
Abstract
Insect cell cultures played central roles in unraveling many insect physiological and immunological processes. Regardless, despite imminent needs, insect cell lines were developed primarily from Dipteran and Lepidopteran orders, leaving many important insects such as Orthopteran locusts under-represented. Besides the lack of cell lines, the slow progress in development of in vitro techniques is attributed to poor communications between different laboratories regarding optimized primary cell cultures. Therefore, we report here about methods developed for primary cell culture of Locusta migratoria hemocyte and phagocytic tissue cells by which we could maintain viable hemocytes in vitro for over 5 d and phagocytic tissue cells for over 12 d. 2-Mercaptoethanol and phenyl-thiourea supplements in Grace’s medium together with addition of fetal bovine serum 30 min after cell seeding resulted in a successful setup of the primary cell cultures and a week-long survival of the hemocytes and phagocytic tissue cells in vitro.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Locusts, chiefly Locusta migratoria and Schistocerca gregaria, are recognized as one of the most notorious crop pests in the world, owing to their swarm forming and voracious feeding behaviors. They exist in two forms either as harmless solitary animals or during food shortages as gregarious wandering pests which could endanger crop harvests (Uvarov 1977; Skaf et al. 1990; Enserink 2004). For this reason, locusts have obtained enormous attention in insect research to develop effective locust control strategies by elucidating the underlying biological mechanisms that control the process of phase transition and ensure their survival. However, despite hitherto achievements, many questions regarding cellular events in the locust which lead to phase change or disease resistance still remain unanswered (Wang et al. 2013, 2014; Wang and Kang 2014). This is mainly attributed to the lack of stable cell lines derived from either of the locust species, which would have otherwise enabled the researchers to explore in vitro many of the underlying molecular and cellular pathways.
As it has been evident in other animal studies, cell lines have been crucial to unravel key molecular players and pathways in many aspects of the insect physiology (Lynn 1996; Sudeep et al. 2005; Smagghe et al. 2009). In case of locusts, most studies were limited to the use of primary cell cultures and there has been a clear lack of effort to isolate continuous locust cell lines. Interestingly, successful reports addressing Acrididae and Tettigoniidae families in cell line development came from a few other grasshopper and cricket species. The most notable examples are cell lines derived from embryos of migratory grasshopper Melanoplus sanguinipes (Tsang et al. 1981; Munderloh et al. 1994) and cricket Gampsocleis gratiosa (Zhang et al. 2011), and from midgut of painted grasshopper Poekilocerus pictus (Kharat et al. 2010). Most reported studies which utilized the primary cell cultures of locust embryonic tissues, muscle, gonads, brain, and neuronal cells targeted rather at elaborating a vertebrate mirrored physiology than at exploring the locust itself (Duce and Usherwood 1986; Rossler and Bickmeyer 1993; Dorn and Dorn 2011; Ostrowski et al. 2011). This is without undermining the intensive past efforts made to establish locust stable cell lines with an intention of studying locust-pathogen interactions, screening locust pathogens, and developing viral bio-pesticides (Khurad et al. 1991; Hernandez-Crespo et al. 2000). Despite the available techniques, the in vitro cultures derived from the locusts, and other Orthopteran insects for that matter, are still very much limited as compared to that of Dipteran or Lepidopteran insects to investigate many of the intriguing issues in the locust research (Lynn 2001).
