
Abstract
Significant progress has been made in developing organic compounds by modifying carbazole (Cz) based donor–spacer–acceptor (D–π–A) type dye molecules. The Cz-1–Cz-3 dyes were theoretically designed and experimentally synthesized successfully. Herein, we report the synthesis, photophysical properties and electrochemical characterization (quasi-reversible oxidation processes) of three Cz-based compounds. The calculated results agree well with the available experimental data of absorption spectra, HOMO–LUMO energy levels and band gaps using the time-dependent density functional theory. Furthermore, the results from natural bond orbital calculations were analyzed with the computational electronic structure and charge transfer (conjugative interaction) in molecular systems. The result shows fluorescence time-correlated single-photon counting between 4.32 ns, 4.25 ns, and 13.2 ns, and green to blue (λPL = 431–881 nm) emission for complexes Cz-1–Cz-3 in DMF solution. The Cz-3 compound showed excellent photovoltaic properties, with power conversion efficiency of 5.68%. These results clearly reveal that modification of the electron-withdrawing capability in D–π–A conjugated metal-free organic dyes is an effective way to improve the optical and electrical properties of organic photovoltaic (PV) devices.
Graphical Abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The global energy crisis stems from the foreseeable end of the cycle of coal, and the reduction of fossil fuels has laid the foundation for the search for alternate energy solutions such as solar energy. Cheap energy generation, pollution-free hydropower, wind energy, geothermal energy, and harnessing solar cells and biomass energy have been identified as hopeful clean alternative renewable energy sources. Dye-sensitized solar cells (DSSCs) have been widely regarded as a new generation of promising light-harvesting devices. DSSCs have drawn great interest since their first report by O’Regan and Gratzel in 1991.1 Furthermore, the high power conversion efficiencies (PCE), chemical versatility, energetic and molecular structure tenability, simple fabrication processes, low-cost manufacturing processes, and environmental friendliness of photovoltaic devices have made DSSCs more popular. To further improve the performance of DSSCs in fulfilling future energy demands, metal-free organic dyes play a pivotal role in increasing light-harvesting efficiency by the electron injection process. The ruthenium (Ru)-complex-based DSSC sensitizer (N719 has been reported as an efficient sensitizer) with a PCE of more than 11% under global air mass 1.5 (AM 1.5G) illumination.2 At present, Zn porphyrin DSSC sensitizers (SM315) have achieved an overall PCE of over 13% using cobalt-based redox electrolyte. Donor-bridge-acceptor (D–π–A) organic sensitizers such as [bis-(dimethylfluorenyl)-amino] phenyl, carbazole, coumarin, indoline, cyanine, triphenylamine, and phenothiazine have been of interest in PCE in recent years, making them attractive candidates for use as sensitizers for DSSCs.3,4,5,6,7,8 The architecture of the metal-free sensitizers possesses a donor–π–acceptor conjugated (D–π–A) structure, where D represents an electron-donating moiety and A is an electron-accepting moiety linked by a π-conjugating spacer, presents promising development in view of their comparably high efficiency, low cost, and easier preparation and purification.
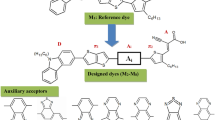
Carbazole (Cz) is a conjugated unit that possesses good hole transporting material in organic devices and can harvest and transfer energy efficiently.9 Cz-based compounds are widely used as building blocks in π-conjugated compounds because of their rigid structures and photovoltaic technology, specific optoelectronic properties, and use as a host material in electroluminescent devices, electronics applications, chemistry and organic photovoltaics. Chemically, Cz is easily functionalized by the five-membered ring at the 3, 6, and 9 (NH) positions of indole (equivalent to the 9a–4a double bond in Cz) and covalently bonded to other molecular compounds. As per a literature review, quite a few bulky Cz and its derivative materials have been reported with increased narrow conjugation through long synthetic procedures, resulting in rather expensive compounds. Moreover, many of these synthetic techniques are quite complex and tedious, including a number of steps that result in low yields. Zhao et al.10 reported the effect of substituents in a series of Cz-based porphyrin dyes employed as sensitizers in dye-sensitized solar cells. In recent years, Sivanadanam et al. reported the synthesis, design, and investigation of three other complexes of similar structures, phenyl-based donors (TBC, TMC, OMC, PC, TBR, TMR, OMR, and PR) or aryl amine donors (OMNC, CNC, and HNC). The calculated outcomes show that their optical, electrochemical, and photovoltaic properties, such as chemical properties, cyclic voltammograms, absorption, and emission spectra, are affected by the different donor and acceptor moieties of these studied derivatives, and have promising potential for application in solar cells. The phenyl compounds show the highest short-circuit current (JSC) of 6.84 mA cm−2 with external stability leading to an efficiency of 3.33%. In the case of the sensitizers with hexyloxy-substituted phenyl amine donors, the increasing bulkiness of the donor group leads to increasing open-circuit potential.11 In the present work, we designed and characterized three dyes in this study by incorporating various acceptors between the carbazole donor and the 2-chlorophenothiazine-linker (the optimized structures of the Cz-1, Cz-2, and Cz-3 molecules are shown in Fig. 1). The UV–Vis experimental and theoretical absorption spectra were compared with the simulated data, photoluminescence (PL) emission character, molecular electrostatic potential (MEP), morphological analysis, chemical reactivity and natural bond orbital analysis (NBO). The spectra were also used to study the charge transfer properties in the complex and the electron analysis. This powerful approach is capable of creating instinctive chemical representations of the complex inhibition mechanical charge transfer within the HOMO–LUMO structure. Further, the design dyes allowed us, in a few steps and with good yields, to obtain dyes with remarkable device performance, achieving a very promising efficiency value of 5.68% with the Cz-3 dye.

