
Abstract
Experiments were performed using a range of test conditions to elucidate the rate controlling step during the reaction of liquid iron-carbon droplets and slags containing manganese oxide. Four conditions were tested in the system: initial MnO content in the slag (5, 10, and 15 wt pct), initial carbon content of the metal (1, 2.5, 4.3 wt pct), initial droplet mass (0.5, 1.0, and 1.5 g), and reaction temperature (1823 K [1550 °C], 1873 K [1600 °C], and 1923 K [1650 °C]). Data were collected using the Constant Volume Pressure Increase (CVPI) technique which tracked the continuous pressure increase in the sealed furnace over time. Samples were quenched at the end of each experiment and chemistry was checked using LECO Carbon Analysis and ICP (Inductively Coupled Plasma) for manganese. The rate of reaction can be broken into a faster initial period related to internal CO formation, and a slower second reaction controlled by a complex mechanism involving transport of oxygen from slag to metal via CO2 and decomposition of the CO2 at the gas–metal interface.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Past studies[1,2] by the authors on the silicothermic reduction of MnO from slag were conducted to better understand the reduction mechanism and provide data to support direct or reductive alloying as a method for manganese addition to 3rd Generation Advanced High-Strength steels (3G AHSS). Should direct alloying processes prove feasible for material addition, an opportunity exists for significant economic improvements related to the production of the 3G AHSS.[3]
Direct or reductive alloying is a proposed method of alloying element addition to steel; in the current case manganese ore can be added to slag in-situ and reduced with reductants such as silicon or carbon.[4,5,6,7] Such additions avoid (or offset) ferroalloy requirements saving producers the need to purchase finished product to add in the ladle.
The current work addresses the fundamental reduction process of MnO by high-carbon iron. Previous work by two of the current authors[1] showed evidence that the reduction of (MnO) by [Si] was under mixed mass transport control in the regime of study; the greatest contributor to transport resistance was MnO mass transport in the slag; however, this alone did not fully explain the rate controlling process for the system. It was proposed that [Si] transport in the metal also had an impact on the reduction rate.
In contrast to the authors’ past work, the carbothermic reduction of MnO is a much more widely studied topic than that of silicothermic reduction. However, across a wide range of studies there is significant variation in the described mechanism for the reaction. Some authors have concluded that chemical reaction controls the rate.[8,9,10,11] Others provided evidence of control by mass transport of manganese ions in the slag.[12,13,14] Another group of authors analyzed the systems assuming mixed mass transport control of manganese in the metal and manganese ions or oxygen in the slag.[15,16,17] All authors ultimately provided evidence for their assumed controlling step. All studies were conducted using experiments where a layer of slag rests atop a layer of metal, and with a greater metal volume than slag volume.
The goal of the present study is to document the rate of reduction of MnO by carbon dissolved in liquid iron droplets. Compared to past literature, where flat bath experiments are more representative of reactions occurring directly between the metal bath and slag layer of a reactor, the present study represents metal droplets ejected into bulk slag and slag foam. Further, findings here will be compared to past work by the authors on the silicothermic reduction of MnO from slag in order to make processing recommendations for the creation of the 3G AHSS.
Experimental Procedure
Material and Preparation
Iron-carbon droplets were prepared from high-purity electrolytic iron and electrode graphite. The electrolytic iron has been measured with a LECO carbon-sulfur analyzer and contains 0.0047 ± 0.0010 wt pct carbon and 0.0032 ± 0.0025 wt pct sulfur. The iron also contains 0.016 ± 0.005 wt pct Mn and 0.072 ± 0.022 wt pct Si. The electrode graphite is 6N purity. The materials were weighed to achieve the composition targets for the specific experiment and melted using a welding arc melter. The partial pressure of oxygen within the arc melter was less than 4 × 10−10 atm and produced metal droplets with less than 50 ppm oxygen.[18]
Slag component purity is presented in Table I. Slag samples were prepared by weighing out an appropriate mass of MnO and mixing with a 2:2:1 weight ratio of SiO2:CaO:Al2O3; total slag mass was 25 g for all experiments. Once combined, the oxide powders were vigorously shaken until the solids were well mixed. In order to ensure homogeneity, slag samples were held for at least 30 minutes in their liquid state prior to the experiment. Based on three measurements of 2:2:1 slag, the average composition of these mixtures after melting was 40.4 pct SiO2, 39.8 pct CaO, and 19.8 pct Al2O3.
To validate mixing MnO with the 2:2:1 oxide mixture, unreacted (but molten) slag samples were quenched and broken apart. Shards of the sample were analyzed using Inductively Coupled Plasma Optical Emission Spectroscopy (ICP-OES); the tested slag pieces varied by less than ± 0.5 wt pct MnO.
Alumina crucibles were used as part of these experiments. Slag samples were not measured for chemistry. In only rare instances was there any noticeable deterioration of the crucible through slag-line attack, which would have resulted in the greatest loss of material to the slag. The maximum observed attack is estimated to be 1.3 g, which corresponds with an average increase in alumina content of 2 wt pct over the course of an experiment.
Equipment
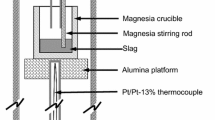
A standing vertical tube furnace with attached pressure transducer was used for the experiments; a schematic is provided in Figure 1. All ceramics used were 99.8 pct alumina. The system is heated with molybdenum disilicide heating elements. The base of the furnace can be detached in order to load slag filled crucibles; these crucibles must be raised up to the hot zone of the furnace over the course of one hour to ensure that the crucible does not crack. The hot zone of the furnace is between the X-ray viewing windows and is checked regularly to ensure there is no drift; the hot zone has less than a ± 1 K (1 °C) temperature difference across a 3 cm height and less than ± 10 K (10 °C) across 10 cm. With Type-B thermocouples, at 1600 °C the error associated with the temperature readout is ± 8 K (8 °C); the thermocouples are checked regularly with a calibrated thermocouple to account for any drift.

