
Abstract
In this research, for the first time, an electrochemical sensor has been introduced by incorporating synthesized graphene nanoparticles (GrNPs) and tungsten disulfide nanoparticles (WS2NPs) into a carbon paste (CP) matrix. This sensor was used for the simultaneous measurement of noscapine (NOS) and papaverine (PAP) as two important alkaloids in opium. The properties of the synthesized nanoparticles were examined by scanning electron microscopy, elemental analysis, and X-ray diffraction. The electrochemical behavior of the modified electrode in aqueous solutions was studied by chronoamperometry, cyclic voltammetry, impedance, and differential pulse voltammetry techniques. A significant enhancement in the peak current response of NOS and PAP was observed at the graphene nanoparticles and tungsten disulfide nanoparticle–modified carbon paste electrode (GrNPs/WS2NPs/CPE) compared to the bare CPE. Also, using differential pulse voltammetry (DPV) method, linear range 5 to 1000 μM and limit of detection (LOD) (S/N = 3.0) 0.5062 μM and 1.0793 μM were obtained for NOS and PAP, respectively. Real-sample analysis results showed a recovery range 93.0–95.0% and 93.0–95.4 for NOS and PAP, respectively, that confirm a powerful ability of GrNPs/WS2NPs/CPE for determination of NOS and PAP in urine and industrial effluent samples.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Recently, the metal sulfides have been received considerable attention because of its promising properties, for example, chemical stability, non-toxicity, electron redox activity, low cost, environmental friendliness, and high electron transfer kinetics which have significant applications [1, 2]. Thus, many research groups studied metal sulfides for various science and technological applications. Therefore, it is widely applied to supercapacitors, corrosion prevention, batteries, hydrogen storage, fuel cells, photo- and electrocatalysts, and electrochemical sensors [3,4,5,6,7]. Currently, tungsten disulfide (WS2) possesses many fascinated properties such as high electrical conductivity, excellent catalytic activity, and high thermochemical stability [8, 9]. Several methods have been introduced for the synthesis of micro-nanoscale WS2 with specific morphologies and startling properties. These methods include the mechanical activation, magnetron sputtering, thermal decomposition, laser ablation, chemical vapor deposition, solid–gas reaction, etc. [10]. However, these methods are less favored due to the involvement of higher time and cost. The synthesis of WS2 via hydrothermal process has multiple advantages as compared with other methods due to the low temperature, an appropriate and simplest fabrication process, and the highly pure end product [11]. In this research, hydrothermal process method was used for WS2NPs synthesis.
Carbon-based solid electrodes are of current widespread use within electroanalysis owing to their broad potential window, low background current, low cost, chemical inertness, and suitability for various sensing applications [12,13,14,15]. Carbon is one of the most abundant nonmetallic elements and is found uniformly distributed throughout the globe. The uniqueness of carbon is due to its catenation property, i.e., the property of an element to form bonds with similar or different kinds of atoms. Because of its catenation property, carbon mainly exists in two different allotropic forms: crystalline and amorphous carbon. The crystalline form has an ordered arrangement which includes diamond (sp3), graphite (sp2), carbine (sp1), and fullerenes (distorted sp2). Graphene is a relatively new 2D material consisting of one atom thick planar layer of sp2 hybridized carbon atoms packed in a hexagonal lattice. Graphene and its composites exhibit good electrochemical performance owing to their superior properties, such as high surface area, high mechanical strength, good conductivity, and chemical stability [16, 17]. On the other hand, nanostructure materials have attracted much attention in the last few years due to their unique properties that are different from the bulk materials [18]. During the past few years, researchers have produced a short analysis and study of graphene preparation literature [19], illustrating the most common graphene. In summary, basic graphene preparation consists of mechanical exfoliation, liquid phase exfoliation, chemical vapor deposition, arc discharge method, and synthesis on SiC. The liquid-phase exfoliation method is an effective method for producing graphene, so in this work, this method was used to produce GrNPs. Many studies have focused on using WS2 along with other nanomaterials as novel modified electrode materials due to their aggregation properties [20, 21]. In this regard, integrating WS2 and high conductive matrix into a nanostructure has been demonstrated as a valuable approach to improve the conductivity [22, 23]. In recent years, various nano- and multilayer–based matrices including graphene and graphene-based high conductive materials have been widely used to construct with WS2 to improve their electrocatalytic performance [24,25,26].