Optimized ground state geometries of the molecules.
Materials
The materials are 9H-carbazole, triethylamine, ethyl thiophene-2-glyoxylate, 2-chlorophenothiazine and 4-nitrophenol. The materials were purchased from Alfa Aesar.
Experimental Section
Scheme 1 depicts the screening of acceptor moieties that play a significant role in donor–π–linker acceptor (D–π–A) type dyes for metal-free organic DSSC performance. The purpose and goals of this research are the quantum-chemical design of a carbazole series of promising photophysical and optoelectronic materials based on carbazole donors and 2-chlorophenothiazine spacers by structurally modifying them with the three different acceptor units and calculating their electrochemical and photophysical properties. Herein, we theoretically designed and synthesized metal-free organic Cz-1 to Cz-3 dyes. Our designed metal-free organic compounds are composed of two main dye units, 9H-carbazole and 2-chlorophenothiazine, which act as donor and spacer parts. Three designed molecules using three acceptor parts, (i) triethylamine, (iii) ethyl thiophene-2-glyoxylate, and (iii) 4-nitrophenol, were carried out theoretically, and the experimental MS data are depicted in Supplementary Fig. 1.

Chemical structures of materials.
N-((7-(9H-Carbazol-9-yl)-8-Chloro-1,2,4a,10,10a-Pentahydro-4aH-Phenothiazin-2-yl)Methyl)-N-Ethylethanamine (Cz-1)
Carbazole (2.4 g) was dissolved in DMF (20 mL) solvent in a 100-mL round-bottom flask at room temperature under an argon atmosphere and stirred at 60°C for 1 h. Then, 2-chlorophenothiazine (0.58 g) was slowly added. Again, the reaction mixture was stirred at room temperature for 1 h under an argon atmosphere at 80°C. The reaction was magnetically stirred for an additional 20 min. After preparing the precursor solution, triethylamine (1.4 g) was added to the reaction mixture at 60°C, and the reaction mixture was allowed to stir at room temperature for 1 h. Then, after cooling to room temperature, the organic layer was washed with brine and dried with Na2SO4. It was crystallized from the chloroform mixture to obtain a pure sample product, Cz-1, as a rose-colored powder with a yield of 83%.
Ethyl 2-(5-(7-(9H-Carbazol-9-yl)-8-Chloro-1,2,4a,10,10a-Pentahydro-4aH-Phenoth Iazin-2-yl)Thiophen-3-yl)-2-Oxoacetate (Cz-2)
In a similar procedure, carbazole (2.4 g) was put into a 100-mL round-bottomed flask equipped with a magnetic stirrer. The 2-chlorophenothiazine (0.58 g) mixture was dissolved in DMF solvent (20 mL) after slow addition, and the reaction mixture was stirred at room temperature for 1 h under 80°C. After preparing the precursor solution and ethyl thiophene-2-glyoxylate, 0.6 mL was added dropwise into the reaction mixture, stirred at 60°C for 1 h, then cooled to room temperature for 1 h. The product was extracted with chloroform solvent and washed to obtain a pure sample product, Cz-2, as a rose-colored powder with a yield of 62%.
7-(3-Nitrophenoxy)-3-(9H-Carbazol-9-yl)-2-Chloro-10H-Phenothiazine (Cz-3)
A mixture of carbazole (2.4 g) was dissolved in DMF solvent (20 mL) followed by the addition of 2-chlorophenothiazine (0.58 g), and the reaction mixture was stirred at 60°C for 1 h. The mixture was warmed to 80°C and cooled to room temperature, followed by the slow addition of 4-nitrophenol (1.085 g). After this addition, the mixture was warmed to 60°C and stirred for 1 h. After completion of the reaction, the solvent was evaporated to dryness. The product was extracted with chloroform solvent and washed to obtain a pure sample product, Cz-3, as a red powder with a yield of 75%.
Dye Sensitized Solar Cell Fabrication
Fluorine-doped tin oxide (FTO) as conducting glass was used for the counter electrode to current (8 mm scattering). The working electrode glass plate was immersed in a 40 mM aqueous TiCl4 solution at 80°C for 30 min to produce 20-nm-sized nanocrystalline TiO2 particles before cleaning with double-distilled water and ethanol. A layer of nanocrystalline TiO2 paste was spin-coated on the electrode glass plate using the doctor blade method and a round mask was made by adhering to a 60-μm-thick piece of polyester tape (3M Magic). The transport dye molecule (Cz-3) was dissolved in the DMF solution by heating and stirring the solution at 80°C for 30 min. The film was sintered at 500°C for 30 min and additional layers of prepared precursor solution were applied, which were gradually heated. The electrode was ready by immersing the 13.0 mm (8.0-mm-thick transparent layer and 5.0-mm-thick scattering) nanocrystalline TiO2 film into the dye solution for 10 h. A photocathode was placed on top of the counter electrode and clipped to form a cell, and a 3-dimethylimidazolium iodide electrolyte solution was injected into the seam between the two electrodes.