Schematic diagram of vertical tube furnace. Not to scale
At the base of the furnace, below the lower water-cooling cap and gas inlet, is a cooling column wrapped in a copper coil circulating cool water. The temperature within the cooling column is approximately 363 K (90 °C). This region ensures that when quenching of the sample occurs a large temperature gradient is achieved from hot zone to cool region.
At the top of the furnace is a metal cap which holds the metal droplet outside of the hot zone of the furnace until the experiment begins; the droplet shares the sealed atmosphere of the furnace. Prior to raising the crucible, the system is evacuated to remove most of the gas, and then backfilled with high-purity (5N) argon. While heating the system is continually purged with argon to maintain a low oxygen content in the furnace.
An X-ray fluoroscope surrounds the furnace such that a video can be taken of the events in the experiment. Density differences between slag, metal, and the surroundings in X-ray images allows for some degree of physical characterization of the reaction over time. Further, X-ray imagining allows for accurate determination of the reaction start time.
The pressure transducer continually tracks pressure change within the sealed furnace. Pressure change can be used to calculate the number of moles of gas produced using a calibration conducted prior to each experiment. The conversion method will be discussed in the procedure.
Procedure
To begin the procedure, the support tube is removed by unscrewing the fastener at the cooling column base. The slag sample (inside the reaction crucible) is loaded onto the platform and support tube and the base is fastened back on. The crucible is now within the cooling column in Figure 1, just below the lower water cooler. The metal cap above the furnace is unscrewed and a metal droplet is loaded in, secured with a magnet. The cap can be screwed back onto the dropper tube thus sealing the furnace. The furnace is evacuated and then the system is backfilled with argon. Once the pressure is equalized, the exhaust is opened allowing for a continual argon flush.
Over the course of approximately 1 hour, the crucible is raised into the hot zone of the furnace. Once the crucible is in position, a timer is started to ensure that the slag is well homogenized (approximately 30 minutes).
In final preparation for the experiment, the inlet argon and exhaust are closed so that any CO gas generated during the reaction will register on the pressure transducer. To start the experiment, the magnet on the upper cap is removed and the droplet falls to the base of the dropper tube. The base of the dropper tube has a hole which is too small for the solid droplet to pass prior to melting; it takes approximately 30 seconds for the droplet to melt and fall into the crucible.
Using X-ray imaging, the zero time of the experiment occurs when the metal droplet contacts the slag; the droplet immediately sinks to the bottom of the crucible. The reaction is observed and tracked with video and Constant Volume Pressure Increase (CVPI) for approximately 20 minutes. After this time the support tube is released from the furnace, quenching the reaction crucible and ensuring the chemistry at the time of quench is locked for ICP analysis.
Prior to evacuating the system, while a crucible is at the base of the cooling column, the system is calibrated to obtain the conversion factor for pressure to moles; this calibration produces identical results whether the crucible is in the hot zone or cooling column. Three known volumes of room temperature air are injected into the hot furnace; the air volume can be converted to moles which do not change with temperature. The CVPI can thus be calibrated to show the pressure response to a known addition of gas moles. This conversion is used to convert continuously recorded pressure data from each experiment into moles of gas generated. Metal droplets are extracted from the slag and used for Mn/Si analysis using ICP.
Observations and Discussion
Observations of CO Evolution
Figure 2 shows how droplet composition changes with time, based on the chemical analysis of quenched droplets; the conditions were 2.5 wt pct carbon in a 1.5 g metal droplet, 15 wt pct MnO, and at 1823 K (1550 °C). The figure presents data for metallic manganese measured by ICP, metallic silicon measured by ICP, CO evolved and measured via CVPT, and CO calculated under the assumption that all Mn and Si added to the metal are a result of carbon oxidation.

Change in moles of [Mn], [Si], and CO gathered through individual quenching experiments for a series of reactions with identical starting conditions (2.5 wt pct C, 15 wt pct MnO, 1823 K (1550 °C), and 1.5 g droplet)
The plot shows that the reaction can be simplified to two stages, before and after 100 seconds; the system appears to reach complete equilibrium between 1100 and 1500 seconds. For consistency throughout the remainder of this analysis, rate or stage 1 refers to the time before 100 seconds and rate 2 encompasses all reaction times afterwards. This split between stage one and two will be further justified with evidence from X-ray video. Carbon oxidation measured by CVPI within the first 100 seconds does not match silicon and manganese gains to the metal; this indicates that an intermediary reaction occurs which buffers the reductant and oxidant. The changes in carbon, manganese, and silicon balance by the end of the reaction. This supports the mechanism proposed by previous researchers that the reaction involves a complex exchange characterized by Reactions 1 to 3.[11,14]
The exchange of MnO and Fe is fast and explains the rapid rise in [Mn]. While not stated in the work of past researchers, it is safe to assume that silica reduction is similar; the initially low values of aFeO and aSi support this reaction.
This rate interpretation corresponds well with X-ray video evidence. Figure 3 demonstrates the physical phenomena occurring over time as seen through X-ray video. Nucleation of CO begins externally before rapidly transitioning over to internal bloating. This period essentially tapers off by 60 seconds, but periodic bloating can still be observed until approximately 100 seconds. This video evidence further supports two stages of reaction. For all the figures showing X-ray images, the upper leftmost image is overlaid with labels for the metal and slag phases. In Figure 3, a scale is provided that applies to all of the figure’s images. In the following images, the white space at the top is the argon atmosphere of the furnace; where the slag is displaced or lighter in color than expected, CO bubbles are moving through the slag layer.