Noscapine and papaverine have been considered as the derivatives of isoquinoline. The second most abundant alkaloid in opium is NOS (in concentrations of 2–8%) without analgesic traits [27] and is often used the pharmaceutical antitussive activity [28]. Moreover, NOS can cause apoptosis in many cell types and has strong antitumor action versus solid murine lymphoid tumors and human breast and bladder tumors fixed in nude mice. Papaverine was first separated from alkaloids of the opium group by Merck in 1848. Its value present in opium is almost 0.5–1% [29]. PAP lowered tonus and reduced the contractile activity of smooth muscles [30]. In significant doses, it decreased the excitability of the cardiac muscle and delayed intracardiac conduction [31]. In spite of many therapeutic effects, it has side effects for example causing prolonged fall in arterial blood pressure, when the intestinal tract is relaxed [29]. Also, PAP has interaction with cellular membrane and influence on affinity of hemoglobin to oxygen. Besides morphine, other alkaloids, such as PAP, NOS, or codeine, are co-extracted together with morphine and therefore reflect both the primary composition of opium and the method of extraction used. The methods of heroin production were experimentally examined by Huizer [32], who compared two main procedures used for illicit isolation of morphine from opium: the lime method (used in Southeast Asia) and the ammonia method (West Asian method). Both methods showed similar yield for morphine, codeine, and thebaine, but the content of PAP and particularly of NOS was strikingly higher in the ammonia extracts. Huizer [32] found 14.99% of NOS and 1.33% of PAP (median values) in 513 non-prescription heroin samples seized in the Netherlands and analyzed in the period 1994–1999. Also, Gough [33] observed that the Pakistani Type 1/Iranian heroin contained approximately 10% NOS and approximately 2% PAP, and in Chinese, Indian, Pakistani Type 2, and Turkish heroin, only traces of these alkaloids were present.
Various methods have been reported for determination of PAP, such as chemiluminescence [34, 35], chromatography [36, 37], spectrophotometry [38, 39], and electrochemistry [40, 41]. Among them, the electrochemical methods have received much interest in recent years due to their rapid, lower cost, disposability, portability, high accuracy, and simplicity of preparation. Also, electrochemical methods in comparison with reported chromatographic methods have lower matrix interferences, so there is no requirement for time-consuming extraction step [42]. The simultaneous utilization of GrNPs/WS2NPs for the fabrication of electrochemical sensors for simultaneous determination of noscapine and papaverine has never been reported in the literature.
In this paper, the graphene nanoparticles (GrNPs) and tungsten disulfide nanoparticles (WS2NPs) were applied for bulk modification of the CPE to fabricate an electrochemical sensor for NOS and PAP measurements. The CPE modified with GrNPs and WS2NPs (GrNPs/WS2NPs/CPE) could remarkably enhance the electrochemical responses of NOS and PAP and improve the sensitivity and selectivity of NOS and PAP detection. Furthermore, the electrochemical sensor was successfully employed to determine the NOS and PAP concentrations in real samples.
Experimental
Materials
All the reagents used in this work including, methanol, ethanol, HCl, HNO3, NaOH, H3PO4, H2SO4, sodium tungstate dihydrate (Na2WO4.2H2O), K2S2O8, P2O5, Na2S, KOH, KCl, NaCl, hydroxylamine hydrochloride, and nujol oil from the Merck company. All solutions were freshly prepared with double-distilled water. NOS and PAP were purchased from the Sigma-Aldrich chemical company.