Model and Computational Details
All first-principles calculations of the B3LYP (Becke’s 3-parameter, Lee, Yang and Parr) exchange–correlation hybrid functional in density functional theory (DFT) were performed with the Gaussian 09W package.12,13,14,15 All calculations, which include the molecular geometries of all complexes studied, were performed on isolated systems using the hybrid functional level of B3LYP theory. A vibrational frequency analysis was carried out in order to confirm the minimum-energy geometries. Electron analyses were carried out using the NBO 3.1 program,16 performed at the hybrid DFT/B3LYP/6-311++G(d,p) level implemented in the Gaussian 09W package. These NBO Fock matrix calculations were accomplished in order to realize various second-order perturbation interactions between the filled orbitals in donor and acceptor moieties and unoccupied orbitals of another subsystem to subsequently obtain a measure of intra- and intermolecular bond delocalization and hyperconjugation systems. The linear-response TD-DFT level model was used to analyze energies, 13C and 1H NMR, UV–Vis spectra, and the oscillator strengths of electronic spin-allowed singlet–singlet transitions. The solvent influence was treated using the polarizable continuum model (PCM) developed by Tomasi and co-workers.17,18,19 The electronic calculations, such as 3D surfaces for frontier molecular orbitals and hyperpolarizability analysis, were estimated and visualized via the GaussView 5 software package.20 Molecular studies such as chemical potential, electronegativity, molecular hardness, global softness, and ionization potential have been realized from HOMO–LUMO and their respective energy gaps, employing quantum chemical TD-DFT at the B3LYP/6-311++G(d,p) levels. The Gaussian 09W package using quantum chemical calculation was used to plot electrostatic potential surface analyses (MEP) for the molecule using the GaussView 5 program package.
Result and Discussion
Effects of Chemical Modifications
Over the years, energies of frontier molecular orbitals (FMOs) have been used to obtain vital parameters of organic molecules with the nature of the transition.21,22,23 The 3D plots of FMO energy levels along with the HOMO–LUMO gaps of compounds Cz-1–Cz-3 are calculated with the DFT/6-311++G(d,p) model quantum mechanism and are furnished in Fig. 2, respectively. The highest occupied MOs, the outermost orbitals containing electrons, have the affinity to give electrons and act as electron donors. On the other hand, in the lowest unoccupied MOs, the innermost orbital contains a vacant electron donor, an electron acceptor.23 In the three compounds, Cz-1–Cz-3, the HOMO were charge-distribution localized over the dye molecules with a high electron density on the acceptor moiety, but the LUMO is characterized by a charge density involving the n–π* transition. Obviously, the HOMO–LUMO perfectly shows the donor–acceptor character of the Cz and methoxy groups. The electronic transition from the HOMO to LUMO energy level is linked with electron charge density from the 10H-phenothiazine parts to the C–C and N–C bond parts. The HOMO shape of Cz-based dye molecules is mostly located both over the acceptor moiety and around C2–C6, C5–C4, C2–C3, C3–C4, C21–C22, C23–C19, and C20–C21 bonds. The LUMO shapes of the ethylethanamine forms are mostly delocalized over N29–C31, C13–C12, C34–O36, C34–O35, N29–O38, and N29–N37 bonds and link atoms between aromatic rings.

Isosurface plots relevant molecular orbitals involved in the excited states of highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) for molecules (green regions (negative), decrease of electron density; red regions (positive), increase of electron density) (Color figure online).
Moreover, the energy gap values of Cz-1, Cz-2 and Cz-3 between the HOMO and LUMO are 3.33 eV, 2.28 eV and 2.68 eV, respectively. The energy gap between MOs has been confirmed to show solar cells based on intramolecular charge transfer (ICT). The results indicated that the HOMO–LUMO excitation induced by light moves the electron distribution from the Cz to the triethylamine/ethyl thiophene-2-glyoxylate/4-nitrophenol moiety.
The calculated HOMO and LUMO energy24,25,26,27,28 values for frontier molecular orbital descriptors help us to identify the chemical potential (μ) = \((IP + EA)/2\), ionization potential (IP) = -EHOMO, electron affinity (EA) = -ELUMO, chemical hardness (ƞ) = \((IP - EA)/2\), global softness (S) = \(1/\eta\), electrophilicity (ω) = \(\mu^{2} /2\eta\) and electronegativity (χ) = \(- (IP + EA)/2\) for all the studied species in the solvent phase at the DFT/B3LYP/6-311++G(d,p) level of theory. The dye molecules obtaining small band gap energy have more polarizability. The smaller gap is generally related to the low kinetic stability and high chemical reactivity of the dye molecules. The computational values of the global hardness, softness, chemical potential, electronegativity, and electrophilicity index of our dye molecules, Cz-1 to Cz-3, are shown in Table 1.