The central focus of the locust research is development of entogmopathogens which can be utilized in locust pest control since chemical insecticides are toxic to humans and the environment (Wang et al. 2013; Fang et al. 2014; Wang et al. 2014; Wang and Kang 2014). To this end, targeted locust-pathogen interaction studies are required to understand both locust defense and pathogen offense mechanisms as well as to develop virulent pathogens against the locusts. The role of in vitro cultures derived from locust immunocytes, particularly hemocytes or cells of phagocytic tissue (also known as hematopoietic tissue), is therefore important in unraveling the regulatory pathways such as pathogen recognition, immune signaling, and immunocyte recruitment during pathogen interaction. Let alone established cell lines, optimized and standardized locust immunocyte primary cell cultures are lacking or at least laboratories which produced such cultures have not communicated them to the wider public. To our knowledge, there is only one documented report about in vitro characterization of locust (S. gregaria) hemocytes in which the hemocytes survived not more than 3 h in the culture (Huxham and Lackie 1988). In addition, the established Orthopteran cell lines or primary cell cultures are derived from tissues other than the immune cells and lack the features necessary for the locust immunity-based studies. For these reasons, optimized in vitro methods aiming at the locust immunocytes are still in demand by communities that are involved in the locust research.
Therefore, we decided to set up primary cell cultures targeting in particular the hemocytes and phagocytic tissue cells of L. migratoria. We investigated further some in vitro behaviors of the locust immune cells, particularly the hemocytes. We were able to incubate in vitro the locust hemocytes for over 5 d and the phagocytic tissue cells for over 10 d. We hope that this optimized method might be utilized for the locust immune-related studies and lead as a startup for a stable cell line transformation.
Materials and Methods
Animals, media, and chemicals.
The locusts were kept in crowded conditions in transparent cages which were placed in a ventilated room with controlled temperature (32 ± 1°C), daylight photoperiod (13 h), and humidity (40–60%). They were fed with grass and dried oat every day. Adult locusts 1–5 d after final molt were used for our experiment, including the hemocyte and phagocytic tissue collections.
Media tested in this study include Grace’s insect medium (11595; GibcoTM-Life Technologies, Paisley, UK), Leibovitz’s L-15 medium (21083; Gibco™-Life Technologies, Paisley, UK), IPL-41 medium (11405; GibcoTM-Life Technologies, Paisley, UK), TC-100 insect medium (13055; Gibco-Life Technologies, Paisley, UK), and EX-CELL 420 medium (Sigma-Aldrich, Saint Louis, Missouri). The following chemicals were used in this study: penicillin-streptomycin and fungizone (GibcoTM-Life Technologies, Grand Island, New York), phenyl-thiourea and β-mercaptoethanol (Sigma-Aldrich, Saint Louis, Missouri), and heat-inactivated fetal bovine serum (FBS) and trypsin (Gibco™-Life Technologies, Grand Island, New York). The Grace’s insect medium (1× concentrated) was used as a basal medium. A simple medium containing the supplements penicillin-streptomycin (111 μg/mL), fungizone (1.11 μg/mL), phenyl-thiourea (16.9 μg/100 mL), and β-mercaptoethanol (5.54 μM) in 1× Grace’s medium, 380 mOsm, pH 6 was used to set up the cell culture and is described in the following parts of the manuscript as “hemocyte medium”. T25 tissue culture flasks and 24-well flat-bottom polystyrene plates from Techno Plastic Products AG (TPP®, Trasadingen, Switzerland) were used for the cell culture. Deckgläser Round glass coverslips (14 mm) were purchased from Waldemar Knittel, Braunschweig Germany.
Cytological staining.
Hemocyte smears were prepared by spreading 20 μL hemolymph on Superfrost®Plus coated glass slides (Thermo Scientific, Newbraunschweik, Germany). They were first briefly air dried and then fixed for 1 h in 2% calcium acetate containing 4% paraformaldehyde at room temperature. After repeated washing with clean water, they were stained with Alcian blue 8GX 300 solution (pH 5.7) as described previously (Costin 1975).
Collection and in vitro culture of locust hemocytes.