Apparatus
Electrochemical data were obtained with a three-electrode system using a Potentiostat/Galvanostat (OrgaFlex 500, Franc). The three-electrode system was employed with an Ag/AgCl (saturated by KCl) electrode as a reference electrode, a Pt wire as a counter electrode, and a CPE modified with graphene and WS2 nanoparticles was used as a working electrode. All of used electrodes were from Metrohm. The electrochemical impedance spectroscopy (EIS) measurements were performed by applying an AC voltage with 5 mV amplitude in a frequency range from 0.01 Hz to 100 kHz under open circuit potential conditions. X-ray diffraction (XRD) patterns were obtained on a Burker AXS (Model B8 Advance). The microstructure and elemental composition of the samples were analyzed by using scanning electron microscope (SEM) (FE-SEM TESCAN MIRA3) and energy-dispersive spectra (EDS) (Mira3 detector SAMx) analysis. A pH meter, PHS-3BWModel (Bell, Italy) with a glass combination electrode was used for pH measurements. An ultrasonic bath (SW3, Switzerland) was used at a frequency of 50/60 kHz.
Synthesis of WS2 nanoparticles
WS2 nanoparticles is synthesized via hydrothermal method [43]. In a typical procedure for the synthesis of WS2 nanoparticles, 329 mg of sodium aqueous tungstate and 156 mg of sodium sulfide are dissolved in 50 mL of deionized water using a magnetic stirrer; then, 347 mg of hydroxyl ammonium chloride is added to this solution at a constant rate. To dissolve completely in the solution, now with the help of 1 M hydrochloric acid or 1 M soda, the pH of the solution is adjusted to 7, and finally, the resulting mixture is placed in an ultrasonic bath at 70 °C for 1 h. The resulting precipitate is then separated by centrifugation and washed several times with deionized water and pure ethanol. Finally, the resulting product is collected and dried under vacuum at 75 °C.
Fabrication of graphene nanoparticles
Graphite was put into a mixture of 12 mL concentrated H2SO4, 2.5 g K2S2O8, and 2.5 g P2O5. The solution was heated to 80 °C and kept stirring for 5 h using oil bath. Next, the mixture was diluted with deionized water (500 mL). The product was obtained by filtering using 0.2-µm Nylon film and dried naturally. The product was re-oxidized by Hummers and Offeman method to produce the graphite oxide [34]. Exfoliation was carried out by sonicating 0.1 mg/mL graphite oxide dispersion for 1 h. Reduction of graphite oxide was carried out by adding 0.5 mL hydrazine into the solution of 50 mg graphite oxide powder in 20 mL water after sonicating for 1 h and kept stirring for 24 h at 50 °C. Finally, black hydrophobic powder of graphene was obtained by filtration and dried in vacuum [44].
Preparation of working electrodes
To prepare the unmodified electrode, a certain amount of graphite and nujol oil as the pasting liquid is weighed and transferred to a mortar. The above materials are ground until a completely homogeneous mixture is prepared. Its kinetics and a small amount of it prevent the mixture from becoming completely homogeneous, thus reducing the reproducibility of the electrode. The prepared carbon paste is then transferred to a double-ended glass or plastic tube and completely compressed. Then, using a copper wire, an electrical connection is established (Scheme 1). Before using the carbon paste electrode, the contact surface of the electrode must be smooth and even. For the preparation of modified electrodes GrNPs/CPE, WS2/NPs/CPE, and GrNPs/WS2/NPs/CPE, the procedure was similar to the preparation of unmodified electrodes; only in the first step, certain amounts of graphene and WS2 nanoparticles are added to graphite and nujol oil.

Schematic procedure of the GrNPs/WS2/NPs/CPE preparation
Prepare a solution of real samples
In this work, industrial effluent and urine were examined and tested as two real samples. The urine samples were collected from candidates without a disease history. The urine samples were filtered through a 0.45-mm pore size cellulose acetate filter and without dilution. The industrial effluent sample prepared was centrifuged for 15 min at 4000 rpm to eliminate the suspended pollutions. Then, each of the samples was brought to the desired pH using sodium hydroxide solution or concentrated hydrochloric acid and was transferred to an electrochemical cell separately for analysis. Finally, the experiments were performed in a linear range with the standard increase method.