X-ray photos of a metal droplet from dataset 1 (initial conditions 2.5 wt pct C, 15 wt pct MnO, 1.5 g droplet, 1873 K [1600 °C]). In order: (0 s) reaction start, (1 s) external nucleation of CO, (1.5 s) partial internal nucleation, (3 to 7 s) majority internal nucleation and small slag foam, (7 s) CO production slowing [collection of metal at base of droplet], (15 s) droplet buoyancy lost, (30 s) periodic CO release from droplet, (60 s) observable reaction essentially complete
While the evolution of [Mn] and [Si] after 100 seconds in Figure 2 appears quite flat, the evolution of CO continues. This occurs because FeO transferred to the slag during the initial stage of reaction is reduced in the second period, a mechanism proposed and measured by previous workers.[11,14] During the fast reaction, FeO has been shown to rise above its final equilibrium value, reverting to metal as the reaction slowed.[11,14] By equilibrium, the level of FeO in the slag will have returned to a very low value.
After 100 seconds, there is an apparent imbalance in the amount of CO generated with respect to metal reduced. The only scenario under which this can occur is if metal oxides are being reduced but not making their way to the metal being measured. This possibility will be discussed in greater detail as part of the discussion.
All CVPI data were calibrated by injecting known quantities of gas into the hot furnace prior to each experiment and measuring the pressure change. Nonetheless, there remained some scatter in the data. The obvious way to correct for this scatter would have been to use the total change in carbon in the droplets over the course of the experiment; however, it was not possible because the droplet to droplet variation in the initial carbon was enough to render this method ineffective. The authors therefore chose to normalize the data using the sum of total silicon and manganese picked up by the droplet. The initial silicon and manganese concentrations in the droplet were negligible and therefore the method was not subject to droplet variation. Regardless of the reaction path, the total number of moles of Si and Mn transferred to the droplet should be related to the total number of moles of CO produced, shown in Reactions 4 and 5.
Based on this assumption, the CVPI data were then normalized using a correction factor as follows: K = (2nSi + nMn)/nCO,Tot. where nSi and nMn are the total number of moles of Si and Mn transferred to the droplet during the experiment and nCO,Tot. is the total number of moles of CO evolved based on pressure transducer data. Corrected nCO is equal to K·nCO,Meas. where nCO,Meas. is the number of moles of CO evolved at a given time as measured using the pressure transducer and nCO is the reported value from the normalization. All curves were duplicated at least once to ensure that gas evolution followed the same trend.
Many past researchers have not considered the reduction of silica by carbon as part of their experiments. It is unknown what the cumulative impact of ignoring these values will be on their experimental data.
Effect of Initial Carbon Content
Figure 4 shows the CO generation curve comparing cumulative CO evolution against time. It is immediately obvious that compared to past research on carbothermic reduction with droplets[19,20,21] these reactions were very slow (by as much as two or three orders of magnitude). This difference is interesting because, using FactSageTM to estimate the equilibrium constant, the ratio of equilibrium constants in this temperature range for decarburization by FeO vs MnO is 200 to 300; this change in driving force accounts for much of the difference in the rate of reaction.

CO evolution vs time for variations in the initial carbon content of the metal droplet (normalized for manganese and silicon gains)
It is important to draw attention to the rapid evolution of CO before 100 seconds and the slower increase following this time. This finding is in reasonable agreement with the trend found in Figure 2. A fast reaction occurs prior to approximately 100 seconds and cedes to a slower second reaction following this time. Assessing the systems for an exact transition time does not yield valuable information. Rate data were obtained by flattening the curves to straight lines, before and after 100 seconds. It is acknowledged that this simplification is not completely accurate as the systems do show signs that they are approaching equilibrium by approximately 1200 seconds (necessitating a decrease in gas generation); however, the simplification holds for the majority of the CO generation plots and the fitting is reasonably good.
The rate of CO generation in the first stage of reaction increases favorably with increasing initial carbon concentration. There is little change in the rates of the second stage. The change in rate with respect to carbon is plotted in Figure 5. This indicates that the rate-determining step is dependent on carbon concentration.

Plot of initial carbon content vs rate of Reaction [8]
When analyzing these plots, two important equations must be considered: a rearranged flux equation (Eq. [6]) and rearranged chemical reaction rate Equation (Eq. [7]). Both equations represent the change in the number of moles of a species with respect to time \( \left( {\frac{{{\text{d}}n^{i} }}{{{\text{d}}t}}} \right) \). Both rates are functions of the mass transfer coefficient or reaction rate constant (\( k_{i} \) or \( k_{i}^{\text{Fwd}} \)), and the interfacial area (AInterface). The mass transport equation also relies on phase volume (VPhase), the number of moles of reacting species in the bulk and at the reacting interface (\( n_{\text{Bulk}}^{i} \) and \( n_{\text{Interface}}^{i} \)), and the term Y is a mass balancing term between phases that changes depending on the specific boundary conditions and rate controlling step. The reaction rate equation considers the product of activities of reactants (\( a_{\text{Interface}}^{i} \)), the product of activities of products (\( a_{\text{Interface}}^{j} \)), and the equilibrium constant of the reaction of interest (Ki).
These equations are useful, even without data, as trends can be predicted between the rates and the parameter of study. Rates must be zero at time zero because no reaction is occurring. In reference to Figure 5, this condition is true for rate 1; the condition need not apply for rate two because this rate does not start from zero.
Past work by the present authors[2] show the mass transfer coefficients of slag species and metal species to be rapid: kSlag equal to 6.7 × 10−4 m/s and kMetal equal to 2.3 × 10−4 m/s. Sain and Belton[22] have summarized the carbon mass transfer coefficient kC as 3.5 × 10−4 m/s in inductively stirred melts. While the carbon mass transfer coefficient is likely to be lower (as the droplet will not be stirred until bloating occurs), these mass transfer coefficients do not correspond well with the rate of reaction (neither Figure 2 nor Figure 4 shows CO evolution rates fast enough to accommodate these mass transfer coefficients). Further, when compared to similar research with metal droplets performed by Gu et al.[20,23] (where FeO was the primary oxygen source in the slag), there is a pronounced difference in the appearance of the bloated metal droplets. The bloated droplets presented in this study are small and appear to have a relatively difficult time bloating.
Figure 6 compares the time it takes for droplets of different initial carbon contents to demonstrate various physical phenomena. The images displayed represent, left to right, the first signs of internal nucleation (low density region appearing at the top of the droplet), the point of maximum expansion during the bloat, and the first point at which the droplet loses buoyancy within the slag. Higher carbon contents exhibit two effects: droplets take longer to present signs of internal nucleation and stay bloated longer. Initial carbon concentration is listed for each row of images, and the other conditions are fixed across rows (15 wt pct MnO, 1.5 g metal, 1873 K [1600 °C]). The scales in the leftmost image of each row apply only to their respective row. The upper leftmost image has an overlay provided to show the appearance of slag and metal within the system.