To collect hemocytes, 500 μL of anti-coagulant solution (98 mM NaOH, 186 mM NaCl, 17 mM Na2EDTA, 41 mM citric acid, pH 4.5) was first injected into each locust as described previously (Huxham and Lackie 1988). The low pH anti-coagulant solution was used for locust hemocyte harvest and processing as recommended before (Matsumoto et al. 2012; Pech et al. 1994). Hemolymph was flushed out into 1 mL ice-cold anti-coagulant solution within 1 min after the injection. Hemolymph collected from up to five locusts was pooled in a 15-mL conical FalconTM tube (TPP®, Trasadingen, Switzerland), and the tube was incubated for 30 min on ice. Afterwards, the cells were washed in succession with ice-cold anti-coagulant solution and Grace’s medium by centrifugation (500×g for 5 min at 4°C) and re-suspension. In parallel, the total number of cells was estimated by using a Burker Chamber hemocytometer. The cells were finally re-suspended in the hemocyte medium (∼500,000 cells/mL).
For in vitro culture, 1 × 106 cells were seeded in the T25 flask in a total volume of 2.7 mL hemocyte medium. After incubation of the cells in vitro for 30 min, 300 μL heat-inactivated FBS was added into the medium containing the cells. Half of the medium was replaced with a new hemocyte medium containing 10% FBS in an interval of 2 d. The flask was incubated at 28°C at all times and was protected from dehydration by placing in the incubator an open container containing clean water. Phase-contrast pictures were taken using a Nikon microscope.
Collection and in vitro culture of locust phagocytic tissue cells.
The locust phagocytic tissue cells were collected and cultured in vitro as follows. First, locusts were disinfected in 70% ethanol for 5 min and in 0.1% sodium hypochlorite for 15 min. Afterwards, they were rinsed three times in sterile water and placed for dissection on a 60-mm Petri dish half-filled with sterile anti-coagulant solution containing antibiotics (200 μg/mL pen-strep and 1 μg/mL fungizone). The dorsal cuticles consisting of the phagocytic tissue, as described previously (Duressa et al. 2015; Hoffmann et al. 1979), were detached carefully from the rest of the body and transferred to another half-filled Petri dish. The detached tissues were then washed in succession with the anti-coagulant solution and NaCl/Pi buffer (8 mM Na2HPO4, 1.5 mM KH2PO4, 137 mM NaCl, and 2.7 mM KCl, pH 7.2). Afterwards, the tissue pieces were incubated in 1 mL of pre-warmed hemocyte medium at 28°C overnight. The following day, the tissues were discarded from the medium and detached cells were further incubated in the hemocyte medium containing 10% heat-inactivated FBS.
Primary Cell Culture Validation Tests.
The viabilities of hemocytes were determined by propidium iodide (PI) dye exclusion method (Chemometec, Allerod, Denmark) according to the manufacturer’s instructions. Briefly, two equal samples containing the locust cells were prepared. The first cell sample was directly stained with PI (Reagent™ A100) to count non-viable cells, and the second cell sample was subjected to cell membrane disintegration using Reagent™ B before PI staining and total cell counting. Cell samples were counted with NucleoCounter® NC-100™. The test was repeated multiple times and cell viability was determined based on differences between the total and non-viable cell counts.
In vitro cell survival.
In vitro survival of cells was determined based on examination of nuclear integrity using terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) detection method (Roche, Almere, The Netherlands) according to the manufacturer’s instructions. Sterile glass coverslips were first inserted into 24-well plates and ∼100,000 cells were seeded in each well. After adjusting the medium volume to 360 μL per well and addition of FBS, the plates were incubated at 28°C for 1–10 d. FBS and media were supplemented in the same manner as it is described in “Collection and in vitro culture of locust hemocytes” section. Empty wells were filled with sterile water to avoid dehydration. For cell survival tests, plates were spun at 500 g for 5 min at 4°C, washed once with NaCl/Pi, and fixed in 4% paraformaldehyde in NaCl/Pi for 10 min at room temperature (RT). After repeated washing steps with NaCl/Pi, the cells were placed in pre-heated (750 W) 10 mM citrate buffer containing Triton X-100 (0.05%) for 1 min and cooled quickly by adding distilled water. After permeabilization with 3% Triton X-100 in TBS (15 min), the cells were incubated in blocking reagent (10 mM Tris-HCl, pH 7.5, containing 3% BSA and 20% FBS) at RT for 1 h. Next, they were rinsed in PBS, incubated in TUNEL reaction mixture in the dark at 37°C in a humid atmosphere for 1 h, rinsed again in PBS, and counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Finally, the coverslips were mounted with anti-fade (Mowiol®4-88) on glass slides for Zeiss Imager.Z1 microscopy.