Results and discussion
Characterization of Gr and WS2 nanomaterials
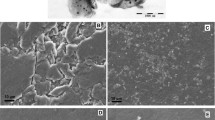
Figure 1A and B show the SEM images of graphene and tungsten disulfide (WS2) nanoparticles, respectively. Graphene nanoparticles are shown as hollow cavities and WS2 nanoparticles as lamellar layers. The size of nanoparticles and cavities was calculated using two software: Digimizer and Origin. The distribution of these two nanoparticles improves the performance of the electrode and increases the speed of electron transfer between the electrode surface and the solution

SEM images of A graphene and B WS2 nanoparticles. EDS results of C graphene and D WS2 nanoparticles. XRD patterns of E graphene and F WS2 nanoparticles
X-ray energy diffraction (EDS or EDX) spectroscopy is an analytical method used to analyze the structural or chemical properties of a sample. This method relies on the interaction between the X-ray excitation source and the sample. The descriptive capabilities of this method are generally based on the general principle that each element has a unique atomic structure that creates a unique set of peaks in the X-ray spectrum. Since the energy of X-rays represents the energy difference between the two layers as well as the atomic structure of the element from which they are emitted, it is possible to measure the composition of the sample elements. The resulting EDX spectrum graphene and WS2 nanoparticles are shown in Fig. 1C and D, respectively. The results about the weight percentage and atomic percentage of the constituent elements of graphene and WS2 are summarized in Tables as insets. These results show the presence of elements C, O, W, and S in the structures of these nanomaterials.
X-ray diffraction (XRD) is used to confirm the phase and crystal structure of Gr and WS2. As shown in Fig. 1E, Gr presents a diffraction peak at 26°, which can be ascribed to (002) reflections of Gr, demonstrating the diffraction characteristics of grapheme [45]. According to Fig. 1F, the pure WS2 sample shows the diffraction peaks at 16.11°, 29.82°, 44.84°, and 59.33° that correspond to the hexagonal 2HWS2 (002), (004), (006), and (008), respectively [46]. To investigate the mean size of graphene and WS2 nanoparticles, the highest and sharpest peaks in the XRD spectrum and Debye–Scherrer Eqs. (1–2) were used.
In this relation, τ is the average particle size in nanometers, K is the crystal shape coefficient which was considered equal to 0.9, λ is the wavelength of the X-ray tube which is equal to 0.154 nm, β is the peak width in Half the maximum height (FWHM), θ is the diffraction angle. In the Scherer relation, K and cosθ have no dimension, and the dimensions λ, β, and τ must be of the same material (unit of length). The following formula is used to convert the β dimension from degrees to units of length.
For graphene in Eq. 1, β is equal to 0.0081 and cosθ is equal to 0.97, which after calculations, the size of graphene nanoparticles was equal to 17.64 nm. Also, for WS2, β is equal to 0.0031 and cosθ is equal to 0.99, which after calculations, the size of WS2 nanoparticles was equal to 45.16 nm.
Impedance measurements of GrNPs/WS2NPs/CPE
Electrochemical impedance spectroscopy (EIS) was also employed to study the Gr/WS2/NPs/CPE. Figure 2 shows the typical results of AC impedance spectra of the bare CPE (curve a), Gr/CPE (curve b), WS2/NPs/CPE (curve c), and Gr/WS2/NPs/CPE (curve d) in 0.1 M KCl solution containing 1.0 mM [Fe(CN)6]3−/4− by applying 5 mV AC voltage in the frequency range of 0–2 Hz. The experimental results for the electrodes were fitted to equivalent circuit (inset of Fig. 2). This comparable circuit encompasses an interfacial capacitance (Cdl), the ohmic resistance of the electrolyte (Rs), the electron-transfer resistance (Rct), and the Warburg impedance (Zw). EIS in full frequency range includes a semicircle part and a linear part. The semicircle part at higher frequencies corresponds to the electron-transfer limited process, and the electron-transfer resistance Rct, which controls the electron transfer kinetics of the redox probe at the electrode interface, is represented by the semicircle diameter. The linear part at lower frequencies corresponds to the diffusion process. It is clear that considerable differences in the electrochemical impedance spectroscopy were observed for these four electrodes. Based on Fig. 2, the Rct of (d) to (a) curves is respectively managed to be 4.9, 32.8, 61.5, and 86.1 kΩ. The value of the Rct also decreases from (d) to (a) due to a rising trend in transfer rate constant, effective surface area, electron, and conductivity. These results show that the surface of CPE was effectively modified by the graphene and WS2 nanoparticles, and the conductivity was significantly increased. Therefore, the modified electrode, the GrNPs/WS2NPs/CPE, was chosen for further electrochemical research.

