
Abstract
The kinetics of silicothermic reduction of manganese oxide from MnO–SiO2–CaO–Al2O3 slags reacting with Fe-Si droplets were studied in the temperature range of 1823 K to 1923 K (1550 °C to 1650 °C). The effects of initial droplet mass, initial droplet silicon content, and initial slag manganese oxide content were studied. Data obtained for 15 pct silicon showed agreement with control by mass transport of MnO in the slag with a mass transfer coefficient (k s) of 4.0 × 10−5 m/s at 1873 K (1600 °C). However, when this rate-determining step was tested at different initial silicon contents, the agreement was lost, suggesting mixed control between silicon transport in the metal and manganese oxide transport in the slag. Increasing the temperature resulted in a decrease in the rate of reaction because of an increase in the favorability of SiO as a product. Significant gas generation was found during all experiments, as a result of silicon monoxide production. The ratio of silicon monoxide to silica formation was increased by factors favoring silicon transport over that of manganese, further supporting the conclusion that the reaction is under mixed control by transports of both silicon and manganese oxide.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Manganese has become an important alloying element in advanced high-strength steels (AHSS) with over 20 wt pct being proposed for certain grades of twinning-induced plasticity (TWIP) steel, and values between 4 and 11 pct being of interest in the third-generation AHSS.[1] Reduction of manganese oxide dissolved in the slag has been proposed as a possible method of adding manganese to the second- and third-generation steels in order to improve the economics of alloy addition.[2,3] The current study seeks to advance the fundamental understanding of the kinetics and mechanism of silicothermic reduction of manganese oxide from slags.
The thermic reduction process promotes solute elements with high oxygen affinities to reduce slag components into the metal. There are two thermodynamically attainable pathways for metallic silicon to reduce manganese oxide. The reduction of manganese in the slag requires the supply of electrons from silicon, either by the formation of tetravalent silicon or, if the supply of Mn2+ is inadequate, by the formation of divalent silicon; the former results in silicon being incorporated into the slag as silicate, while the latter requires the net transfer of one O2− ion from the slag to form SiO gas with the divalent silicon. While the authors do not make any claims to the detailed mechanism, conceptually and stoichiometrically, these reactions can be written as Eqs. [1] and [2]. Bracket notation of [metal], (slag), and {gas} is used in this study. The free energies for these reactions were taken from FactSage Reaction Module and the FTDemo database,[4] which take the following data from the NIST JANAF Tables.[5] At 1873 K (1600 °C, Eq. [1] yields −103 kJ/mol, while Eq. [2] yields −14.7 kJ/mol; under standard conditions, it is clear that while SiO2 may be more favorable, SiO can still form.
Some of the earliest quantitative studies regarding the reduction of MnO using Si in nongraphite crucibles have come from Daines and Pehlke.[6] Their study showed that manganese mass transport in the metal was the rate-limiting step, where the mass-transfer coefficient (k m) was equal to 7 × 10−6 m/s. The system showed significant rate enhancement with stirring, confirming mass transport control. The system underwent a two-stage reaction, with the first stage being faster than the second.
Shibata et al.[7] performed a series of experiments involving multiple simultaneous reactions. MnO was reduced by silicon and carbon simultaneously; those authors concluded that MnO reduction was more likely to be controlled by mass transport in the metal than in the slag. They further noted that desiliconization appeared to finish within the first 300 seconds of reaction.
The study of Heo et al.[8] shows interesting correlations with CaF2 addition to the slag; a ferromanganese slag reduced using metallic silicon was shown to produce a stoichiometrically balanced amount of SiO2 and Mn in the presence of CaF2, whereas in its absence, a greater than stoichiometric quantity of silicon was consumed. These authors proposed SiO generation as the alternate manganese reduction pathway. Such a result would appear to indicate that mass transport in the slag may play a role in rate control of the system. Those authors concluded that silica mass transport in the slag was controlling. However, they acknowledged the study of Sohn et al.[9] who suggested for high-viscosity slags, mass transport of MnO would be the rate-controlling step.
Similar studies can be found in the ferromanganese production literature; however, most of these studies are directed toward understanding carbothermic reduction of manganese oxide. Early authors like Tarby and Philbrook[10] were the first to show a two-stage reaction of MnO with C, with the first stage exhibiting significant gas generation and the second producing nearly no gas. The initial fast stage of reaction was proposed to be controlled by manganese transport in the slag. Daines and Pehlke[11] claimed that the reaction was under chemical reaction control at the slag–metal interface.
Pomfret and Grieveson[12] claimed that the observations of Daines and Pehlke were for the slower second stage of the reaction. Pomfret and Grieveson plotted the manganese partition over time, defined in their case as the activity of MnO in the slag (a MnO) divided by the activity of Mn (a Mn) in the metal, and showed that a critical ratio of a MnO/a Mn characterized the transition from faster to slower rates. Ashizuka et al.[13] were able to demonstrate results similar to those of Daines and Pehlke. Xu et al.[14] used X-ray fluoroscopic techniques to observe nucleation sites for carbon monoxide along the slag–metal interface. Here it was concluded that the rate of generation of CO along the interface was the rate-controlling step.
A more recent study by Kononov et al.[15] demonstrates the importance of gaseous species in affecting the rate of manganese reduction. Different temperatures and different ratios of C/MnO were used in hydrogen, helium, and argon environments. The results indicate increasing reaction rates with the increasing temperature. Further, the densest gas (argon) had the lowest CO diffusivity which was reflected in MnO reduction rates. Helium did not reduce MnO but has a higher CO diffusivity than argon, resulting in a faster rate. Hydrogen participated in the reduction and had a high CO diffusivity and so had the fastest rate of all.
Sun et al.[16] reacted slag with a carbon substrate where it generated a reduced metal product. Energy dispersive spectroscopy (EDS) revealed a manganese concentration gradient in the slag near the slag–metal boundary; such a gradient may imply that MnO transport contributes to reduction control. The rate of reduction was said to increase with the increasing MnO content in the slag. Their final conclusion was that chemical or mixed-mode control may exist in the system.