Photophysical Properties
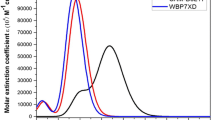
Figures 3 and 4 show the simulated absorption spectra (DMF) overlaid with experimental spectra, and transitions were compared well with the experimental spectra of Cz compounds. The spectra were recorded in DMF at room temperature, and the λmax values were found by using TD-DFT calculation at the 6-311++G(d,p) level basis set.29,30 The optical and electrochemical parameters are tabulated in Table II. The Cz-1–Cz-3 dye molecules with two important electronic transitions were described for each compound for calculated and experimental bands. One has the largest absorption band, and the other electronic transition has a maximum wavelength λmax of 461–554 nm. An experimental band appears at 336–530 nm and the other is associated with an intramolecular charge transfer (ICT) excitation from the Cz donor group based on the triethylamine, ethyl thiophene-2-glyoxylate, and 4-nitrophenol acceptor units of the present compounds. The theoretical absorption spectra (λmax) are in the order of 461 nm, 424 nm and 554 nm, and the experimental values of 525 nm, 336 nm and 442 nm for Cz compounds can be ascribed to a π–π* transition, indicating a better light-harvesting ability of these Cz metal-free organic compounds. The higher value of absorption wavelength for Cz-2 and Cz-3 is due to the extended conjugation with 2-chlorophenothiazine linker and triethylamine, ethyl thiophene-2-glyoxylate, and 4-nitrophenol units. An increased number of Cz units leads to a higher molar extinction coefficient due to the large oscillator strength of the electronic charge transition.

Simulated absorption spectra of the molecules.

Experimental UV–Vis spectra on oxidation states by carbazole molecules (a) Cz-1, (b) Cz-2, (c) Cz-3.
The experimental fluorescence spectra of compounds Cz-1–Cz-3 in the solid state at room temperature are shown in Fig. 5. The emission profiles of all compounds are similar, with almost superimposable emissions observed at 487–881 nm with the redshifted region. In addition, the fluorescence spectra of Cz molecules demonstrate a significant redshift with increasing solvent polarity, which is indicative of the CT character of the fluorescent state. The fluorescence lifetimes of all Cz functionalized compounds determined from the TCSPC ranged between 4.32 ns and 13.2 ns. The essential photophysical properties in solid states are tabulated in Table III for solar cell applications.

Normalized fluorescence emission spectra of (a) Cz-1, (b) Cz-2, (c) Cz-3.
Electrochemical Properties
Cyclic voltammetry (CV) was performed to determine the ground state oxidation-redox potential corresponding to the HOMO, while the LUMO was obtained from the values of (Eox) of the Cz-1–Cz-3 compounds using DMF solution (Fig. 6a, b, and c).31,32,33 As can be seen, the cyclic voltammograms of all Cz-1–Cz-3 compounds show reversible oxidation waves, indicating stable electrochemical properties. The CV of Cz-1–Cz-3 shows the first quasi-reversible oxidation wave with an anodic peak potential (E1/2ox) at −2.891 V, −2.378 V, and −2.29 V corresponding to the formation of the cation-radical and the related data are furnished in Table III and also graphically depicted in Fig. 6d. The HOMO level of the Cz complexes was determined from the onset of the redox (short oxidation reaction) potentials with regard to the energy level (4.3 eV), and the LUMO level was deduced from the HOMO energy levels determined by the onset of absorption. The HOMO levels determined from the half-wave oxidation potentials were −5.0 eV for Cz-1, −4.3 eV for Cz-2, and −3.08 eV for Cz-3, respectively. Calculation of the LUMO level of Cz compounds was exhibited at −1.67 eV for Cz-1, −1.47 eV for Cz-2, and 0.4 eV for Cz-3. The results reveal that the introduction of the Cz-3 dye molecule leads to their better hole-transporting properties.

Cyclic voltammograms for (a) Cz-1, (b) Cz-2, (c) Cz-3 recorded in DMF solution, and (d) computed electronic distribution of the first occupied/unoccupied molecular orbitals of the studied species and the CB energy level of LiF/AI.
Natural Bond Orbital Analysis
NBO analysis is one of the most efficient approaches to the total electronic wave function of the system. It is expressed first as a set of natural atomic orbitals (NAOs), which are the natural orbitals of isolated atoms, defined as eigenfunctions of the first-order density matrix.34 However, the NBO is derived from a quantum mechanical calculation on the system at some level of theory, typically DFT or HF, using a suitable basis set. They are classified in the NBO analysis as core electron pairs (CR), lone-pair (LP), anti-bond pairs (BD*), and bond pairs of electrons (BD), the first two extending over a single atom and the last over two atoms. They are orthogonalized to each other to form the NBO occupied orbital set, the "Lewis" orbitals (donor) NBOs, with reference to the Lewis bonding theory.16 There are also unoccupied or "non-Lewis" orbitals (acceptors), comprising anti-bond and "Rydberg" extra valence natural atomic orbitals. This remaining delocalization does not take place in the natural Lewis-type structure representation and can be quantified in terms of the intra-molecular interactions between donor NBOs (i) and acceptor NBOs (j). The determination of the stabilization energy E(2) is connected with electron delocalization between donor–acceptor interaction, which is estimated as
where \(q_{i}\) is the donor orbital occupancy, \(\varepsilon_{i}\) and \(\varepsilon_{j}\) are diagonal elements (orbital energies) and \(F(i,j)\) is the off-diagonal NBO matrix element. In the present study, taking into account NBO analysis, we report some of the electron donor–acceptor charge transfer orbital interactions and the interaction stabilization energy. The NBO calculation has been performed on the title molecules at the DFT/B3LYP/6-311++G(d,p) level to explain the intra-molecular hybridization and delocalization of charge density within the detailed data that are summarized in Table IV.