Nyquist plots of the bare CPE (a), GrNPs/CPE (b), WS2NPs/CPE (c), and GrNPs/WS2NPs/CPE (d) in 0.1 M KCl solution containing 1.0 mM [Fe(CN)6]3−/4−. Conditions: Eac, 5 mV; frequency range, 0.1 to 10,000 Hz. Inset: Equivalent circuit used in the fit procedure of the impedance spectra
Electrochemical behavior of NOS and PAP at GrNPs/WS2NPs/CPE
Cyclic voltammograms obtained at different electrodes in phosphate buffer solution (pH 7.0) containing 0.5 mM NOS and 0.5 mM PAP as mixture within the potential window of 0.3–1.7 V and at a scan rate of 50 mV s−1 are shown in Fig. 3. The results indicated that the graphene and WS2 nanoparticles had great improvement to increase peak currents and decrease anodic peak potentials of NOS and PAP at the GrNPs/WS2NPs/CPE (curve d) compared the bare CPE (curve a), GrNPs/CPE (curve b), and WS2NPs/CPE (curve c). From these results, it can be concluded that the GrNP/WS2NP-modified electrode displayed a synergistic catalytic effect of GrNPs and WS2NPs towards NOS and PAP oxidations, because GrNPs provided a large surface area for decoration of WS2NPs and thereby enhanced the electrocatalytic activity of the GrNPs/WS2NPs. Thus, the simultaneous determination of NOS and PAP is feasible o the GrNPs/WS2NPs/CPE surface.

Cyclic voltammograms of the CPE (a), GrNPs/CPE (b), WS2NPs/CPE (c), and GrNPs/WS2NPs/CPE (d) in in the phosphate buffer solution (pH 7.0) containing 500 µM NOS + PAP at a scan rate 50 mV s.−1
Chronoamperometric measurements of NOS and PAP at the GrNPs/WS2NPs/CPE were carried out by setting the working electrode potential at 0.7 and 1.2 V vs. Ag/AgCl/KCl (3.0 M), respectively, for 300, 500, and 700 µM NOS and the same concentrations of PAP in 0.05 M phosphate buffer (pH 7.0) (Figs. 4A and 5A). For an electroactive material (NOS and PAP in this case) with a diffusion efficient of D, the current observed for the electrochemical reaction at the mass transport limited condition is described by the Cottrell equation [47].
where D and Cb are the diffusion coefficient (cm2 s−1) and the bulk concentration (mol cm−3), respectively. Experimental plots of I vs. t−1/2 were employed, with the best fits for NOS Fig. 4A1) and PAP (Fig. 5A1). The slopes of the resulting straight lines were then plotted vs. NOS and PAP (Figs. 4A2 and 5A2, respectively). From the resulting slopes and Cottrell equation, the mean values of the D were found to be 5.32 × 10−6 and 1.86 × 10−5 cm2 s−1 for NOS and PAP, respectively.

(A) Chronoamperograms obtained at the GrNPs/WS2NPs/CPE surface in the presence of (a) 300, (b) 500, and (c) 700 µM NOS in the buffer solution (pH 7.0) at setting of the working electrode potential at 0.7 V vs. Ag/AgCl/KCl (3.0 M). Insets: (A1) Cottrell’s plots for NOS based on the data obtained from the chronoamperograms and (A2) curves for the slope of Cottrell’s plots versus concentration of NOS

(A) Chronoamperograms obtained at the GrNPs/WS2NPs/CPE surface in the presence of (a) 300, (b) 500, and (c) 700 µM PAP in the buffer solution (pH 7.0) at setting of the working electrode potential at 1.2 V vs. Ag/AgCl/KCl (3.0 M). Insets: (A1) Cottrell’s plots for PAP based on the data obtained from the chronoamperograms and (A2) curves for the slope of Cottrell’s plots versus concentration of PAP
Influence of experimental parameters
Voltammetric experiments were carried out with four different modified electrodes at graphene nanoparticles content 5.0%, containing WS2 nanoparticles ratio of 2.0, 5.0, 7.0, and 10.0% (w/w) with respect to the carbon paste in phosphate buffer solution (pH 7.0) containing 250 μM NOS and PAP. The value of anodic peak currents of differential pulse voltammograms of NOS and PAP increased with increasing of WS2 percentage, and the maximum value was obtained at 5.0% then decreased (Fig. 6A and B). It may be caused by the fact that the presence of WS2 increases the number of sites for NOS and PAP adsorptions; subsequently, the oxidation peak current increases. However, the excess of WS2 increases the resistance of the electrode and the anodic peak currents of these compounds on the GrNPs/WS2NPs/CPE surface decreases. Hence, a WS2 nanoparticle (5.0%, w/w)–modified carbon paste electrode was chosen as optimal.