In vitro phagocytosis.
FITC-labeled yeast Candida albicans prepared as described previously (Duressa et al. 2015) were used for the phagocytosis assays. The locust cells seeded on glass coverslips as stated above were incubated at 28°C for 30 min. After incubation, 10% FBS and 10 μL FITC-labeled C. albicans were added into each well and the plates were further incubated for 3 h. Afterwards, the cells were washed three times with NaCl/Pi by spinning at 500×g for 5 min. The wells were then loaded with 400 μL of 0.2% trypan blue in NaCl/Pi for quenching of extracellular FITC labeled yeast. After 5 min of incubation at RT, excess trypan blue was removed, and the cells were washed three times with NaCl/Pi and fixed in 4% paraformaldehyde. Finally, the coverslips were washed three times with NaCl/Pi and mounted with anti-fade (Mowiol®4-88) on glass slides for microscopy (Zeiss Imager.Z1). In parallel, wells containing only FITC-labeled C. albicans were processed in the same way and used as a negative control.
Results and Discussion
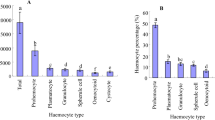
We observed that locusts contain different types of hemocytes in the circulation (Fig. 1). The different hemocytes include prohemocytes, plasmatocytes, granulocytes, coagulocytes, spherule cells, and oenocytoids as illustrated previously (Costin 1975). The majority of the locust hemocytes form spindle-like shapes when observed under an optical microscope while the remaining few cells take round shapes (Fig. 2a ). It was demonstrated earlier that locusts similar to some other insects contain the different hemocyte types with varied concentrations, plasmatocytes, and granulocytes dominating the hemocyte composition (Gupta 1979). The spindle-like cells represent mainly plasmatocyte and granulocyte cells while round cells might represent all kinds of hemocytes. We noticed that use of the low pH anticoagulant solution as described in the “Materials and Methods” section is essential to harvest the locust hemocytes as it avoids risks of acute cell clumping. After harvest, the hemocytes if placed on ice in either anticoagulant solution, Grace’s, or EX-CELL 420 media maintain their viabilities (over 85%) for over 5 h, providing a sufficient time window to set up a primary cell culture.

Morphological features of locust hemocytes. Locusts contain different types of circulating hemocytes. The images represent prohemocytes (a), plasmatocytes (b), granulocytes (c), coagulocytes (d), oenocytoids (e), and spherule-like cells (f).

Locust hemocyte collection and in vitro requirements for establishing primary cell culture. Each experiment was done in four replicates by seeding 106 cells in a T25 flask and was repeated at least five times. Locust hemocytes in anti-coagulant solution (a). The majority of hemocytes, as displayed in this image in the anticoagulant solution, form spindle-like shape while few cells have spherical features. Locust hemocytes in Grace’s medium without FBS (b). Addition of FBS immediately after hemocyte seeding (c). Immediate exposure of seeded hemocytes to FBS (10%) triggers acute and irreversible aggregation. Introduction of FBS in locust hemocyte culture 30 min post-cell seeding (d).