The obtained intermolecular interaction results for the three BD-Pairs BD (2) C1-C2, BD (2) C11-C12, BD (2) C13-C14 of Lewis orbitals or electron donors (i), and BD*(2) C5-C6, BD*(2) C8-C9, BD*(2) C18-C19 in non-Lewis interaction (j), as well as the three BD*-BD* orbitals of carbon. NBO analysis assumes BD*(2) C1-C2, BD*(2) C8-C9, and BD*(2) C11-C12 of carbon. The most important conjugative charge transition state mode is for Cz-1 between the electron donor interaction of the N7 lone pair orbital and the C8–C9 anti-bonding acceptor orbital σ* with smaller E(2) value (36.21) in comparison to the Cz-2 value (19.39) and the Cz-3 (13.75). These results indicate reduced charge transfer and intermolecular orbital interaction for Cz-1. The electron donates from the bonding orbitals of the C–C anti-bonding orbital and gives BD (2) C11–C12. The weaker intermolecular orbital interaction and compounds Cz-1 and Cz-2 have BD (2) C11-C12 values of 10.26 and 25.25, respectively, which contributes to weak interactions where the charge transition state of Cz-3 is very high. Compound Cz-3 had the highest E(2) (179). This was ascribed to the strong charge transfer and intermolecular interactions between the anti-bonding of C11–C12 of the benzene ring and the anti-bonding orbital of the C13–C14 ring.
Structure and Working Principle of Dye-Sensitized Solar Cells (DSSC)
An advanced DSSC, the Grätzel cell, contains five mechanisms: conductive glass, solid-state semiconductor, photosensitizers, a counter electrode and a photoanode. The device structure of DSSCs is illustrated in Fig. 7. In brief, the solar cell devices are composed of a transparent conducting oxide (TCO) substrate, a mesoporous semiconductor (predominately TiO2) film adsorbed with photosensitizer dyes, an electrolyte layer containing a redox couple, and a counter electrode. The photosensitizer dye can absorb sunlight and create a high-energy state from which a photo-excited electron is injected into the conduction band of the TiO2 semiconductor. After percolation through the semiconductor film (mesoscopic), the excited dye and injected electrons are collected by the conduction band edge of the oxide material. Subsequently, the oxidized dye sensitizers (S+) generated after excited electron injection are compacted to their neutral state by the reducing species (generally iodide ions) in the electrolyte solution. The species in the electrolyte diffuse into the counter electrode and receive the external circuit electrons to complete the whole circuit. This cycle is repeated indefinitely with no material being consumed but with energy being transformed from light to electricity. Among the three dye molecules, more light harvesting efficiency has been obtained in the particularly designed dye molecule.

Schematic diagram of DSSC basic structure. Principle of operation and energy level scheme of the dye-sensitized nanocrystalline solar cell. Photo-excitation of the sensitizer (S) is promoted into an electronically excited state (S*) is followed by electron injection into the conduction band of the TiO2 semiconductor.
SEM Analysis
Scanning electron microscope (SEM) images are used to analyze the morphology of the synthesized material. As shown in Fig. 8, the diameters of Cz compounds were in the range of 50–150 nm. It is observed from the image that the sample contains some irregularly shaped particles and a clear change in particle size was observed on making the particles in DMF solvent. The Cz-1, Cz-2 and Cz-3 with D–π–A character with high emission would find a variety of applications in blue organic photovoltaic devices.

Scanning electron microscope (SEM) images.
Electrostatic Potential Surface Analysis
The concept of molecular electrostatic potential (MEP) is extremely useful in understanding electrophilic and nucleophilic attack sites, as well as hydrogen bonding interactions.35,36 The total electron density map with electrostatic potential in the background of quantum chemical calculation results from the 3D image visualization in the GaussView 5 program. The different values of the electrostatic potential on the surface are represented by different colors; the most positive potential region (blue color) is spread over the carbazol group which indicates the nucleophilic reactions, and the most electronegative potential region (red color) of the MEP surface represents the electrophilic reactions of charged point-like reagents on organic molecules, as shown in Fig. 9. These regions can understand the location of interactions of these complexes when they get involved in electrophilic or nucleophilic interactions.

ESP surface plot of molecules (a) Cz-1, (b) Cz-2, (c) Cz-3 (Color figure online).