The dependence of the anodic peak currents of cyclic voltammograms of A NOS and B PAP on WS2 nanoparticles content and the effect of graphene amount on C NOS and D PAP responses at the GrNPs/WS2NPs/CPE surface
The effect of graphene nanoparticle content on response of the GrNPs/WS2NPs/CPE was studied using cyclic voltammetry technique at WS2 nanoparticles content 5.0% by varying the graphene percentage (2.0, 5.0, 7.0, and 10.0%) in phosphate buffer solution (pH 7.0) containing 250 μM NOS and PAP (Fig. 6C and D). With increasing the graphene over 5.0% in the modified electrodes, the anodic peak currents of NOS and PAP decrease. It may be due to electrode conductivity decrease by the excessive amount of graphene on the electrode surface.
The effect buffer pH (pH range from 6.0 to 9.0) on the response of 500 µM NOS and PAP as mixture in 0.05 M phosphate buffer was studied at the surface of GrNPs/WS2NPs/CPE (Fig. 7). It was shown that with pH increasing from 6.0 to 9.0, the NOS and PAP anodic peak currents (Ipa) reach a maximum value at pH 7.0 and then decreases gently with increasing of pH (inset of Fig. 7). As the dissociation constants (pKa) of NOS and PAP were 7.8 [48] and 8⋅07 [49], respectively, the drugs presented as the protonated forms in acidic media in fact. In pHs > 7, the concentration of protonated these compounds were decreased and as a result the peak currents are decreased. Therefore, the accumulation of NOS and PAP on the GrNPs/WS2NPs/CPE film performed out through electrostatic interaction between GrNPs/WS2NPs with negative charge and pH dependent protonated NOS and PAP forms. So, pH value of 7.0 was selected for the determination of NOS and PAP. On the other hand, as the pH increased, the anodic peak potentials (Epa) of the drugs at the surface of GrNPs/WS2NPs/CPE shifted to more positive values. The NOS and PAP peaks shift to more positive potentials in the pH range of 6.0 to 9.0 from 781 mV (pH 6.0) to 1060 mV (pH 9.0) for NOS and from 1327 mV (pH 6.0) to 1550 mV (pH 9.0) for PAP. It may be caused by the effect of proton (H+) on these compound peaks at the GrNPs/WS2NPs/CPE surface (Scheme 2) [48, 50].

Cyclic voltammograms of a NOS (500 µM) and b PAP (500 µM) in various pH values of buffer solutions: 6.0, 7.0, 8.0 and 9.0. Inset: Ipa-pH curves for oxidation of a NOS and b PAP at the GrNPs/WS2NPs/CPE surface (scan rate 50 mV s.−1)

Electrooxidation mechanisms of noscapine and papaverine
Effect of scan rate
The effect of scan rate on the simultaneous oxidation of 500 μM NOS and PAP at the GrNPs/WS2NPs/CPE investigated in 0.05 M phosphate buffer (pH 7.0) at different potential scan rates (Fig. 8A). It can be seen that the oxidation peak currents for both NOS and PAP linearly increased with the square root of the scan rates (ν1/2) (Fig. 8B and C, respectively), suggesting that the redox reaction at the electrode surface is predominantly diffusion-controlled process for both NOS and PAP [47].

