results obtained revealed that the standard deviation for the results depicted in Fig. 6(a)-(b) is approximately ±3% of the average value, while for the peak positions and peak magnitudes shown in Fig. 11(a)-(c), the standard deviation is approximately ±2 and ±3%, respectively. Thus, the results obtained cannot be considered as being very sensitive to the starting configuration of the target plate. Unfortunately, sensitivity of the results obtained to the potential variation in the force-field parameters could not be investigated since within the Discover (Ref 25) molecular-modeling program, one has the ability to use but not to modify the COMPASS force-field parameters.
Relative Stability of Fused Silica, α-Quartz, Stishovite, and Coesite
To further help rationalize the changes in the partial pair correlation functions induced by the ballistic impact, the quantum-mechanical DFT calculation results pertaining to the relative stability of fused silica, α-quartz, stishovite, and coesite, as well as of the energy barriers associated with the fused-silica → α-quartz, fused-silica → stishovite, and fused-silica → coesite transition states, are presented and discussed in this section. The relative room-temperature potential energies of the fused silica, α-quartz, stishovite, and coesite as a function of pressure are presented in Fig. 12(a). Examination of the results displayed in this figure reveals that, at the ambient pressure, α-quartz is the most stable form of SiO2, followed by coesite, fused silica, and then by stishovite. At a pressure of 50 GPa, on the other hand, the relative positions of α-quartz, fused silica, and coesite, with respect to thermodynamic stability, have changed. That is, fused silica becomes the most thermodynamically stable form of SiO2 and is followed closely by α-quartz and coesite (which possess almost identical thermodynamic stabilities).

Relative room-temperature potential energies of fused silica, α-quartz, stishovite, and coesite, as a function of pressure, in the (a) absence and (b) presence of 5% shear
In Fig. 12(b), the relative room-temperature potential energies of the fused silica, α-quartz, and stishovite as a function of pressure, in the presence of 5% shear, are presented. A comparison of the results displayed in Fig. 12(a)-(b) reveals that the application of 5% shear strain did not change the relative stability of fused silica, α-quartz, stishovite, and coesite at the ambient pressure. In sharp contrast, at 50 GPa pressure, the application of 5% shear has been found to make the stishovite the most stable phase, followed by coesite, α-quartz, and then fused silica. This finding suggests that, in the presence of shear, fused silica is more likely to undergo a devitrification conversion into stishovite and, to a lower extent, into coesite and α-quartz. This finding is important since the fused-silica region surrounding the projectile is generally subjected to high shear, in addition to high pressures. This explains why in Fig. 8 this region was found to undergo an extensive fused-silica → stishovite conversion.
The likelihood for the aforementioned devitrification processes is affected not only by the relative stabilities of fused silica, α-quartz, stishovite, and coesite, but also by the size of the energy barrier associated with the respective transition state. Variations in the fused-silica → α-quartz, fused-silica → stishovite, and fused-silica → coesite transition-state energy barriers with pressure, in the absence and the presence of 5% shear, are shown, respectively, in Fig. 13(a)-(b). Examination of the results displayed in these figures, at a pressure of 50 GPa, reveals that the energy barrier for fused-silica devitrification, in the absence of shear, is slightly higher for the fused-silica → stishovite and fused-silica → coesite conversion than for the fused-silica → α-quartz conversion. On the other hand, the energy barrier for fused-silica devitrification at a pressure of 50 GPa, in the presence of shear, is the lowest for the fused-silica → stishovite conversion and the highest for the fused-silica → coesite conversion. These findings suggest that, in the presence of shear, and at pressures as high as 50 GPa, fused silica is most likely to convert to stishovite and least likely to convert to coesite. In other words, under the applied loading and the attendant (high) local temperatures, fused silica is most likely to convert into the state, the transition to which is associated with the lowest energy barrier. This finding is fully consistent with the molecular-level results presented in section 3.