From an analysis of the above literature, it would appear that there are two control steps in the carbothermic reduction of manganese oxide. The initial, fast step is controlled by MnO transport through the slag, and the slower step appears to be controlled by CO production at the interface. These conclusions, and specifically the effect of gas formation at the interface, may also be relevant to silicothermic reduction should Eq. [2] play a significant role.
Many reacting systems have been found to exhibit dynamic interfacial phenomena influenced by the transfer of elements across the interface[17,18,19,20,21] where spontaneous increases in surface area can be observed; surface area recovers as the reaction slows and nears equilibrium. Many explanations for these observations have been provided, though none have fully explained the phenomena. Most explanations incorporate an apparent decrease in surface tension[18,21] caused by one or more capillary effects: solutocapillarity based on surface active elements, electrocapillarity from charge differences, and thermocapillarity from localized temperature gradients. Surface tension decreases can manifest as dimpling, flattening, and in the most extreme case, emulsification.
Previous authors have documented reaction rates associated with a wide range of reaction couples[22,23,24]—of particular interest is the study of Rhamdhani and the aluminothermic reduction of silica. This reaction (Al/SiO2) has a similar Gibbs energy of reaction to the current system per mole of metal solute but can only generate liquid products. Rhamdhani’s study was shown to generate a metal–slag emulsion that improved the reaction kinetics in terms of increase in interfacial area.
Possibly, even more relevant to the current study is the recent study of White and Sichen[25] where the rates of mass transfer in a stirred system of Si and CaO–SiO2 were investigated. During the course of the reaction, rapid transfer of calcium across the interface decreased the interfacial tension to a level low enough such that mechanical agitation caused emulsification. Such observations imply that systems which do not normally exhibit spontaneous emulsification may be induced to do so with stirring.
Recently, Assis et al.[26] and Spooner et al.[27] showed examples of emulsification during reactions in confocal microscopes; it should be noted that Spooner identified some issues related to the heating regimen applied during the second set of experiments. While this changed the replicability of the study of Assis, it still led to emulsification. Those authors concluded that the effect on surface tension of interfacial oxygen transfer rather than that of phosphorous is likely to be the dominant cause of emulsification. Given that reductive alloying will result in a high rate of transfer of oxygen across the interface, it is reasonable to expect that emulsification or similar surface phenomena will occur.
There is considerable disagreement in the literature regarding silicothermic reduction of manganese oxide from slag; there have been a number of different conclusions about the rate control and reaction mechanism. As it stands, no model exists to piece together the various findings. The current study is part of an ongoing study to further the collective understanding of silicothermic reduction kinetics of MnO from slag and to rationalize the apparently disparate findings in the published literature. This will be addressed first by collecting experimental data in the untested mid-range of silicon and manganese oxide concentrations, and second by analyzing not only these experimental data but also that of other authors. By doing so, the mechanism can be detailed, and a model developed to describe changes in the rate-controlling steps under changing reaction conditions. The current issue presents the first step toward this goal. The experimental data are presented and analyzed for the mid-range concentrations, and a tentative mechanism is proposed. A subsequent publication, currently under preparation, will offer a more detailed analysis of these data on the basis of mixed control involving mass transport of silicon in the metal and manganese in the slag.
Experimental Procedure
Usage of Materials
All ceramic components used were of 99.8 pct alumina refractory material. Table I shows the compositions of all raw materials used in the experiments.
Sample Preparation
Slag samples were first prepared by compressing a mixture having 2:2:1 weight ratio of SiO2:CaO:Al2O3 into pellets, and melting them in a platinum crucible using a resistance-heated muffle furnace at 1873 K (1600 °C). Melts were quenched by pouring onto a steel I-beam. The shattered pieces of quenched slag were crushed and remixed; the melting/quenching/crushing procedure was repeated twice. Appropriate amounts of MnO and preprepared master slag were mixed for use in each experiment. Homogeneous mixing of MnO with the pre-mixed slag was ensured in three ways: vigorous shaking of the powdered slag and MnO mixture prior to adding to the crucible, X-ray observation of the molten slag once in the furnace, and another after allowing for a minimum of 30 minutes homogenizing time in a liquid state, once in the furnace. Owing to the X-ray opacity differences between the MnO and other slag components, it was quite clear when a slag was not homogenized; a lighter band of slag was observed on top of a darker band of MnO-rich slag. ICP analysis of a blank slag, where all steps in the experimental procedure were followed except for the addition of a droplet, indicates a maximum variation in the composition of ±0.5 wt pct MnO across the entire slag. The total mass for all slag samples was fixed at 25 g; no variations in slag mass were tested during these experiments.
Metal droplets were produced by mixing appropriate quantities of silicon and iron and melting in a vacuum arc melter. The melting atmosphere contained less than 4 × 10−10 atmospheres of O2; droplets were melted 2 to 3 times for homogeneity; a total oxygen content of less than 50 ppm[28] is expected.
Procedure
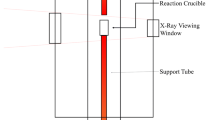
All reduction experiments were conducted using a vertical tube furnace heated using molybdenum disilicide elements. Reaction temperatures up to 1923 K (1650 °C) measured using a type-B platinum-rhodium thermocouple could be held constant within ±8 K (8 °C). The 8.9-cm diameter alumina furnace tube was sealed using water-cooled stainless steel caps at either end, fitted to allow either inert gas to flow through the furnace or to evacuate the furnace using a vacuum pump. The furnace was equipped for X-ray imaging of the slag and metal inside the crucible to allow for the observation of interfacial phenomena and gas bubble formation during the experiment. A diagram of the furnace setup is provided in Figure 1.