We tested five different insect culture media, which included Grace’s, IPL-41, TC100, L-15, and EX-CELL 420 media, to screen basal medium for the locust hemocyte culture. The GibcoTM basal media other than Grace’s medium support cell growth in lower osmomolar ranges (below 350 mOsm) while Grace’s and the EX-CELL media provide extracellular environment with higher osmomolar ranges (between 370 and 400 mOsm). In our tests, Grace’s and EX-CELL media provided the most suitable environment for the hemocyte primary cell culture while the other media seemed hypotonic to many of the hemocytes. Given that the hemolymph of L. migratoria has slightly acidic pH and osmolality in a range of 370–390 mOsm (Van Der Horst et al. 1980; Brone et al. 2003), one would actually foresee the fate of the hemocytes in the basal media. Therefore, while we did our experiments with the Grace’s medium, we suggest that locust hemocytes might also be similarly maintained in vitro using the EX-CELL medium.
We observed that, when setting up the primary cell culture, an immediate transfer of locust hemocytes from anticoagulant solution to the hemocyte medium containing heat-inactivated FBS (10 %) induces strong and an irreversible aggregation, leading to hemocyte lysis and death (Fig. 2b, c ). Hence, we found out that the cells once transferred to the hemocyte medium need to be seeded and briefly warmed at 28°C before adding the FBS (Fig. 2d ). We observed also that the FBS supplement has a rather positive effect on the hemocytes if introduced in the hemocyte medium after 30 min of warming the seeded cells. In this time window, many hemocytes settle at the base of the culture plate, and subsequent addition of the serum seems to enhance the attachment of the hemocytes to the culture plate (Fig. 3a ).

In vitro behavior and survival of locust hemocytes. The locust hemocytes attach to the cell culture plate and form a monolayer of cells within 24 h post-seeding (a). Locust hemocytes primary cell culture 3 and 5 d post seeding (b, c). Each experiment was done in four replicates by seeding 106 cells in a T25 flask and was repeated at least five times. Phagocytized yeast cells (green) by hemocytes in vitro (d). Display of surviving (DAPI, blue) and dying (TUNEL-FITC, green) hemocytes in the hemocyte primary cell culture 24 h and 5 d post seeding (e, f). The phagocytosis and survival tests were done in three replicates by seeding 105 cells in 24-well plates and were repeated at least three times.
We noticed that many of the locust hemocytes when incubated in vitro in a basal medium undergo melanization, which lead to observed change in the color of some cells and eventually of the medium (blackening of the cells and the medium). The melanization reactions in insect hemocytes occur through a series of enzymatic pathways, which involves the production of quinones and free radicals (Ratcliffe et al. 1991; Ling and Yu 2005). Due to this problem, it is well recognized that insect hemocytes are difficult to grow in vitro without using anti-oxidants and melanization inhibitors (Lynn 2001). This challenge extends to other arthropods wherein phenyl-thiourea (PTU) and β-mercaptoethanol (β-ME) are successfully applied in the hemocyte cultures to address the problem (Soderhall et al. 2005; Jose et al. 2010; Jayesh et al. 2013). In our locust hemocyte cultures, the antioxidant (β-ME) and melanization inhibitor (PTU) supplements were also able to effectively block the melanization reaction which in turn ensured extended in vitro survival of the hemocytes (Fig. 3). Other melanization inhibitors, glutathione and cysteine which were previously proposed for insects (Lynn 2001), were however much less effective than PTU in our locust hemocyte cultures.
The locust hemocytes which we cultured in vitro as described above were able to survive for over 5 d as long as half of the medium was replaced every 2 d with fresh hemocyte medium containing 10% FBS (Fig. 3). We observed that with survival, some hemocytes appeared to maintain their in vivo phagocytic behavior as they were able to endocytose the yeast cells inoculated into the medium (Fig. 3d ). In addition, the hemocytes attached predominantly to the culture plate and formed a monolayer of cells within 24 h post-seeding (Fig. 3a ). The cells displayed also elongated cytoplasmic features which mostly contained the nucleus at the center. As we have demonstrated using the TUNEL tests, cells losing viability showed nuclear disintegration and positive staining while healthy cells maintained intact nucleus and remained unstained with the TUNEL reaction (Fig. 3e, f ). Apart from this, despite notions that PTU might be toxic to insect cells, we observed a rather positive effect of this compound in stabilizing the hemocytes during the 5-d primary cell culture. The hemocytes, however, start to lose viability from 6 d onwards which might be due to various reasons such as cell aging or possible long-term toxicity effects of some media supplements (e.g., PTU, fungizone, and β-ME).