Comparison of 3 different initial carbon concentration droplets. Droplets with higher carbon content take longer to bloat and stay bloated longer. The left image column shows the time where internal nucleation first become visible, the center where the droplet is maximally bloated, and the right column when droplet buoyancy is lost
Based on the suggested mass transfer coefficients, mass transport is not a likely controlling phenomenon within this system. The findings in Figure 6 explain the initial few seconds of reaction. Per (Eq. [7]), if a chemical reaction is the rate-limiting step, there is an expectation that increasing carbon content will increase the driving force of the reaction and subsequently the rate; this is true for Figures 4 and 6. The transition period before the onset of droplet bloating may be controlled by a chemical reaction at the interface: if more carbon is present in the melt, more oxygen is consumed at the interface; it is obvious however that some oxygen passes through the interface as a precursor condition to internal CO nucleation. This incubation period has been reported by past authors.[24]
For a droplet to experience bloating, oxygen flux must exceed carbon flux to the slag–metal interface; CO bubbles cannot form inside the metal droplet without dissolved oxygen. In order for CO to nucleate pCO must exceed one atmosphere. Droplet buoyancy is a physical manifestation of the reaction rate: droplet bloating is a function of the rate of internal CO generation and rate at which CO is lost through the metal droplet surface. In most cases, the droplets triple their surface area upon bloating (which is consistent across most tested conditions); assuming CO loss through the interface is proportional to the surface area means that the rate of CO loss is similar in all systems. Thus, the length of time a droplet remains bloated is related to the rate of internal CO generation. Since high-carbon droplets remain bloated longer, the rate-limiting mechanism must be proportional to carbon in the metal; this agrees well with the changes in reaction rate presented in Figure 5.
The mole generation plot presented in Figure 4 shows that the rate of gas generation is essentially linear in the first stage of reaction. Contrast this to Figures 3 and 6 where an area change in the system obviously occurs. This indicates that the rate of reaction for internal CO generation is independent of the droplet surface area and the only reaction mechanism that the authors can imagine independent of droplet surface area is internal CO nucleation.
Nucleation of new CO bubbles essentially ceases before the end of stage 1 but growth of pre-existing bubbles continues. The growth of pre-existing bubbles and nucleation of fresh bubbles are both driven by chemistry within the metal droplet. Nucleation is an energetically expensive reaction when compared to growth, so the appearance of larger CO bubbles over time is not surprising. Once these bubbles form, any oxygen remaining inside of the metal is likely relegated to the task of bubble growth. All of the above-mentioned findings indicate that the system is controlled by the nucleation and growth of internal CO bubbles.
Effect of Initial Manganese Oxide Content
Figure 7 shows the gas evolution associated with changes in initial manganese oxide content; rates of each stage are listed in the figure. Interestingly, the curves are in reverse order of what one might expect, where lower manganese oxide contents produce faster rates of CO evolution. It is important to remember that these curves are representative of CO evolution from both SiO2 and MnO reduction. Previous work by the authors[2] has shown that the mass transfer coefficient for silica is essentially the same as for manganese oxide in this slag system; however, the concentration of silica in the slag is much higher than MnO. Per (Eq. [6]), if the system was limited by mass transport, the rate of reaction would be proportional to the mass transfer coefficient times driving force and for a fixed mass transfer coefficient, one would expect systems with lower MnO to react more quickly as more carbon is available to reduce silica. What is likely, since internal nucleation is occurring, is that there is no impediment to oxygen transport and the oxygen content of the metal droplet is determined from the equilibrium of manganese and manganese oxide at the interface. This is further complicated by the apparent simultaneous reduction of silica and the associated equilibrium associated with silicon and silica at the interface. Ultimately, the confounding effect of oxygen sources makes isolating the effects of initial manganese oxide content difficult to interpret.

CO evolution vs time for variations in the initial manganese oxide content in slag (normalized for manganese and silicon gains)
Figure 8 compares bloated droplets in slags ranging from 5 to 15 wt pct MnO. There is apparently very little difference in reaction rates between reference (15 wt pct) and the 10 wt pct MnO samples, per Figure 8, but the 5 wt pct trials show interesting evidence for the influence of oxygen on the system. Even though the quantified rate of decarburization is faster in the 5 wt pct MnO system the rate of internal decarburization must be slower as evidenced by the degree of bloating. This suggests that the rate of external decarburization is faster than internal decarburization.