Schematic diagram of the vertical tube furnace, not to scale
The following paragraph uses names taken from Figure 1 to better explain the stepwise procedure. An alumina crucible containing slag was placed in the cold zone of the furnace, located inside the quench collar at the base of the furnace and supported by an alumina support rod; the O-ring connections at the base were sealed. The holding cap above the furnace was removed, and a metal droplet was placed inside, supported by an external neodymium magnet. The cap was attached and sealed to the dropper tube such that the droplet can fall when the magnet is removed (without unsealing the furnace). With all the seals closed, the system was evacuated and backfilled with argon. Thereafter, the system was continuously flushed with argon for the duration of the experiment. The crucible was raised from the bottom of the furnace into the hot zone over the course of approximately one hour to prevent thermal shock to the crucible. The hot-zone temperature was kept uniform at approximately ±1 K (1 °C) for 4 cm, or ±10 K (10 °C) for 10 cm. The experiment was started by removing the magnet from the exterior of the holding cap, thus allowing the metal droplet to fall to the base of the dropper tube which was situated near the mouth of the crucible. This tube had a small hole drilled in the end, too small for a solid metal droplet to fall through but large enough for liquid metal to pass. This was done to ensure that the metal was fully liquid prior to the start of the reaction; the duration of melting process was dependent on the size of the droplet. The X-ray equipment was used to determine the precise time when the metal droplet melted, moved through the hole, and entered the slag which established precise zero times for each reaction. The locations of the droplet at various times are demonstrated in Figure 2. The crucible that contained both slag and metal could be quenched in less than one second by removing the support tube, which allowed for accurate end times to be established. In order to generate a concentration versus time profile for a specific set of initial conditions, several of these experiments were conducted; the collection of many discrete data points allowed for the fitting of the data to a continuous curve for each initial reaction condition.