The locust phagocytic tissue (hematopoietic tissue) represents a major arsenal in the locust defense against intruding pathogens (Duressa et al. 2015). We were able to successfully isolate cells of this phagocytic tissue and culture them in vitro for over 12 d (Fig. 4). After incubation of the phagocytic tissue in the hemocyte medium overnight, many cells which resemble predominantly the spindle-like locust hemocytes were released from the tissue into the medium (Fig. 4a ). The tissue was subsequently discarded and the released cells were further incubated and supplemented with the growth medium containing FBS as described for the hemocytes (Fig. 4b–d ). We noticed that the majority of the cells eventually attach to the culture plate like the hemocytes (Fig. 4d ), displaying also elongated cytoplasmic structures. However, while the cells derived from the hemocytes started to detach from the culture plate from 6 d onwards, possibly due to loss of viability, the phagocytic tissue cells remained attached to the plate for at least 12 d.

Isolation and primary cell culture of locust phagocytic tissue cells. Each experiment was done in five to seven replicates in 24-well plates, each replicate representing isolated phagocytic tissue cells from a single locust. The experiment was repeated at least five times. Cluster of cells released from the phagocytic tissue in 24 h (a) gradually attached to culture plate (b, c) and developed elongated cytoplasmic feature (c), finally forming a monolayer of cells in the primary cell culture (d). Hours/days of cell incubation are indicated on the pictures.
Insect cell cultures played central roles in unraveling many insect physiological and immunological processes. They have been utilized beyond insect studies such as in virus and recombinant protein productions, and in investigations of key mammalian molecular pathways. Regardless, development of insect cell lines favored primarily the Dipteran and Lepidopteran orders for some reasons and many insect orders including the Orthopteran left under-represented (Lynn 2001). The orthopteran order includes species like L. migratoria and S. gregaria which are two of the most recognized pest insects to date. Strikingly, despite imminent needs for various pest control targeted studies, there have been only limited sporadic attempts to develop either primary cell cultures or stable cell lines using tissues derived from the locusts or other Orthopteran insects as such (Tsang et al. 1981; Zhang et al. 2011). Therefore, we hereby presented our recent advancement to set up a primary cell culture using the locust hemocytes and cells derived from the locust phagocytic tissue. As reported for other insects (Pech et al. 1994; Lynn 2001), we faced challenges in containing hemocyte clumping and melanization during the different stages of in vitro cell culture. Following multiple attempts by employing various techniques described previously for insect hemocyte collection (Huxham and Lackie 1988; Pech et al. 1994) and for inhibition of melanization (Lynn 2001; Soderhall et al. 2005), we were able to formulate a simple Grace’s-based hemocyte growth medium which contains supplements such as FBS, β-ME, and PTU that can be utilized to start up the locust hemocyte primary cell cultures. Working with the locust hemocytes has advantages as they are easier to harvest in contrast to cells derived from other body tissues. For primary cell cultures, sufficient hemocytes can be also easily harvested from few locusts as individual locusts have relatively large body sizes in comparison to other insects. We hope that our targeted effort on the primary cell culture will benefit locust researchers who study various cellular and molecular processes in the locust immune system and locust-pathogen interaction. Besides, our method might contribute to lasting efforts that are still to be made for developing locust cell lines.