Variations in the fused-silica → α-quartz, fused-silica → stishovite, and fused-silica → coesite transition-state energy barriers with pressure, in the (a) absence and (b) presence of 5% shear
Figure 14 shows the conversion of an initially amorphous SiO2 structure into a stishovite-like structure under the influence of high pressure and shear. The transition state associated with this conversion is also shown in this figure. For clarity, only a small SiO2 region consisting of three Si and 15 O atoms is displayed in Fig. 14. Due to such a small size of the region, some O atoms appear to be non-bonded. These O atoms are bonded to Si atoms, but the Si atoms that they are bonded to are not shown in this figure. The Si atoms in question are also bonded to some of the bonded O atoms displayed in Fig. 14. Examination of this figure reveals that, as expected, the fused-silica state contains only fourfold coordinated Si and twofold coordinated O atoms, while the stishovite state contains sixfold coordinated Si and threefold coordinated O atoms. The Si and O atom coordination in the transition state involves fourfold and fivefold coordinated Si and twofold and threefold coordinated O atoms. To help with the interpretation of the results displayed in Figure 14, the same three Si atoms appearing in the three configurations are denoted using labels “Si 1,” “Si 2,” and “Si 3.” It should be recalled that the results presented in Fig. 13(a)-(b) reveal that the maximum energy associated with the transition state is lowered, and thus the conversion of the fused-silica → stishovite becomes feasible, under high pressure and shear.

Conversion of an initially amorphous SiO2 structure into a stishovite-like structure under the influence of high pressure and shear. The associated transition state is also shown
Comparison of Present and Recent Work
In this section, a brief comparison is made between some of the results obtained in the present work and the corresponding results reported recently (Ref 37-41).
Sato et al. (Ref 37, 39, 40) and Brazhkin (Ref 38) developed synchrotron x-ray absorption and diffraction techniques for measuring the density and structure of noncrystalline materials at high pressures and applied them to studying the behavior of fused silica. The density, coordination number, and Si-O bond length at a pressure of 50 GPa were found to be 4.63 g/cm3, 6.30, and 1.71 Å, respectively. These results are quite comparable to the ones obtained in the present work but for the region surrounding the penetration hole: density 4.60 g/cm3, coordination number 6.21, and Si-O bond length 1.76 Å. Sato et al. (Ref 37-40) established the presence of a hexahedron network environment in fused silica at high pressures, but could not conclusively establish if the structure is a highly distorted crystalline stishovite or amorphous glass. Our molecular-level computational results presented in section 3 established also the presence of a sixfold coordinated (highly distorted) Si network (but only locally in the region surrounding the penetration hole). Adopting the semantics used in Ref 14, the network structure in question is referred to as stishovite.
Zeidler et al. (Ref 41) carried out a combined neutron-diffraction experimental and molecular dynamics computational analysis of the structure of fused silica under high pressures. The results obtained clearly revealed that at pressures of 40 GPa and higher, the average size of the primitive Si-O rings (that is, the rings which cannot be decomposed into smaller rings, Ref 42-44) begins to decrease from six Si-O pairs, in fused silica, to five Si-O pairs, found in stishovite. These results are fully consistent with the ones obtained in the present work and displayed in Fig. 9(a)-(b).
Summary and Conclusions
Based on the results obtained in the present work, the following summary remarks and main conclusions can be drawn:
-
1.
To investigate the fused-silica devitrification under impact loading conditions and to provide a thermodynamics basis for the observations made, novel combined all-atom molecular-level and density functional theory quantum-mechanical calculations are carried out. The focus of these calculations was identification of the microstructural changes experienced by fused silica during impact and explanation of these changes using the concept of relative thermodynamic stability of different fused-silica polymorphs and the transition-state energy barriers, the barriers which control the rate of different devitrification processes.
-
2.
By determining the Si-Si, Si-O, and O-O partial radial distribution functions, and the coordination numbers for Si and O atoms, before and after a ballistic impact experienced by a fused-silica target plate, it was found that the target-plate regions surrounding the penetration hole have undergone intrinsic topological changes manifested in the acquisition of a hexahedron-type network (in contrast to the tetrahedron type of network present in the initial state of fused silica). This observation was found to be fully consistent with a recent combined experimental/computational study (Ref 41).
-
3.
Closer examination of the impacted fused-silica internal microstructure revealed the presence of stishovite-like regions characterized by sixfold coordinated Si and threefold coordinated O atoms.
-
4.