Comparison of bloated droplets vs initial MnO content; 5 wt pct MnO slags do not lead to complete bloating of the metal droplets
Effect of Reaction Temperature
Figure 9 shows the CO evolution as it relates to temperature increases. The rate data are quantified in the figure; there is an apparent discrepancy in the initial rate data, but the data for the second stage of reaction show that the two lower temperatures have the same rate of reaction and the higher temperature is faster.

CO evolution vs time for variations in reaction temperature (normalized for manganese and silicon gains)
Figure 10 shows X-ray observations from the droplets. The lower temperature system does not bloat as completely as higher temperature droplets. The free energy change associated with the reaction of carbon and manganese oxide is more favorable with increasing temperature.[25] The lower temperature reaction has similarities to the low MnO system. At higher temperatures, CO nucleation is more favorable and the reaction proceeds quickly.

Comparison of 1823 K (1550 °C) and 1923 K (1650 °C) maximally bloated droplets; in all cases droplets at the lower temperature had a metal pool at their base indicating slower reactions rates
Effect of Metal Droplet Mass
Figure 11 shows the relationship between CO evolution and droplet mass; the rate data are included in the figure. There is an increase in the absolute rate of reaction as the droplet mass rises for both the first and second stages of reaction.

CO evolution vs time for variations in metal droplet mass (normalized for manganese and silicon gains)
It has already been established that a relationship does not exist between area and rate during the first stage of reaction. Figure 12 shows the relationship between the rate of reaction and metal volume. The smaller droplet is somewhat of an outlier, but the volumes can be reasonably fit through zero. Interestingly, the volume relationship for stage two has an even stronger relationship with the rate.

Relationship between rate of CO generation and metal volume
An analysis of X-ray video evidence suggests that smaller droplets spend less time bloated than the larger droplets. If MnO mass transfer (and oxygen supply) were rate controlling, this should not have happened because no changes were made to the oxygen supply.
Changes in droplet mass show an interesting observation regarding the switch from external to internally nucleated CO. Figure 13 shows the progression of a 1.0 g metal droplet in the early stages of reaction. At 2 seconds, the droplet is suspended in the slag by an external cloud of CO. At 3 seconds, the droplet is seen to blur as it loses buoyancy and falls. At 3.5 seconds, the droplet has lost density on its upper surface, characteristic of internal CO formation. The droplet once again ascends by 5 seconds and is fully bloated. This suggests that early in the reaction, the supply of oxygen and carbon to the droplet surface are sufficient to sustain an external halo of CO. After some time, the external gas nucleation slows as carbon concentration at the interface falls. This is supported by the observation of subsequent internal nucleation of CO. Once internal nucleation starts, it seems likely that the droplet will be well stirred and that the reaction rate will be controlled by either supply of oxygen to the droplet or the rate of nucleation and growth of CO bubbles. The fact that the bubble loses buoyancy in the transition step from external to internal nucleation supports a conclusion that the two decarburization stages are distinct and that there is an incubation time associated with the internal CO generation. The two reactions may occur in parallel, but if they do it is at significantly different rates such that one appears to stop when the other begins.

Transition from primarily external to primarily internal CO nucleation in a droplet from dataset 8 (initial conditions 2.5 wt pct C, 15 wt pct MnO, 1.0 g droplet, 1873 K [1600 °C]) showing clear evidence that the external CO nucleation slows dramatically and that there is an incubation period for internal nucleation
Formation of Metal in the Slag
An interesting phenomenon is found in the systems when compared to the work of past authors who studied the carbothermic reduction of FeO using iron-carbon alloys. Sommerville et al.[9] discovered iron globules on the surface of their quenched slag samples. Two alternatives were proposed: they were a product of the reduction reaction of FeO from slag or that they originated in the bulk material and were carried to the top of the slag. The same phenomenon was discovered when they analyzed nickel oxide reduction, where nickel was found on the slag surface, leading them to believe that the reduction reaction was the primary source of this material.
In the present work, there is evidence supporting both droplet dispersion and oxide reduction as a possible mechanism. Figure 14 comes from a sample where the metal droplet had 4.3 wt pct carbon. There is no obvious evidence that any metal has broken away from the main droplet up to 10 seconds. Little happens until 60 seconds where a fragment of metal is detected in the bottom right of the crucible. The next four images are important and demonstrate how fragmentation of the bulk is at least partially responsible for the metal scattered about the slag. At 61 seconds, the fragment bloats internally and begins to rise into 61.5 seconds. At 62 seconds, the bloated droplet pops and the metal begins to fall reaching the bottom by 62.5 seconds. If these fragments were composed of reduced metal oxide, there should not be any carbon in the metal, and thus they could not bloat.

Appearance of metal fragment in a high-carbon sample
Comparatively, Figure 15 shows a sample where the slag contained 5 wt pct MnO; this system produces much more CO externally than the high-carbon sample. The release of externally generated CO bubbles is much less violent than the release of internal CO from the metal, reducing the degree to which dispersion would be a viable mechanism. At 30 seconds, a small droplet of metal can be detected along the slag foam’s surface. This metal moves and grows over time, seen from 50 to 150 seconds. A second piece of metal is detected at 180 seconds as well, immediately above the main droplet. This sample may support the conclusion of Sommerville et al.[9] as the secondary droplet appears to be growing with CO evolution and CO bubbles are not large enough to “burst” and scatter the main droplet. There is still the possibility that the CO is forming in or along the metal side of the interface causing a metal film to be pulled away with each CO bubble.