References
Brone B, Tytgat J, Wang DC, Van KE (2003) Characterization of Na(+) currents in isolated dorsal unpaired median neurons of Locusta migratoria and effect of the alpha-like scorpion toxin BmK M1. J Insect Physiol 49:171–182
Costin NM (1975) Histochemical observations of hemocytes of Locusta migratoria. Histochem J 7:21–43
Dorn DC, Dorn A (2011) Structural characterization and primary in vitro cell culture of locust male germline stem cells and their niche. Stem Cell Res 6:112–128
Duce JA, Usherwood PNR (1986) Primary cultures of muscle from embryonic locusts (Locusta migratoria, Schistocerca gregaria)—developmental, electrophysiological and patch-clamp studies. J Exp Biol 123:307–323
Duressa TF, Vanlaer R, Huybrechts R (2015) Locust cellular defense against infections: sites of pathogen clearance and hemocyte proliferation. Dev Comp Immunol 48:244–253
Enserink M (2004) Entomology. Can the war on locusts be won? Science 306:1880–1882
Fang WG, Lu HL, King GF, St Leger RJ (2014) Construction of a hypervirulent and specific mycoinsecticide for locust control. Scientific Reports 4:
Gupta AP (1979) Hemocyte types: their structures, synonymies, interrelationships, and taxonomic significance. Insect hemocytes development, forms, functions, and techniques. Cambridge University Press, Cambridge, pp 85–128
Hernandez-Crespo P, Lopez-Blachere C, Bergoin M, Quiot JM (2000) Establishment of two new orthopteran cell lines. In Vitro Cell Dev Biol Anim 36:559–562
Hoffmann JA, Zachary D, Hoffmann D, Brehelin M, Porte A (1979) Postembryonic development and differentiation: hemopoietic tissues and their functions in some insects. In: Gupta AP (ed) Insect hemocytes development, forms, functions, and techniques. Cambridge University Press, Cambridge
Huxham IM, Lackie AM (1988) Behavior in vitro of separated fractions of hemocytes of the locust Schistocerca gregaria. Cell Tissue Res 251:677–684
Jayesh P, Jose S, Philip R, Bright Singh IS (2013) A novel medium for the development of in vitro cell culture system from Penaeus monodon. Cytotechnology 65:307–322
Jose S, Mohandas A, Philip R, Bright Singh IS (2010) Primary hemocyte culture of Penaeus monodon as an in vitro model for white spot syndrome virus titration, viral and immune related gene expression and cytotoxicity assays. J Invertebr Pathol 105:312–321
Kharat KR, Sawant MV, Peter S, Hardikar BP (2010) Development and characterization of new cell line BPH22 from midgut epithelial cells of Poekilocerus pictus (Fabricius, 1775). In Vitro Cell Dev Biol Anim 46:824–827
Khurad AM, Raina SK, Pandharipande TN (1991) In vitro propagation of Nosema locustae using fat body cell line derived from Mythimna convecta (Lepidoptera: Noctuidae). J Protozool 38:91S–93S
Ling EJ, Yu XQ (2005) Prophenoloxidase binds to the surface of hemocytes and is involved in hemocyte melanization in Manduca sexta. Insect Biochem Mol Biol 35:1356–1366
Lynn DE (1996) Development and characterization of insect cell lines. Cytotechnology 20:3–11
Lynn DE (2001) Novel techniques to establish new insect cell lines. In Vitro Cell Dev Biol Anim 37:319–321
Matsumoto H, Tsuzuki S, Date-Ito A, Ohnishi A, Hayakawa Y (2012) Characteristics common to a cytokine family spanning five orders of insects. Insect Biochem Mol Biol 42:446–454
Munderloh UG, Kurtti TJ, Liu Y, Chen Ch (1994) Grasshopper cell culture. In: Maramorosch K, McIntosh AH (eds) Arthropod cell culture systems. CRC Press Inc, pp. 51-64
Ostrowski D, Ehrenreich H, Heinrich R (2011) Erythropoietin promotes survival and regeneration of insect neurons in vivo and in vitro. Neuroscience 188:95–108