Diagram of droplet location at various times within the furnace setup, not to scale
Inductively Coupled Plasma-Optical Emission Spectroscopy (ICP-OES) was used to analyze the quenched metal droplets for manganese and silicon. Approximately 0.1 g sample of metal was dissolved using a microwave digester and a solution of 3 mL HCl, 2 mL HNO3, and 2 mL HBF4. Samples from the digester were then diluted to 100 mL twice to get them into a measurable concentration range for the elements of interest (0 to 40 ppm). Experimental slags were not analyzed using ICP as the concentration gradients formed within the slag rendered bulk concentration measurements meaningless.
Results
Figures 3 through 11 show the changes in metallic silicon and manganese contents as a function of time observed for the various reaction conditions. The solid lines in these figures have been drawn through the data using exponential functions by the author. The dashed line shows the predicted manganese values by converting the change in wt pct Si to wt pct Mn using Eq. [1], while the dotted line shows the same conversion using Eq. [2]. During the initial seconds of reaction, the measured data appear to be almost entirely described by Eq. [1]. Beyond this time, deviation can be found, where the amount of manganese reduced to the metal is less than that calculated from the stoichiometry from Eq. [1], implying a contribution from Eq. [2]. A depletion of Mn2+ ions would improve the favorability of the SiO reaction, which may explain this behavior. The time to achieve equilibrium for these reactions was between 240 and 360 seconds. This value is in agreement with the desiliconization data of Shibata et al.[7]