Growth of metal in the slag corresponding with largely external CO evolution
Among the other presented information, this supports the conclusions about a transition in the rate-limiting step of the reaction. When the decarburization is primarily internal, there is an explicit requirement that oxygen (and charge balancing metal ions) is reaching the interface of the metal and entering the metal droplet; thus, the only fragments of metal found in the slag away from the bulk droplet during the initial period are those caused by the ejection of a metal film. When the reaction is primarily external, and the metal interface is essentially blocked by gas, there is a high likelihood that gas shuttling across the bubbles occurs and thus metal oxides are reduced at the slag–gas interface, becoming trapped inside the slag. These findings coincide with the findings in Figure 2, and it seems possible that some degree of metal oxide reduction can occur in the slag.
Evidence of Slag Transport Control from Failed Experiments
With the experimental design and crucibles used in this work, it is possible for the alumina crucible to crack as it is being elevated in the furnace. Upon liquification of the slag, the leak becomes obvious and the experiment must be aborted. In a few outlying cases, the crucibles may fail during the experiment as well. These few experiments produced very interesting results as shown in Figure 16. Up until 1400 seconds, the experiment proceeds as normal, and the rate curve can be divided into an initial fast period and a slower second period where pressure rise decays over time. At 1400 seconds, the crucible fails, slag begins to leak, and a large rise in moles of CO generation occurs. At 1600 seconds, the rise stops and holds (as the crucible runs out of slag) until the system is quenched (breaking the seal on the furnace caused the pressure drop).

Plot of CO evolution measured from the CVPT when a crucible failed after 23 min; at 1400 s a sharp pressure increase can be seen in the plot
The above trend is repeated in several failed experiments where the same pressure rise can be documented. When a crucible fails, slag begins to flow from the crucible (but the metal remains largely stagnant). Unintentionally, flowing slag mimics some of the properties of stirred slag; thus, if the system were under transport control in the slag, one would expect an increase in the rate of reaction if the transport limitation were altered via slag stirring.
Alternatively, this sharp rise in pressure can be explained by the wiping of gas bubbles from the metal surface by agitated slag. There is strong evidence to support this claim in the absence of slag mass transport limitations.
Discussion
Several rate controlling mechanisms have been supported with the preceding data, and others have been dismissed. The first stage of reaction encompassing times before 100 seconds appears to involve internal nucleation of CO: the reaction rate remains higher for longer with increasing carbon content, droplets reacting with low MnO slags do not bloat as fully as those with higher MnO concentration, and a relationship may exist between the reaction rate and metal volume. For the second stage of reaction the system returns to a reaction occurring on the surface of the metal droplet: there is a low likelihood that a reaction within the metal controls this stage of reaction. In order to better understand the system, the work of past authors will be analyzed in more detail.
The following section offers a summary of some of the past reported rate-limiting steps and rate constants. Table II summarizes these values. A brief commentary on comparisons between the literature is provided afterwards.
There are two groups of authors who deserve special attention in this study and are detailed here. Wei et al.[26] originally studied desiliconization from iron containing carbon and with a slag containing FeO. The system was described to be mixed transport control, where kSi was 5 × 10−4 and kFeO was 3 × 10−5.
These authors also studied decarburization from the same system.[10] Their analysis is similar to work done by Sommerville et al.[9] so their work is discussed together. Sommerville et al.[9] laid the foundation for the chemical reactions occurring within the carbothermic reduction system, where Wei et al.[10] emphasized the significance of gas phase transport. Experiments and modeling showed three likely mechanisms become or share the rate-limiting step of the reaction. These authors concluded that contact between slag and metal is obstructed by gas, and thus the gas acts as an oxygen transport intermediary.
At the slag–gas interface, (Eq. [8]) occurs, subdivided into (Eqs. [8a] and [8b]) for a better physical understanding of the interface; ■ and ■O represent a vacant site and oxygen filled site on the interface, subscripts GS and GM represent the gas–slag and gas–metal interfaces.
Iron oxide dissociates to iron and oxygen along the interface; for this condition to be true, iron must be found in the slag as it is assumed it has no way of traveling to the metal; the work of Sommerville et al.[9] substantiates this claim. Wei et al.[10] have reported that the reaction has a relative independence on pCO, and thus Reaction [8] is assumed limited by iron oxide dissociation, (Eq. [8a]). The oxygen is retrieved by CO in the bubble and transported to the gas–metal interface by CO2 where the reaction described by (Eq. [9]) proceeds via Eqs. ([9a] and [9b]).
Wei et al.[10] had success when applying this mechanistic understanding of the reacting system to their experimental data. When CO bubbles at the interface are large, the system shifts to CO2 transport control across the bubbles. When the slag oxygen supply is low (less than 2.5 wt pct FeO[9]), Reaction [9a] is the rate-limiting step because there is an insufficient partial pressure of CO2; this is supported by studies noting a drop in reaction rate with increasing [S] content.[9,22] When the oxygen content in the slag is high, the rate-limiting step is Reaction [8a]; Reaction 8b was eliminated because research shows that the system is relatively independent of gas pressure above 0.5 atm.[10] Wei et al.[10] also tested the effect of phosphorus in the slag on the reaction rate and showed that the decarburization slowed down with increasing phosphorus content, confirming that the system can experience rate limitation along the gas–slag interface. This model can be applied to the second stage of reaction of the present study. This model is very similar to findings of Min and Fruehan[24] who studied decarburization with iron droplets and FeO in the slag. This idea will be returned to when the second stage of reaction of the current study is discussed.
First Contact of Slag and Metal
In the moments after the metal droplet contacts the slag, externally nucleated CO is obvious and likely corresponds with a relative balance between carbon supply from the metal and oxygen supply from the slag. Mass transfer coefficients from the literature[2,22] are too fast for the documented rate of CO generation. When comparing FeO and MnO, the equilibrium constant for FeO reduction[21,24] is 200 to 300 greater than MnO which may correspond well with the difference in reaction rate of this system. It seems likely that this brief stage of reaction is related to the chemical reaction of MnO and C at the interface.
Internal Nucleation and Droplet Bloating
Evidence suggests that the rate of CO generation is the rate-limiting step during this stage of reaction, a combination of nucleation and growth phenomena. Evidence to support this claim is listed below.
-
Every condition tested shows droplet bloating (X-ray video).
-
The initial rate of reaction is linear despite changes in area; thus, the rate of reaction is independent of the interfacial area.
-
The rate of reaction is strongly linked to initial carbon content; the rate of nucleation and growth would improve with increasing carbon activity. Further, droplets take longer to bloat and remain bloated for longer as carbon content rises; all of this suggests that the carbon supply is a rate limitation to CO generation.
-
The rate of reaction is linked to droplet volume; the rate of nucleation rises with increasing nucleation sites explaining the volume relationship.
-
When temperature is low or MnO content is low, bloating is reduced; this indicates a chemical reaction with oxygen dependence as part of the rate-limiting step.
-
A rapid exchange of manganese and silicon with iron occurs here; this reaction is believed to occur outside of the decarburization mechanism.
External CO Nucleation Post Bloating
Figure 17 helps describe the mechanism. This droplet is from dataset 1 with 2.5 wt pct C, 15 wt pct MnO, 1.5 g droplet, 1873 K [1600 °C]. The highly facetted interface is covered in a halo of CO bubbles, which likely block any direct contact of slag and metal; this differs significantly from SiO formed in previous work[1] which only blocks a fraction of the interface. This faceting is not present in samples that are low in reactants (i.e., 1 wt pct C or 5 wt pct MnO) implying that these reactions have nearly reached equilibrium by the time of quench, or that the CO bubbles are leaving before they have a chance to facet the interface.