Pech LL, Trudeau D, Strand MR (1994) Separation and behavior in vitro of hemocytes from the moth, Pseudoplusia includens. Cell Tissue Res 277:159–167
Ratcliffe NA, Brookman JL, Rowley AF (1991) Activation of the prophenoloxidase cascade and initiation of nodule formation in locusts by bacterial lipopolysaccharides. Dev Comp Immunol 15:33–39
Rossler W, Bickmeyer U (1993) Locust medial neurosecretory cells in vitro—morphology, electrophysiological properties and effects of temperature. J Exp Biol 183:323–339
Skaf R, Popov GB, Roffey J (1990) The desert locust—an international challenge. Philos Trans R Soc Lond Ser B-Biol Sci 328:525–538
Smagghe G, Goodman CL, Stanley D (2009) Insect cell culture and applications to research and pest management. In Vitro Cell Dev Biol Anim 45:93–105
Soderhall I, Kim YA, Jiravanichpaisal P, Lee SY, Soderhall K (2005) An ancient role for a prokineticin domain in invertebrate hematopoiesis. J Immunol 174:6153–6160
Sudeep AB, Mourya DT, Mishra AC (2005) Insect cell culture in research: Indian scenario. Indian J Med Res 121:725–738
Tsang KR, Freeman FA, Kurtti TJ, Brooks MA, Henry JE (1981) New cell lines from embryos of Melanoplus sanguinipes (F) (Orthoptera, Acrididae). Acrida 10:105–111
Uvarov BP (1977) Grasshoppers and locusts. Centre for Overseas Pest Research, London
Van Der Horst DJ, Houben NMD, Beenakkers AMTh (1980) Dynamics of energy substrates in the haemolymh of Locusta migratoria during flight. J Insect Physiol 26:441–448
Wang X, Fang X, Yang P, Jiang X, Jiang F, Zhao D, Li B, Cui F, Wei J, Ma C, Wang Y, He J, Luo Y, Wang Z, Guo X, Guo W, Wang X, Zhang Y, Yang M, Hao S, Chen B, Ma Z, Yu D, Xiong Z, Zhu Y, Fan D, Han L, Wang B, Chen Y, Wang J, Yang L, Zhao W, Feng Y, Chen G, Lian J, Li Q, Huang Z, Yao X, Lv N, Zhang G, Li Y, Wang J, Wang J, Zhu B, Kang L (2014) The locust genome provides insight into swarm formation and long-distance flight. Nat Commun 5:2957
Wang XH, Kang L (2014) Molecular mechanisms of phase change in locusts. Annu Rev Entomol 59:225–244
Wang Y, Yang P, Cui F, Kang L (2013) Altered immunity in crowded locust reduced fungal (Metarhizium anisopliae) pathogenesis. PLoS Pathog 9:e1003102
Zhang X, Feng Y, Ding WF, Chen XM, Wang CY, Ma T (2011) Establishment and characterization of an embryonic cell line from Gampsocleis gratiosa (Orthoptera: Tettigoniidae). In Vitro Cell Dev Biol Anim 47:327–332
Acknowledgments
This work was supported by KU Leuven Research Foundation (GOA/11/002). The authors gratefully thank Roger Jonckers and Evelien Herinckx for caring the locust breeding and Ria Vanlaer for technical assistance. We thank the laboratory of Prof. Lieve Van Mellaert (Rega Institute, KU Leuven) for providing us with heat-deactivated Candida albicans. We would like to extend also our appreciation to the anonymous reviewers and associate editor for their constructive comments which made this article acceptable for publication in this journal.
Author information
Authors and Affiliations
Corresponding author
Additional information
Editor: Tetsuji Okamoto
Rights and permissions
About this article

Cite this article
Duressa, T.F., Huybrechts, R. Development of primary cell cultures using hemocytes and phagocytic tissue cells of Locusta migratoria: an application for locust immunity studies. In Vitro Cell.Dev.Biol.-Animal 52, 100–106 (2016). https://doi.org/10.1007/s11626-015-9952-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11626-015-9952-5