Concentration vs time plot for the 15 wt pct MnO, 5 wt pct Si, 1.5 g droplet, and 1873 K (1600 °C)

Concentration vs time plot for the 15 wt pct MnO, 10 wt pct Si, 1.5 g Droplet, and 1873 K (1600 °C)

Concentration vs time plot for the 10 wt pct MnO, 15 wt pct Si, 1.5 g droplet, and 1923 K (1650 °C)

Concentration vs time plot for the 15 wt pct MnO, 15 wt pct Si, 0.5 g droplet, and 1873 K (1600 °C)

Concentration vs time plot for the 15 wt pct MnO, 15 wt pct Si, 1.0 g droplet, and 1873 K (1600 °C)

Concentration vs time plot for the 15 wt pct MnO, 15 wt pct Si, 1.5 g droplet, and 1823 K (1550 °C)

Concentration vs time plot for the 15 wt pct MnO, 15 wt pct Si, 1.5 g Droplet, and 1873 K (1600 °C)

Concentration vs time plot for the 15 wt pct MnO, 15 wt pct Si, 1.5 g droplet, and 1923 K (1650 °C)

Concentration vs time plot for the 20 wt pct MnO, 15 wt pct Si, 1.5 g droplet, and 1923 K (1650 °C)
Visual observation of quenched slags, as shown in Figure 12, shows that a distinct color gradient exists within the sample taken at approximately three minutes. Given that MnO is the only species to cause significant pigmentation of the slag, this gradient appears to be related to manganese depletion of the slag and is not found in slags near time zero or slags that are at equilibrium. Hence, it is reasonable to assume that transport of manganese oxide in the slag contributes to control of the reaction for a large portion of the reaction time. Figure 13, taken after twenty seconds of reaction, is consistently dark in both the interface and bulk slag, implying that no significant gradient exists. Figure 14 taken at twenty minutes of reaction is consistently lighter in color. In all cases, gas bubbles can be observed forming along the slag–metal boundary, although each image has a different bubble shape associated with it. This is believed to be associated with how the gas bubbles evolve over time.

Color gradients surrounding the droplet (lower middle of the image) and gas at 185 s of reaction; the lightest area is to the left and darkest to the bottom right

Negligible color gradients found surrounding the droplet at 20 s of reaction; note the distinct outline of spherical gas bubbles surrounding the large central droplet