Quenched slag (near 22 min) and droplet from dataset 1 (initial conditions 2.5 wt pct C, 15 wt pct MnO, 1.5 g droplet, 1873 K [1600 °C]); initial conditions 2.5 wt pct C in 1.5 g metal, 15 wt pct MnO, and 1873 K [1600 °C]
Under the assumption that the interface between slag and metal is blocked by gas, the model and findings of Sommerville et al.[9] and Wei et al.[10] can be discussed. These authors described the stoichiometry at the gas–slag interface with (Eq. [8]); for the present system Eqs. ([10] or [11]) are their equivalents. Silicon transport could also occur by SiO transport in place of CO2; metallic silicon is unlikely to remain in the slag when (MnO) is present.
For these reactions to occur, there must be evidence of metal buildup in the slag, away from the primary metal droplet. Figure 15 demonstrates how metal builds up in the slag as the system reacts, and that this metal is not the result of fragmentation of the main droplet. Figure 18 shows more faceting and shows that there is some degree of MnO reduction from the slag where CO is in direct contact with the slag. Further, it should be added that the final mass of metal droplets was less than the initial mass of the droplet: because [C] is exchanged with heavier elements, the mass would be expected to rise with time. This decrease in mass can be accommodated by this mechanism.

Quenched slag (approximately 4 min) surrounding metal droplet from dataset 6 (initial conditions 2.5 wt pct C in 1.5 g metal, 15 wt pct MnO, and 1823 K [1550 °C]). Significant faceting is present on the metal surface, CO bubbles are leaving the metal surface to the top of the slag, and left and right edges show homogeneous bulk slag
The CO2 produced by these reactions ferries oxygen across the gas bubbles where it reacts with carbon at the interface to regenerate CO. The system could be controlled by metal oxide decomposition at the gas–slag interface, CO2 transport, or CO2 decomposition at the gas–metal interface. Wei et al.[10] quantified the critical diameter at which transport in the gas bubble became negligible to be 2 mm.[10] The figures presented here show bubble diameters ranging from 0.5 to 4 mm, and thus CO2 transport may play a role in the rate-determining step as well as a chemical reaction at either the gas–metal or slag–metal interfaces.
The findings of Sommerville et al.[9] can be extrapolated to the present system to determine which of the chemical reactions is more important. They stated that because the CO2/CO ratio created by FeO decomposition was low with respect to that created by either NiO or PbO, the rate-limiting chemical reaction was the gas–metal interface; Reaction [8] produced little CO2 creating a scarcity of CO2 in Reaction [9]. NiO and PbO are significantly more reducible than FeO, and FeO is significantly more reducible than MnO or SiO2, and thus the logical extrapolation is that the chemical reaction responsible for rate control in the present system is the decomposition of CO2 at the gas–metal interface (Eq. [9a]).
This proposed mechanism may also explain the stage 2 reaction rate results from Figure 7 where the slag with lowest MnO content has the highest rate of CO evolution. If a stable layer of gas cannot be formed along the metal interface, then the slag and metal are not separated, and thus the system does not experience a rate limitation association with CO2.
The apparent increase in reaction rate in Figure 16 associated with crucible breakage can be explained because stirring of the slag agitates and detaches gas bubbles from the surface of the metal, which brings fresh slag into contact with the metal. This will allow for an exchange reaction such as in (Eq. [1]) to occur or for direct carbon oxidation to occur such as in Eqs. ([4] or [5]); neither reaction is limited by the transport or reaction kinetics of CO2.
In summary, post bloated droplet the system is rate limited by CO2 transport across the bubble layer that forms around the metal droplet and by decomposition of CO2 once it reaches the gas–metal interface.
Conclusions
The carbothermic reduction of manganese oxide from slag to metal droplets was studied across a range of conditions. Initial concentrations of carbon and manganese oxide were varied to 1, 2.5, and 4.3 and 5, 10, and 15 wt pct, respectively. Temperatures were varied from 1823 K to 1923 K (1550 °C to 1650 °C). Droplet masses were varied from 0.5 to 1.5 g. Plots for total CO gas generation can be simplified to two reaction regimes, the first described by internal CO nucleation (demonstrated in X-ray imaging) and the second which is limited by the formation and transport of CO2. The reacting system is highly complex and explains why a variety of documented rate-limiting mechanisms have been discussed by past authors. Compared to the reduction of MnO by silicon, the rates found in these experiments are significantly slower. Further research is required to quantify the dynamic reaction mechanisms and rate constants for the proposed controlling steps. In summary, the following statements can be made.