To provide justification for the formation of stishovite under ballistic-impact conditions, and for the lack of formation of coesite and α-quartz, quantum-mechanical calculations of the relative stability of different fused-silica polymorphs are carried out and of the transition-state energies associated with the conversion of fused silica into the three possible crystalline phases (identified as α-quartz, stishovite, and coesite). The results obtained clearly revealed that, at high pressures (50 GPa) and in the presence of shear (5% shear strain), stishovite becomes thermodynamically the most stable SiO2 polymorph and the energy barrier associated with the fused-silica → stishovite conversion is the lowest. Thus, both on the thermodynamics and kinetics grounds, the fused-silica → stishovite conversion is preferred, as supported by the molecular-level computational results obtained.
References
W.D. Kingery, H.K. Bowen, and D.R. Uhlmann, Introduction to Ceramics, 2nd ed., Wiley, New York, 1976, p 91–124
C.S. Alexander, L.C. Chhabildas, W.D. Reinhart, and D.W. Templeton, Changes to the Shock Response of Quartz Due to Glass Modification, Int. J. Impact Eng., 2008, 35, p 1376–1385
M. Grujicic, B. Pandurangan, N. Coutris, B.A. Cheeseman, C. Fountzoulas, P. Patel, and E. Strassburger, A Ballistic Material Model for Starphire®, A Soda-Lime Transparent Armor Glass, Mater. Sci. Eng. A, 2008, 492, p 397–411
M. Grujicic, B. Pandurangan, W.C. Bell, N. Coutris, B.A. Cheeseman, C. Fountzoulas, and P. Patel, An Improved Mechanical Material Model for Ballistic Soda-Lime Glass, J. Mater. Eng. Perform., 2009, 18, p 1012–1028
M. Grujicic, B. Pandurangan, N. Coutris, B.A. Cheeseman, C. Fountzoulas, and P. Patel, A Simple Ballistic Material Model for Soda-Lime Glass, Int. J. Impact Eng., 2009, 36, p 386–401
M. Grujicic, W.C. Bell, P.S. Glomski, B. Pandurangan, B.A. Cheeseman, C. Fountzoulas, P. Patel, D.W. Templeton, and K.D. Bishnoi, Multi-length Scale Modeling of High-Pressure Induced Phase Transformations in Soda-Lime Glass, J. Mater. Eng. Perform., 2011, 20, p 1144–1156
M. Grujicic, W.C. Bell, B. Pandurangan, B.A. Cheeseman, C. Fountzoulas, and P. Patel, Molecular-Level Simulations of Shock Generation and Propagation in Soda-Lime Glass, J. Mater. Eng. Perform., 2012, 21, p 1580–1590
M. Grujicic, W.C. Bell, B. Pandurangan, B.A. Cheeseman, C. Fountzoulas, and P. Patel, The Effect of High-Pressure Densification on Ballistic-Penetration Resistance of Soda-Lime Glass, J. Mater. Des. Appl., 2011, 225, p 298–315
M. Grujicic, B. Pandurangan, Z. Zhang, W.C. Bell, G.A. Gazonas, P. Patel, and B.A. Cheeseman, Molecular-Level Analysis of Shock-Wave Physics and Derivation of the Hugoniot Relations for Fused Silica, J. Mater. Eng. Perform., 2012, 21, p 823–836
M. Grujicic, W.C. Bell, B. Pandurangan, B.A. Cheeseman, P. Patel, and P.G. Dehmer, Effect of the Tin- vs. Air-Side Plate-Glass Orientation on the Impact Response and Penetration Resistance of a Laminated Transparent-Armor Structure, J. Mater. Des. Appl., 2012, 226, p 119–143
M. Grujicic, W.C. Bell, B. Pandurangan, B.A. Cheeseman, P. Patel, and G.A. Gazonas, Inclusion of Material Nonlinearity and Inelasticity into a Continuum-Level Material Model for Soda-Lime Glass, J. Mater. Des., 2012, 35, p 144–155
M. Grujicic, B. Pandurangan, and N. Coutris, A Computational Investigation of the Multi-Hit Ballistic-Protection Performance of Laminated Transparent Armor Systems, J. Mater. Eng. Perform., 2012, 21, p 837–848