Small light to dark gradient from left to right across the image at 20 min of reaction; note the destabilization of bubbles seen at long times resulting in elongated shapes
Note that these images have had small adjustments made to their color and contrast in order to make the changes in the slag more obvious to readers. This action has not changed the position of the MnO-depleted zone. An overlay has further been added to point out the key areas of interest. Although only three images have been presented, similar observations by the author were found for nearly all the samples.
In order to quantify the correlation between color and (MnO) concentration, EDS was performed at different distances from the slag/metal interface. These data show concentration increasing from approximately 4 wt pct Mn to 6 wt pct Mn over a 0.7-mm distance in the direction perpendicular to the interface. Without a standard to reference in the scanning electron microscope, these values cannot be taken at face value, although their trend can be, where the manganese content increases by 50 pct. This further reinforces the idea that manganese transport in the slag controls the reaction rate.
Based on the above qualitative evidence for mass transport control in the slag, Eq. [3] was used to plot the experimental data. The mass transfer coefficients are represented as k s and k m for slag and metal, respectively, in units of m/s; similarly, volume is given as V s or V m in m3. Time is represented as ‘t’ in seconds, and A as the interfacial area in m2. The number of moles of a particular species in either the slag or metal is given as n s or n m, respectively (and can have the desired species substituted); the superscripts denote whether the value is taken at equilibrium ‘e’ or at the initial composition ‘o’, and the superscript-free variable is for any arbitrary time. Masses of slag and metal are represented as m s or m m. Molar mass is represented as capital M. Equation [3] was developed assuming control by mass transport in the slag employing a molar balance such that the equation could be written in terms of number of moles of the relevant species in the metal. Equation [4] is Eq. [3] where an appropriate conversion of moles to mass has been applied. Further, this derivation includes the initial moles of manganese in the metal as part of the expression; in the present case this is a negligible non-zero value that can be ignored.
Further, in order to verify that metal transport is not the rate control, Eq. [5] for metal control is shown below. Note that the significant differences are the used metal vs slag volume and the pre-logarithmic term. The molar terms in Eq. [5] have been converted to mass terms as shown in Eq. [6].
If mass transport of manganese oxide in the slag is assumed to control the reaction rate, the change in the manganese concentration in the metal over time is given by Eq. [4], whereas if mass transport of manganese in the metal controls the reaction rate, then Eq. [6] would apply. This information is displayed in Table II for clarity.
In the current study, different droplet sizes, different starting [Si], and different starting (MnO) contents have been used to alter the value of Y. The effect of temperature has also been studied. Figures 15 and 16 are plotted for different droplet sizes according to Eqs. [4] and [6], respectively. The data plotted according to control by MnO mass transport in the slag can be fitted well to a single straight line, whereas the data plotted according to control in the metal do not fit so well. This suggests that the manganese oxide reduction is controlled by mass transport in the slag. Figure 15 shows an excellent fit of the data to a single straight line, with significant deviation only occurring only after crossing the 300 second mark. Meanwhile, Figure 16 shows that the equivalent plot for mass transport in the metal appears to diverge at a much earlier time. The mass transfer coefficient k s is approximately 4.0 × 10−5 m/s.

[Mn] Concentration data plotted as a function of droplet size and time, using Eq. [4] for mass transport of MnO in the slag; initial conditions 15 wt pct Si, 15 wt pct MnO, 1873 K (1600 °C), Variable Mass

[Mn] Concentration data plotted as a function of droplet size and time, using Eq. [6] for mass transport on Mn in the metal; initial conditions 15 wt pct Si, 15 wt pct MnO, 1873 K (1600 °C), variable mass
This result does not agree with the study of Daines and Pehlke who stated that mass transport of manganese in the metal was the rate-controlling step[6]. Daines and Pehlke used much lower amounts of [Si] and (MnO) in their system, and made the bulk of their observations at times greater than the five to seven minute mark; this could explain the discrepancy. The more recent study of Heo et al. is in agreement with slag control findings, even though their claim is for a silica-controlled pathway and not for manganese oxide.[8]
Carrying forward the conclusion that the system is under mass transport control in the slag, the following figures show the results of changing initial (MnO) concentrations and initial [Si] concentrations. Figure 17 shows that changing the initial concentration of (MnO) in the system does not change the rate constant of reaction. This provides further evidence to the idea of control by transport of manganese oxide in the slag. Note that, as expected, the ks value here is near that established in Figure 15 (approximately, a ten percent difference).

[Mn] Concentration data plotted as a function of initial (MnO) and time, using Eq. [4] control by mass transport of MnO in the slag; initial conditions 15 wt pct Si, variable MnO, 1923 K (1650 °C), 1.5 g droplet
Figure 18 shows that by increasing the initial concentration of silicon in the metal, an increase in the slope of the rate plot can be observed. This observation appears to be in contradiction to the previous evidence of mass transport control by manganese oxide in the slag, as the pre-logarithmic term is not successfully normalizing the data with changes in silicon. This finding is contradictory to the previous findings, and may imply that mixed control is controlling this system.

[Mn] Concentration data plotted as a function of initial [Si] and time, using Eq. [4] for control by mass transport of MnO in the slag; initial conditions variable Si, 15 wt pct MnO, 1873 K (1600 °C), 1.5 g droplet
Figure 19 compares changes in temperature among reactions. Strangely, it appears as if increasing temperature may result in a decline in the rate constant. This is atypical of most systems under mass transport control, as an increase in temperature typically corresponds to an increase in the mass transfer coefficient. This anomalous change in the apparent mass transfer coefficient could be because of a shift in balance between two rate-determining steps in a mixed control system or because of different levels of bubble formation on the metal surface. Further, near time zero, it appears as if the higher temperature starts from a lower YlnZ than that of the lower temperatures. This may imply that a reaction occurs near time zero, without the presence of any gas, corresponding well to the bulk mass transfer and the increased rate constant that temperature typically brings.