-
(1)
The rate of reaction for the studied systems can be broken into an initial fast stage and slower second stage; the difference between these rates is less than a factor of four.
-
(2)
The initial rate is controlled by internal CO nucleation and growth, demonstrated with the X-ray video evidence and by analyzing plots of increasing carbon content and droplet volume against the CO evolution rate. Oxygen from the slag must transport through the interface faster than carbon from the metal reaches the interface for droplet bloating to occur.
-
(3)
The second rate is limited by CO2 mass transport through stagnant or partially stagnant bubbles, from the gas–slag interface to the gas–metal interface, and the corresponding decomposition of CO2 at the gas–metal interface. This mechanism agrees well with past authors studying the carbothermic reduction of FeO.
-
(4)
With respect to its use in direct alloying applications, a fast period of material exchange may occur at the beginning of reactions which could contribute some net manganese to the melt within a reasonably short period of time; this gain is less than 0.5 wt pct. The kinetics of the slower second step of reaction do not support bulk manganese additions to metal.
References
B.J. Jamieson and K.S. Coley: Metall. Mater. Trans. B, 2017, vol. 48, pp. 1613–24.
B.J. Jamieson, Y. Tabatabaei, M. Barati, and K. S. Coley: Metall. Mater. Trans. B, 2019, vol. 50, pp. 192–203.
R. Elliott, K. Coley, S. Mostaghel, and M. Barati: Jom, 2018, vol. 70, pp. 1–11.
O.S. Bobkova and V.V. Barsegyan: Metallurgist, 2006, vol. 50, pp. 463–68.
L.N. Kologrivova, A.Ya. Nakonechnyi, Z.G. Trofimova, O.V. Nosochenko, and N.N. Kulik: Metallurg, 1987, vol. 5, pp. 28–29.
O.I. Nokhrina, V.P. Komshukov, and V.I. Dmitrienko: Metallurgist, 2004, vol. 48, pp. 264–65.
M. Eissa, H. El-Faramawy, and G. Farid: Steel Res., 1998, vol. 69, pp. 373–80.
W.L. Daines and R.D. Pehlke: Metall. Trans., 1971, vol. 2, pp. 1203–11.
I.D. Sommerville, P. Grieveson, and J. Taylor: Ironmak. Steelmak., 1980, vol. 7, pp. 25–32.
P. Wei, M. Sano, M. Hirasawa, and K. Mori: ISIJ Int., 1991, vol. 31, pp. 358–65.
K. Xu, G. Jiang, W. Ding, L. Gu, S. Guo, and B. Zhao: ISIJ Int., 1993, vol. 33, pp. 104–8.
S.K. Tarby and W.O. Philbrook: Trans. Metall. Soc. AIME, 1967, vol. 239, pp. 1005–17.
T. Yagi and Y. Ono: Trans. Iron Steel Inst. Japan, 1970, vol. 10, pp. 36–37.
R.J. Pomfret and P. Grieveson: Ironmak. Steelmak., 1978, vol. 5, pp. 191–97.
Y. Kawai, N. Shinozaki, and K. Mori: Can. Metall. Q., 1982, vol. 21, pp. 385–91.
N. Shinozaki, K. Ishido, K. Mori, and Y. Kawai: Tetsu-to-Hagane, 1984, vol. 70, pp. 73–80.
H. Sohn, Z. Chen, and W. Jung: Steel Res., 2000, vol. 71, pp. 145–52.
M.A. Rhamdhani: PhD Thesis—McMaster Univ., 2005.
C.L. Molloseau, and R.J. Fruehan: Metall. Mater. Trans. B. 2002, vol. 33, pp. 335–44.
K. Gu, N. Dogan, and K.S. Coley: Metall. Mater. Trans. B 2017, vol. 48, pp. 2984–3001.
E. Chen and K.S. Coley: Ironmak. Steelmak., 2010, vol. 37, pp. 541–45.
D.R. Sain and G.R. Belton: Metall. Trans. B, 1976, vol. 7, pp. 235–44.
K. Gu, N. Dogan, and K.S. Coley: Metall. Mater. Trans. B, 2018, vol. 49, pp. 1119–35.
D. J. Min and R. J. Fruehan: Metall. Trans. B, 1992, vol. 23, pp. 29–37.
C.W. Bale, E. Bélisle, P. Chartrand, S.A. Decterov, G. Eriksson, A.E. Gheribi, K. Hack, I.H. Jung, Y.B. Kang, J. Melançon, A.D. Pelton, S. Petersen, C. Robelin, J. Sangster, and M.A. Van Ende: Calphad, 2016, vol. 54, pp. 35–53.
P. Wei, M. Sano, M. Hirasawa, and K. Mori: Trans. Iron Steel Inst. Japan, 1988, vol. 28, pp. 637–44.
Acknowledgements
The authors thank the National Science and Research Council of Canada (NSERC, STPGP463252-14) for funding support. Special thanks to ArcelorMittal Dofasco, Stelco, Praxair, and Hatch Ltd. for their in-kind support, technical expertise, and their many helpful discussions.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Manuscript submitted February 19 2019.
Rights and permissions
About this article

Cite this article
Jamieson, B.J., Barati, M. & Coley, K.S. Kinetics of the Carbothermic Reduction of Manganese Oxide from Slag. Metall Mater Trans B 50, 2733–2746 (2019). https://doi.org/10.1007/s11663-019-01696-9
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11663-019-01696-9