R. Chakraborty, A. Dey, and A.K. Mukhopadhyay, Loading Rate Effect on Nanohardness of Soda-Lime-Silica Glass, Metall. Mater. Trans. A, 2010, 41, p 1301–1312
O. Tschauner, S.-N. Luo, P.D. Asimow, and T.J. Ahrens, Recovery of Stishovite-Structure at Ambient Conditions Out of shock-Generated Amorphous Silica, Am. Miner., 2006, 91, p 1857–1862
A. Salleo, S.T. Taylor, M.C. Martin, W.R. Panero, R. Jeanloz, T. Sands, and F.Y. Génin, Laser-Driven Formation of a High-Pressure Phase in Amorphous Silica, Nat. Mater., 2003, 2, p 796–800
B. Mantisi, A. Tanguy, G. Kermouche, and E. Barthel, Atomistic Response of a Model Silica Glass Under Shear and Pressure, Eur. Phys. J. B, 2012, 85, p 304–316
A. Kubota, M.-J. Caturla, L. Davila, J. Stolken, B. Sadigh, A. Quong, A. Rubenchik and M. D. Feit, Atomistic response of a model silica glass under shear and pressure. Laser-Induced Damage in Optical Materials 2001, Proceedings of SPIE, Vol 4679, G.J. Exarhos, A.H. Guenther, K.L. Lewis, M.J. Soileau, and C.J. Stolz, Ed., 2002, p 108–116
http://accelrys.com/products/datasheets/materials-visualizer.pdf, accessed August 20, 2014.
Y.H. Tu, J. Tersoff, G. Grinstein, and D. Vanderbilt, Properties of a Continuous-Random-Network Model for Amorphous Systems, Phys. Rev. Lett., 1998, 81, p 4899–4902
S. Nosé, Constant Temperature Molecular Dynamics Methods, Prog. Theor. Phys. Suppl., 1991, 103, p 1–46
M. Grujicic, G. Cao, and R. Singh, The Effect of Topological Defects and Oxygen Adsorbates on the Electronic Transport Properties of Single-Walled Carbon Nanotubes, Appl. Surf. Sci., 2003, 211, p 166–183
H. Sun, COMPASS: An Ab Initio Force-Field Optimized for Condensed-Phase Applications Overview with Details on Alkane and Benzene Compounds, J. Phys. Chem. B, 1998, 102, p 7338–7364
H. Sun, P. Ren, and J.R. Fried, The COMPASS Force Field: Parameterization and Validation for Phosphazenes, Comput. Theor. Polym. Sci., 1998, 8, p 229–246
M. Grujicic, Y.P. Sun, and K.L. Koudela, The Effect of Covalent Functionalization of Carbon Nanotube Reinforcements on the Atomic-Level Mechanical Properties of Poly-Vinyl-Ester-Epoxy, Appl. Surf. Sci., 2007, 253, p 3009–3021
http://accelrys.com/products/datasheets/discover.pdf, accessed August 21, 2014.
http://accelrys.com/products/datasheets/dmol3.pdf, accessed August 21, 2014.
W.J. Hehre, Ab Initio Molecular Theory, Wiley, New York, 1986
W. Kohn and L.J. Sham, Self-Consistent Equations Including Exchange and Correlation Effects, Phys. Rev., 1965, 140, p A1133–1138
M. Grujicic, G. Cao, A.M. Rao, T.M. Tritt, and S. Nayak, UV-Light Enhanced Oxidation of Carbon Nanotubes Through Adsorption of Polar Molecules, Appl. Surf. Sci., 2003, 214, p 289–303
J.P. Perdew, K. Burke, and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77(3865), p 3868 (Erratum Phys. Rev. Lett., 1997, 78, 1396)
T.A. Halgren and W.N. Lipscomb, The Synchronous-Transit Method for Determining Reaction Pathways and Locating Molecular Transition States, Chem. Phys. Lett., 1977, 49, p 225–232
G. Henkelman and H. Jonsson, Improved Tangent Estimate in the Nudged Elastic Band Method for Finding Energy Paths and Saddle Points, J. Chem. Phys., 2000, 113, p 9978–9985
H.J.C. Berendsen, J.P.M. Postma, W.F. van Gunsteren, A. DiNola, and J.R. Haak, Molecular Dynamics with Coupling to an External Bath, J. Chem. Phys., 1984, 81, p 3684–3690
M. Grujicic, K.M. Chittajallu, G. Cao, and W.N. Roy, An Atomic Level Analysis of Conductivity and Strength in Poly (Ethylene Oxide) Sulfonic Acid Based Solid Polymer Electrolytes, Mater. Sci. Eng. B, 2005, 117, p 187–197