1H and 13C NMR Spectral Analysis
The computational and experimental chemical shifts (1H and 13C NMR) of designed dyes were recorded. The theoretically calculated 1H and 13C NMR spectra were obtained using GIAO methods and optimized molecular structures, with a hybrid B3LYP functional method and a 6-311++G(d,p) basis set. The experimental 1H and 13C NMR spectra of designed dyes were obtained in CDCl3 shown in Fig. 10a and b, and the theoretical chemical shifts were given in Supplementary Fig. 2a and b. In the present investigation, the aromatic ring protons (such as benzene) give rise to 13C NMR signals in frequently overlapping areas of the spectrum through the chemical shift range of 100–150 ppm. The observed 13C NMR chemical shifts (in ppm) of the hetero-aromatic carbons (C1, C3, C4, C5, and C6) are in the range of 114.7–139.8 ppm. The carbonyl functional group is the more electronegative of the carbon atoms and also polarizes the electron density distribution. Therefore, the calculated 13C NMR peaks of C1, C3, C4, C5 and C6 atoms fused to the carboxyl bonded group are too high, with experiential chemical shift values of 128.3 ppm, 117.6 ppm, 136.0 ppm, and 116.7 ppm for Cz-1 dye, at 129.5 ppm, 138.8 ppm, 117.4 ppm, 119.9 ppm for Cz-2 dye and at 129.8 ppm, 137.9 ppm, 118.0 ppm, and 120.3 ppm for the Cz-3 molecule, simulated at 129.4 ppm, 114.7 ppm, 139.8 ppm, 115.9 ppm and 120.5 ppm. Similarly, the chemical shift (δ) values of C12 (NH) and C2 (Cl) atoms are large, compared to donor unit carbon groups. The nitrogen and chlorine atoms show a de-shielding effect of the carbon nucleus in the NMR signals of 129.5, 143.0, 158.6 ppm, and 130.4 (Cz-1), 134.0 (Cz-2), 134.4 (Cz-3) ppm to downfield. The values were theoretically predicted at 129.3, 141.5, 123.3 (C12) and 132.1 (C2) ppm. Furthermore, other carbon atoms in the Cz donor unit appear at much larger peak values of NMR shift at 136.3 (C26), 138.7 (C21), and 137.4 (C20) ppm, which is the double bond of the amine group. The acceptor units found at 57.1 (C17), 15.4 (C21) ppm for Cz-1 dye, 140.5 (C32), 164.5 (C39), 57.8 (C36), 19.4 (C34) ppm for Cz-2 dye, and at 158.2 (C30), 125.1 (C31), 132.7 (C32), 146.3 (C34), ppm for Cz-3 dye display that the carbon atoms of ethyl thiophene-2-glyoxylate, 2-chlorophenothiazine and 4-nitrophenol parts are in different environments. These peaks are in good agreement with the experimental data. The 13C NMR data are listed in Table V, and atom positions were chemically shifted according to supplementary Fig. 3, respectively.

(a) Experimental 13C-NMR spectra for designed dyes. (b) Experimental 1H-NMR spectra for designed dyes.
The chemical shift (δ) value provides information on the chemical environment/magnetic properties of the protons. Proton donating groups are shielded, and proton withdrawing groups are de-shielded. In the designed dyes, the experimental δ peaks of hydrogen atoms in the benzene and pyrrole rings were measured in the region of 3.42–7.55 ppm and theoretically calculated in the region of 3.83–7.63 ppm. The proton numbers H37, H45 (1), H37, H59, H60 (2), and H58, H55 and H41 (3) atoms were observed with values of 4.0, 2.51 (1) ppm, 3.8, 1.42 (2) ppm, and 3.99, 7.14, and 7.64 (3) ppm, which mark the aromatic hydrogens of the amino and oxygen atoms. The observed H37 proton chemical shift in the secondary Cz group appears at the highest peaks of 4.05 (1), 3.83 (2), and 3.99 (3) ppm due to the electronegative amino group. Furthermore, the 1H NMR spectra of the dye materials show proton triplets in the high shift values of 1.42–3.48 ppm for H45, H60, and H43, and the proton multiples in the observed peaks at 1.30–3.42 ppm are shown in Table VI. These experimental values are in good agreement with the calculated data. The 1H NMR spectra of the atom positions and shift values are shown in Supplementary Fig. 4.
Photovoltaic Properties
Figure 11 shows the high power conversion efficiencies (PCE) of the organic DSSCs sensitized by Cz dye materials calculated by measuring the J-V characteristics at 100 mW cm−2 intensity under simulated AM 1.5G irradiance (i.e., 1 sun) in solar light. The important electrical parameters used to characterize the photovoltaic performance of DSSCs are the short-circuit current density (Jsc), open-circuit voltage (Voc), fill factor (FF), and conversion efficiency (η) and are listed in Table VII. The DSSC device sensitized by the Cz-3 dye has achieved the highest energy conversion efficiency of 5.68% with Jsc value of 11.69 mA cm−2, Voc of 0.726 V, and FF of 0.761, compared to the other dyes. Furthermore, organic solar cells based on Cz-1 and Cz-2 dyes have efficiencies of 3.73% (7.08 mA cm−2 (Jsc), 0.714 V (Voc) and 0.691 (FF), and 3.97% (8.37 mA cm−2 (Jsc), 0.720 V (Voc) and 0.713 (FF), respectively. Therefore, the Cz-3 dye shows better photovoltaic performance than the Cz-1 and Cz-3 dyes due to the implanted electron-withdrawing acceptor unit (4-nitrophenol), which significantly increases the photo-conversion efficiency.

J–V characteristics of the photovoltaic device is using the molecule Cz-1–Cz-3.
Conclusion
In summary, we have successfully designed and synthesized a D–π–A series of dyes containing carbazole donor, 2-chlorophenothiazine spacer, and conjugation pathway with different acceptor units, namely triethylamine, ethyl thiophene-2-glyoxylate and 4-nitrophenol. The donor exhibited intense absorptions with high molar extinction coefficients, while the 4-nitrophenol-linked dye showed a redshift in intramolecular charge transfer transition. Theoretical calculations using TD-DFT have shown the contribution of π-type transitions to longer wavelength absorption. Surprisingly, the dye with the highest contribution from π-type transitions to visible region absorption has resulted to efficient photocurrent generation. The synthesized compounds exhibit smaller HOMO–LUMO energy gaps, stronger absorption with the redshift, driving force of electron injection, dye regeneration and a long excited-state lifetime. The DSSC based on Cz-3 (Voc = 0.726 V, Jsc = 11.69 mA cm−2, FF = 0.761 and η = 5.68%) dye yielded higher power conversion efficiency (PCE) as compared to Cz-1 and Cz-2 dyes. In summary, our results show that the designed dyes, particularly the Cz-3 dye have been identified as promising candidate for high-efficiency DSSC.