[Mn] Concentration data plotted as a function of temperature and time, Using Eq. [4] for control by mass transport of MnO in the slag; initial conditions 15 wt pct Si, 15 wt pct MnO, variable temperature, 1.5 g Droplet
One final observation from this study is about the marks left by bubbles on the metal surface. Figure 20 shows a metal droplet that has been extracted from slag with large, flattened areas. These areas appear to be gas nucleation and growth sites, where the pressure exerted by {SiO} manages to flatten the interface. From thermodynamic calculations, silicon monoxide is the most likely gas present. This flattening is likely to happen when gas generation is at a maximum and would exert the greatest pressure on the interface. In some cases, the X-ray equipment allowed for the in situ observation of large gas bubbles in the slag, apparently breaking away from the metal. It is believed that these larger bubbles are the result of coalescence of many small bubbles into a single large bubble that can break away from the surface of the metal droplet. Unfortunately, the image quality is not sufficient to measure the volume of the bubble film around the metal surface.

Localized flattening on a droplet interface at 4 min of reaction
Meanwhile, Figure 21 shows marks left by bubbles attached to the metal interface with a network of slag surrounding the sites. These circular areas of apparently clean metal show how the gas sticks directly to the metal and displaces slag around the nucleation and growth sites. This observation provides credible evidence to the theory that a dynamic volume/area term may exist over the course of the reaction relating the ability of (MnO) to diffuse past the bubble layer and react with the metal in the slag regions; this is discussed in more detail below.

Cleared metal surface surrounded by slag deposits in a web-like pattern at 2 min of reaction
It is worth noting that none of the dynamic interfacial phenomena (flattening, roughening, or emulsification) as identified by previous researchers[20,22,26] were found during the experiments. The localized flattening shown in Figure 20 is likely attributed to bubble nucleation and not surface tension breakdown. Despite exceeding the 0.1 mol/m2/s interfacial oxygen transfer rate criterion proposed by Riboud and Lucas,[20] the system did not exhibit interfacial breakdown; the formation of a stable bubble film around the droplet may inhibit spontaneous emulsification. This hypothesis is purely speculative, although other researchers in the authors’ laboratory studying iron carbon droplets have found that when bubbles are present, the interface remains remarkably stable even at high oxygen flux.[29]
Discussion
The results presented above offer strong evidence for control by mass transport of manganese oxide in the slag. A gradient in slag pigmentation (indicative of depletion of manganese oxide close to the slag/metal interface) can be observed by visual observation. Rate plots assuming control by mass transport of MnO show remarkable consistency for a variety of droplet sizes and initial MnO concentration. Contrary to these findings, changing the starting metallic silicon content of the metal disagrees with mass transport control by MnO; silicon transport in the metal may also impact the rate and control steps for the reaction. The curves from Figures 3 through 11 show that the concentration change of metallic manganese follows neither Eq. [1] nor [2], falling somewhere in between, but that the conditions likely to cause a greater deficit in the transport of Mn2+ to the interface relative to that of silicon will more closely follow the theoretical SiO curve.
Taken in their entirety, the above observations imply some element of mixed control between silicon in the metal and manganese oxide in the slag, which may explain the apparent discrepancies between different researchers.[6,7,8] Initial concentrations are likely to impact the dominant reaction pathway.
These findings are best summarized in Table III; all MnO changes are matched with 15 wt pct Si droplets, and all Si changes matched with 15 wt pct MnO slags. From Figures 3 through 11, it is possible to compare the measured changes in manganese concentration with the theoretical values that would be obtained through the SiO2 or SiO pathways. At a minimum, one mole of silicon will reduce one mole of manganese oxide according to Eq. [2], and at a maximum, one mole of silicon will reduce two moles of manganese oxide according to Eq. [1]. One can calculate the ‘silicon efficiency ratio’ (SER) using Eq. [7], where wt pct Mn represents the measured concentration of manganese, wt pct MnSiO represents the concentration of manganese assuming that all silicon reduces manganese via Eq. [2], and \( {\text{wt pct Mn}}_{{{\text{SiO}}_{ 2} }} \) represents the concentration of manganese assuming that all silicon becomes silica according to Eq. [1]; all concentrations are taken from the metal.
Values of SER calculated using Eq. [7] are shown in Table III and these follow the trend one would expect given the stated mechanism. High initial MnO content results in a higher initial ratio indicating that Mn2+ flux is high and more able to oxidize the available silicon. Low initial silicon similarly results in a slower demand for Mn2+. With the noted exception of 10 wt pct MnO, there appears to be a slight downward trend in the efficiency with time, indicating as expected that depletion of Mn2+ near the interfaces results in more production of SiO. The other data sets do not show observable trends. From this analysis, it appears that the early stages of reaction are dominated by kinetic factors which eventually become somewhat stable and approach a ratio close to the equilibrium of the system; despite differences in the ratio near the 300 second mark, eight of the nine systems (with the exception of 10 wt pct MnO) closely approach their expected equilibrium. The analysis shows that in order to meet the demands of the mass balance within the system, the ratio of [Mn] to [Si] reacted over time must change.
A gas layer has been found in almost every experiment carried out. This has not been incorporated into the rate assumptions presented above and is likely also to have complicated the picture, particularly with regard to the effect of temperature on reaction rate. This result implies that mass transport of manganese oxide in the slag might not be the lone rate-controlling step in this reaction. Given that all of the data for MnO differences came from droplets with 15 wt pct starting silicon, it is possible that at lower values of silicon, the control shifts away from manganese to silicon instead. Such a result may help explain the issues authors have had in explaining the rate controlling step ranging from mass transport of manganese in the metal to mass transport of manganese or silicon in the slag. Given the apparent gas generation in the system, and the increasing favourability SiO with temperature, it is possible that higher temperatures result in more gas generation around the interface leading to blocking of the reaction pathways; this is complicated by diffusivity increases in slag with the increasing temperature.
One area of concern surrounding the gas is how it affects the volume and area terms employed in any rate equation. For example, the assumption that the area of the slag-metal interface is constant is unlikely to be true if it is partially covered by gas bubbles. Figure 22 provides an illustration of how bubbles may impact the area of the reaction interface and thereby the rate of reaction. The extent of the bubble layer and the volume of slag between the bubbles may change over time depending on a number of factors, such as temperature and the balance between silicon and manganese oxide transports. A gas layer would be expected to impact the interfacial area in two possible ways: first by taking up space along the interface and preventing the reaction of slag and metal in those areas, as well as forcing the transported material to funnel between the bubbles. Given the spherical geometry, the point where the neck between bubbles is at its narrowest should impact the transport more greatly than coverage of the slag–metal interface. If one considers an imaginary sphere around the metal droplet, separated by one bubble radius from the slag–metal interface, prior to formation of gas bubbles would have a defined area ‘A’, occupied entirely by slag. Upon the formation of a bubble layer, the area occupied by slag would decrease thereby reducing the cross-sectional area of the transport path. Assuming the spherical bubbles pack tightly in a hexagonally close-packed monolayer would reduce the available transport path by 89 pct.