A. Tilocca, N.H. deLeeuw, and A.N. Cormack, Shell-model Molecular Dynamics Calculations of Modified Silicate Glasses, Phys. Rev. B, 2006, 73, p 104209(14)
E.W. Dijkstra, A Note on Two Problems in Connexion with Graphs, Numer. Math., 1959, 1, p 269–271
T. Sato and N. Funamori, Sixfold-Coordinated Amorphous Polymorph of SiO2 Under High Pressure, Phys. Rev. Lett., 2008, 101, p 255502
V.V. Brazhkin, Comment on ‘Sixfold-Coordinated Amorphous Polymorph of SiO2 Under High Pressure, Phys. Rev. Lett., 2009, 102, p 209603
T. Sato and N. Funamori, Reply to Comment by V. V. Brazhkin, Phys. Rev. Lett., 2009, 102, p 209603(1)
T. Sato and N. Funamori, High-Pressure Structural Transformation of SiO2 Glass up to 100 GPa, Phys. Rev. B, 2010, 82, p 184102(5)
A. Zeidler, K. Wezka, R.F. Rowlands, D.A.J. Whittaker, P.S. Salmon, A. Polidori, J.W.E. Drewitt, S. Klotz, H.E. Fischer, M.C. Wilding, C.L. Bull, M.G. Tucker, and M. Wilson, High-Pressure Transformation of SiO2 Glass from a Tetrahedral to an Octahedral Network: A Joint Approach Using Neutron Diffraction and Molecular Dynamics, Phys. Rev. Lett., 2014, 113, p 135501(5)
M. Grujicic, K.M. Chittajallu, and J.T. Pukrushpan, Control of the Transient Behavior of Polymer Electrolyte Membrane (PEM) Fuel Cell Systems, J. Automob. Eng., 2004, 218, p 1239–1250
M. Grujicic, P.S. Glomski, T. He, G. Arakere, W.C. Bell, and B.A. Cheeseman, Material Modeling and Ballistic-Resistance Analysis of Armor-Grade Composites Reinforced with High-Performance Fibers, J. Mater. Eng. Perform., 2009, 18(9), p 1169–1182
M. Grujicic, B. Pandurangan, W.C. Bell, C-F. Yen, and B.A. Cheeseman, Application of A Dynamic-Mixture Shock-Wave Model to the Metal-Matrix Composite Materials, Mater. Sci. Eng. A, 2011, 528(28), p 8187–8197
Acknowledgments
The material presented in this paper is based on the work supported by the Office of Naval Research (ONR) research contract entitled “Reactive-Moiety Functionalization of Polyurea for Increased Shock-Mitigation Performance,” Contract Number N00014-14-1-0286.
Author information
Authors and Affiliations
Corresponding author
Additional information
The Publisher has retracted the following article from the Journal of Materials Engineering and Performance at the request of the Editor-in-Chief, following an investigation that revealed extensive duplication of previous publications.
M. Grujicic, J.S. Snipes, S. Ramaswami, R. Yavari and R.S. Barsoum, “All-Atom Molecular-Level Analysis of the Ballistic-Impact-Induced Densification and Devitrification of Fused Silica”, J. Mater. Eng. Perform. (2015), 24, 2970.
The above article is a copy of M. Grujicic, J.S. Snipes, S. Ramaswami, R. Yavari and B. Cheeseman, “Densification and Devitrification of Fused Silica Induced by Ballistic Impact: A Computational Investigation”, J. Nanomater. (2015), Article ID 650625, DOI:10.1155/2015/650625.
We regret the inconvenience caused to our readers.
An erratum to this article is available at http://dx.doi.org/10.1007/s11665-017-2585-y.
About this article

Cite this article
Grujicic, M., Snipes, J.S., Ramaswami, S. et al. RETRACTED ARTICLE: All-Atom Molecular-Level Analysis of the Ballistic-Impact-Induced Densification and Devitrification of Fused Silica. J. of Materi Eng and Perform 24, 2970–2983 (2015). https://doi.org/10.1007/s11665-015-1590-2
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11665-015-1590-2