Data Availability
The raw/processed data required to reproduce these findings cannot be shared at this time as the data also form part of an ongoing study.
References
B. O’Regan and M. Grätzel, A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO2 films. Nature 353(6346), 737–740 (1991).
X. Zhang, F. Gou, J. Shi, H. Gao, C. Xu, Z. Zhu, and H. Jing, Molecular engineering of new phenothiazine-based D-A–π–A dyes for dye-sensitized solar cells. RSC Adv. 6(108), 106380–106386 (2016).
Z.S. Wang, Y. Cui, K. Hara, Y. Dan-oh, C. Kasada, and A. Shinpo, A high-light-harvesting-efficiency coumarin dye for stable dye-sensitized solar cells. Adv. Mater. 19(8), 1138–1141 (2007).
B. Liu, W. Zhu, Q. Zhang, W. Wu, M. Xu, Z. Ning, Y. Xie, and H. Tian, Conveniently synthesized isophorone dyes for high efficiency dye-sensitized solar cells: tuning photovoltaic performance by structural modification of donor group in donor–π–acceptor system. Chem. Commun. 13, 1766–1768 (2009).
C. Zafer, M. Kus, G. Turkmen, H. Dincalp, S. Demic, B. Kuban, Y. Teoman, and S. Icli, New perylene derivative dyes for dye-sensitized solar cells. Sol. Energy Mater. Sol. Cells 91(5), 427–431 (2007).
X. Ma, J. Hua, W. Wu, Y. Jin, F. Meng, W. Zhan, and H. Tian, A high-efficiency cyanine dye for dye-sensitized solar cells. Tetrahedron 64(2), 345–350 (2008).
S. Hayashi, M. Tanaka, H. Hayashi, S. Eu, T. Umeyama, Y. Matano, Y. Araki, and H. Imahori, Naphthyl-fused π-elongated porphyrins for dye-sensitized TiO2 cells. J. Phys. Chem. C 112(39), 15576–15585 (2008).
P. Shen, Y. Liu, X. Huang, B. Zhao, N. Xiang, J. Fei, L. Liu, X. Wang, H. Huang, and S.T. Tan, Efficient triphenylamine dyes for solar cells: effects of alkyl-substituents and π-conjugated thiophene unit. Dyes Pigm. 83(2), 187–197 (2009).
F. Sanda, T. Nakai, N. Kobayashi, and T. Masuda, Synthesis of polyacetylenes having pendant carbazole groups and their photo-and electroluminescence properties. Macromolecules 37(8), 2703–2708 (2004).
L. Zhao, P. Wagner, A.B.S. Elliott, M.J. Griffith, T.M. Clarke, K.C. Gordon, S. Moric, and A.J. Mozer, Enhanced performance of dye-sensitized solar cells using carbazole-substituted di-chromophoric porphyrin dyes. J. Mater. Chem. A 2, 16963–16977 (2014).
J. Sivanadanam, P. Ganesan, P. Gao, M.K. Nazeeruddin, A. Emeline, D. Bahnemann, and R. Rajalingam, Impact of strength and size of donors on the optoelectronic properties of D–π–A sensitizers. RSC Adv. 6(44), 37347–37361 (2016).
M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G.A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H.P. Hratchian, A.F. Izmaylov, J. Bloino, G. Zheng, J.L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, J. Honda, O. Kitao, H. Nakai, T. Vreven Jr., J.A. Montgomery, J.E. Peralta, F. Ogliaro, M. Bearpark, J.J. Heyd, E. Brothers, K.N. Kudin, V.N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J.C. Burant, S.M. Iyengar, J. Tomasi, M. Cossi, R. Rega, J.M. Millam, M. Klene, J.E. Knox, J.B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R.E. Stratmann, O. Yazyev, A.J. Austin, R. Cammi, C. Pomelli, J.W. Ochterski, R.L. Martin, K. Morokuma, V.G. Zakrzewski, G.A. Voth, P. Salvador, J.J. Dannenberg, S. Dapprich, A.D. Daniels, O. Farkas, J.B. Foresman, J.V. Ortiz, J. Cioslowski, and D.J. Fox, Gaussian 09 Revision A02 (Wallingford: Gaussian Inc, 2009).
A.D. Becke, Density-functional thermochemistry. I. The effect of the exchange-only gradient correction. J. Chem. Phys. 96(3), 2155–2160 (1992).
A.D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 38, 3098 (1988).
C. Lee, W. Yang, and R.G. Parr, Development of the Colle–Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785 (1988).
A.E. Reed and F. Weinhold, Natural localized molecular orbitals. J. Chem. Phys. 83(4), 1736–1740 (1985).