Diagram illustrating how gas generation may affect the diffusion pathway and change both volume and area terms in the rate equation
Such an assumption may help to explain what appears to be a dramatic decrease in reaction rate after the first six seconds. Close examination of the plots in Figures 3 through 11 shows that the y-axis intercept at time equal to zero is always less than zero. This observation implies an early rate—much faster than that evident for times greater than 6 seconds. Based on the data available, it is not possible to determine the exact point at which the rate changes but, if for the sake of discussion one assumes six seconds, we can estimate a slope and therefore a mass transfer coefficient for this very early time period. This process is illustrated in Figure 23 as ‘k s,0’ plotted alongside the calculated k s value from Figure 15. The mass transfer coefficient calculated based on times greater than six seconds is approximately 11 pct of the estimated initial slope. The authors are not claiming that the predicted initial mass transfer coefficient can be determined quantitatively but merely propose that it is of the correct order of magnitude to support the idea of blockage of transport paths by a relatively stable bubble film. The slope plotted for time less than six seconds is a very reasonable estimate; these first few seconds of reaction can account for up to 10 pct of the total reaction (as is the case with Figure 11).

Zoomed version of Fig. 15 where the initial Y-axis jump is displayed
The above findings indicate that there are two major areas for future study: derivation of a rate equation involving both manganese oxide transport from the slag and silicon transport from the metal, as well as the incorporation of the change in interfacial area over time. The interfacial area appears to change over a relatively short period of time as the result of a relatively stable bubble film, and once formed, does not change much over the course of the reaction.
Conclusions
-
1.
MnO mass transfer in the slag is the rate-controlling step for experiments using 15 wt pct silicon, and the mass transfer coefficient is 4.0 × 10−5 m/s.
-
2.
Decreasing the initial silicon content changes the rate control to mixed control by means of mass transport of silicon in the metal and mass transport of MnO in the slag.
-
3.
Lowering the temperature appears to increase the reaction rate because of the decreasing favorability of SiO formation at lower temperatures.
References
Z.H. Cai, H.Ding, X.Xue, and Q.B. Xin: Mater. Sci. Eng. A, 2013, vol. 560A, pp. 388–95.
O.S. Bobkova and V.V. Barsegyan: Metallurgist, 2006, vol. 50, pp. 463–68.
L.N. Kologrivova, A.Ya. Nakonechnyi, Z.G. Trofimova, O.V. Nosochenko, and N.N. Kulik: Metallurg, 1987, vol. 5, pp. 28–29.
C.W. Bale, E. Bélisle, P. Chartrand, S.A. Decterov, G. Eriksson, A.E. Gheribi, K. Hack, I.H. Jung, Y.B. Kang, J. Melançon, A.D. Pelton, S. Petersen, C. Robelin, J. Sangster, and M.A. Van Ende: Calphad, 2016, vol. 54, pp. 35–53.
M.W. Chase, Jr., C.A. Davies, J.R. Downey, Jr., D.J. Frurip, R.A. McDonald, and A.N. Syverud: NIST Stand. Ref. Database 13, 1985, vol. 1.
W.L. Daines and R.D. Pehlke: Trans. Met. Soc. AIME, 1968, vol. 242, pp. 565–75.
E. Shibata, H. Sun, and K. Mori: Metall. Mater. Trans. B, 1999, vol. 30B, pp. 279–86.
J.H. Heo, Y. Chung, and J.H. Park: Metall. Mater. Trans. B Process Metall. Mater. Process. Sci., 2015, vol. 46B, pp. 1154–61.
H. Sohn, Z. Chen, and W. Jung: Steel Res., 2000, vol. 71, pp. 145–52.
S.K. Tarby and W.O. Philbrook: Trans. Metall. Soc. AIME, 1967, vol. 239, pp. 1005–17.
W.L. Daines and R.D. Pehlke: Trans. Met. Soc. AIME, 1971, vol. 2, pp. 1203–11.
R.J. Pomfret and P. Grieveson: Ironmak. Steelmak., 1978, vol. 5, pp. 191–97.
M. Ashizuka, A. Moribe, and K. Sawamura: Tetsu-to-Hagane, 1975, vol. 61, pp. 36–45.
K. Xu, G. Jiang, W. Ding, L. Gu, S. Guo, and B. Zhao: ISIJ Int., 1993, vol. 33, pp. 104–8.
R. Kononov, O. Ostrovski, and S. Ganguly: Metall. Mater. Trans. B Process Metall. Mater. Process. Sci., 2008, vol. 39B, pp. 662–68.
H. Sun, M Yaser Lone, S. Ganguly, and O. Ostrovski: ISIJ Int., 2010, vol. 50, pp. 639–46.
A. Sharan and A. W. Cramb: Metall. Mater. Trans. B, 1995, vol. 26B, pp. 87–94.
H. Gaye, L. D. Lucas, M. Olette, and P. V. Riboud: Can. Metall. Q., 1984, vol. 23, pp. 179–91.
A. Jakobsson, Du Sichen, S. Seetharaman, and N.N. Viswanathan: Philos. Trans. R. Soc. Lond. A, 1998, vol. 356A, pp. 995–1001.
P.V.V. Riboud and L.D. D. Lucas: Can. Metall. Q., 1981, vol. 20, pp. 199–208.
Y. Chung and A.W. Cramb: Metall. Mater. Trans. B, 2000, vol. 31B, pp. 957–71.
M.A. Rhamdhani, K.S. Coley, and G.A. Brooks: Metall. Mater. Trans. B, 2005, vol. 36B, pp. 219–27.
R.J. Pomfret and P. Grieveson: Can. Metall. Q., 1983, vol. 22, pp. 287–99.
Y. Chung and A.W. Cramb: Philos. Trans. R. Soc. A. Math. Phys. Eng. Sci., 1998, vol. 356, pp. 981–93.
J. F. White and D. Sichen: Metall. Mater. Trans. B Process Metall. Mater. Process. Sci., 2014, vol. 45B, pp. 96–105.
A.N. Assis, J. Warnett, S. Spooner, Richard J. Fruehan, M.A. Williams, and S. Sridhar: Metall. Mater. Trans. B, 2015, vol. 46B, pp. 568–76.
S. Spooner, Andre N. Assis, J. Warnett, R. Fruehan, Mark A. Williams, and S. Sridhar: Metall. Mater. Trans. B, 2016, vol. 47B, pp. 2123–32.
M.A. Rhamdhani: PhD Thesis - McMaster Univ., 2005.
E. Chen and K. S. Coley: Ironmak. Steelmak., 2010, vol. 37, pp. 541–45.
Acknowledgments
The authors thank the Natural Science and Engineering Research Council of Canada (NSERC, STPGP463252-14) for the funding support. The authors offer their special thanks to Arcelor Mittal Dofasco, US Steel Canada, Praxair, and Hatch Ltd. acknowledging their in-kind support, technical expertise, and their many helpful discussions.
Author information
Authors and Affiliations
Corresponding author
Additional information
Manuscript submitted September 30, 2016.
Rights and permissions
About this article

Cite this article
Jamieson, B.J., Coley, K.S. Kinetics of Silicothermic Reduction of Manganese Oxide for Advanced High-Strength Steel Production. Metall Mater Trans B 48, 1613–1624 (2017). https://doi.org/10.1007/s11663-017-0967-z
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11663-017-0967-z