S. Miertuš, E. Scrocco, and J. Tomasi, Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys. 55(1), 117–129 (1981).
S. Miertus and J. Tomasi, Approximate evaluations of the electrostatic free energy and internal energy changes in solution processes. Chem. Phys. 65(2), 239–245 (1982).
M. Cossi, V. Barone, R. Cammi, and J. Tomasi, Ab initio study of solvated molecules: a new implementation of the polarizable continuum model. Chem. Phys. Lett. 255(4–6), 327–335 (1996).
R.I. Dennington, T. Keith, and J. Millam, GaussView, Version 508 (Shawnee: Semichem. Inc, 2009).
R.G. Pearson, Absolute electronegativity and hardness correlated with molecular orbital theory. Proc. Natl. Acad. Sci. 83(22), 8440–8441 (1986).
R.V. Solomon, P. Veerapandian, S.A. Vedha, and P.A. Venuvanalingam, Tuning nonlinear optical and optoelectronic properties of vinyl coupled triazene chromophores: a density functional theory and time-dependent density functional theory investigation. J. Phys. Chem. A 116(18), 4667–4677 (2012).
Y. Xue, Y. Dou, L. An, Y. Zheng, L. Zhang, and Y. Liu, Electronic structure and spectral properties of aurones as visible range fluorescent probes: a DFT/TDDFT study. RSC Adv. 6(9), 7002–7010 (2016).
G. Gece, The use of quantum chemical methods in corrosion inhibitor studies. Corros. Sci. 50(11), 2981–2992 (2008).
R.G. Parr, L.V. Szentpaly, and S. Liu, Electrophilicity index. J. Am. Chem. Soc. 121(9), 1922–1924 (1999).
P.K. Chattaraj, B. Maiti, and U. Sarkar, Philicity: a unified treatment of chemical reactivity and selectivity. J. Phys. Chem. A 107(25), 4973–4975 (2003).
R.G. Parr, R.A. Donnelly, M. Levy, and W.E. Palke, Electronegativity: the density functional viewpoint. J. Chem. Phys. 68(8), 3801–3807 (1978).
R.G. Parr and R.G. Pearson, Absolute hardness: companion parameter to absolute electronegativity. J. Am. Chem. Soc. 105(26), 7512–7516 (1983).
R.G. Parr and P.K. Chattaraj, Principle of maximum hardness. J. Am. Chem. Soc. 113(5), 1854–1855 (1991).
T. Iijima, A. Momotake, Y. Shinohara, T. Sato, Y. Nishimura, and T. Arai, Excited-state intramolecular proton transfer of naphthalene-fused 2-(2′-hydroxyaryl) benzazole family. J. Phys. Chem. A 114(4), 1603–1609 (2010).
X. Ren, J. Li, R.J. Holmes, P.I. Djurovich, S.R. Forrest, and M.E. Thompson, Ultrahigh energy gap hosts in deep blue organic electrophosphorescent devices. Chem. Mater. 16(23), 4743–4747 (2004).
C. Fan, Y. Wei, D. Ding, and H. Xu, Linkage engineering in hosts for dramatic efficiency enhancement of blue phosphorescent organic light-emitting diodes. Opt. Express 23(10), 12887–12899 (2015).
T. Daeneke, T.-H. Kwon, A.B. Holmes, N.W. Duffy, U. Bach, and L. Spiccia, High-efficiency dye-sensitized solar cells with ferrocene-based electrolytes. Nat. Chem. 3(3), 211–215 (2011).
S.J. Su, C. Cai, and J. Kido, RGB phosphorescent organic light-emitting diodes by using host materials with heterocyclic cores: effect of nitrogen atom orientations. Chem. Mater. 23(2), 274–284 (2010).
E. Scrocco and J. Thomasi, Electronic molecular structure, reactivity and intermolecular forces: an euristic interpretation by means of electrostatic molecular potentials. J. Adv. Quantum Chem. 11, 115–193 (1978).
S.S. Amiri, S. Makarem, H. Ahmar, and S. Ashenagar, Theoretical studies and spectroscopic characterization of novel 4-methyl-5-((5-phenyl-1,3,4-oxadiazol-2-yl) thio) benzene-1, 2-diol. J. Mol. Struct. 1119, 18–24 (2016).
Acknowledgments
We are greatly thankful to I. Ragavan, Research Scholar, Department of Physics, Periyar University, Salem-11, Tamilnadu, India, for his valuable suggestions and strong encouragement to the work. The authors extend their appreciation to the Research Center for Advanced Materials Science (RCAMS), King Khalid University, Saudi Arabia, for funding this work under Grant Number RCAMS/KKU/020-22.
Author information
Authors and Affiliations
Contributions
KP, IR, AA and CV: Design work, experimental processes, conceptualization; computational investigation; methodology; data curation; formal analysis; writing—original draft. PS and PMA: Supervision; software; review and editing; conceptualization; validation; data curation, formal analysis. MS, VRMR and VB, WKK: English language in the revisions to the manuscript; funding acquisition, writing—review and editing.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Periyasamy, K., Sakthivel, P., Ragavan, I. et al. Design, Synthesis, and Optical and Electrochemical Properties of D–π–A Type Organic Dyes with Carbazole-Based Donor Units for Efficient Dye-Sensitized Solar Cells: Experimental and Theoretical Studies. J. Electron. Mater. 52, 2525–2543 (2023). https://doi.org/10.1007/s11664-023-10210-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11664-023-10210-6