A Cyclic voltammograms for oxidation of NOS (500 µM) and PAP (500 µM) at the GrNPs/WS2NPs/CPE surface in the buffer solution (pH 7.0) at different scan rates: 5, 10, 30, 50, 100, 150, 200, and 300 mV s−1. Dependence of anodic peak currents B NOS and C PAP on the square root of scan rate. Linear relationship between logarithm of anodic peak potentials D NOS and E PAP and logarithm of scan rate
Moreover, the oxidation peak potential (Epa) of both NOS and PAP shifted in the positive direction with increasing the scan rate. Also, the Tafel slope for NOS and PAP (Fig. 8D and E, respectively) can be obtained from the slope of Epa vs. log ν using Eq. (4) [51]:
Based on the Eq. (4), the slope of Ep vs. logν curve is b/2, where b indicates the Tafel slope and b = 2 ×\(\partial\) Epa/\(\partial\)(logν). The Tafel slopes were found to be 0.123 and 0.0951 V for NOS and PAP, respectively. According to Laviron, b = 2.303RT/n(1 − α)F, for anodic peak, where F is the Faraday’s constant (96,485 C mol−1) [52] and the other symbols have their usual meanings. In addition, based on the documents [51], the electron-transfer number (n) both for NOS and PAP oxidations is 2. Therefore, the values of the electron-transfer coefficients of NOS and PAP (α) are 0.531 and 0.38, respectively.
Analytical performance
The quantitative analysis of nitrite on GrNPs/WS2NPs/CPE was carried out by differential pulse voltammetry (DPV). The DPV parameters, such as step potential of 10 mV, modulation amplitude of 60 mV, modulation time of 40 ms, and interval time of 200 ms, represent the optimum values with satisfactory height of current responses of NOS and PAP. In Fig. 9, it can be observed that the DPV responses of GrNPs/WS2NPs modified electrode to different additions of NOS and PAP at the potential range (0.3 to 1.7 V) in 0.05 M phosphate buffer (pH 7.0). The linear dynamic ranges (LDRs) for determination of both NOS and PAP were 5–1000 µM. The detection limits (3σ) were 0.51 for NOS and 1.08 µM for PAP (insets A and B of Fig. 9, respectively). These values are comparable with values reported by other research groups for oxidations of NOS and PAP at the surface of chemically modified electrodes by other modifiers (Table 1).

Differential pulse voltammograms of the GrNPs/WS2NPs/CPE in the buffer solution (pH 7.0) containing different concentrations of NOS and PAP in the ranges of (a) 5, (b) 10, (c) 25, (d) 50, (e) 100, (f) 300, (g) 500, (h) 750, and (i) 1000 µM. Insets show calibration curves for simultaneous determination of (A) NOS and (B) PAP
Analysis of real samples
The practicality of the GrNPs/WS2NPs/CPE was investigated by DPV method towards the detection of NOS and PAP in urine and industrial effluent samples using standard addition method. Prior to the analysis (sampling procedure given in the “Experimental” section), the urine and industrial effluent samples were prepared and diluted with buffer. Then, certain concentrations of NOS and PAP were added to the samples. Finally, as-prepared real samples were analyzed by DPV method and the practicality of the modified sensor has proved. In addition, the obtained peak current responses have been calculated by standard addition method and the results are given in Tables 2 and 3. The relative error and recovery percentages summarized show the successful performance of this technique in measuring the analytes present in real samples.
Selectivity, reproducibility, and stability
The effects of potential interferents on the voltammetric responses of NOS (200 μM) and PAP (200 μM) were evaluated at pH 7.0. The potential interfering substances were chosen from the group of substances commonly found with NOS and PAP in pharmaceuticals and/or in biological fluids. The corresponding test results are listed in Table 4. The results show that the peak current change is less than 5%, which suggests the good selectivity of the created GrNPs/WS2NPs modified electrode toward the determination of NOS and PAP.
To test the repeatability of the Fe3O4/MWCNTs-CPE, the response of a mixture of NOS (250 μM) and PAP (250 μM) was measured successively five times at per day in 3 days (Table 5). Also, as shown in Table 5, the stability of the electrode was evaluated by probing of the electrode responses to electrocatalytic oxidation of NOS and PAP after being put away at room temperature for 3 days. The relative standard deviations were obtained to be 1.37% and 0.79% for NOS and PAP, respectively. Subsequently, the results showed that the proposed sensor has very good repeatability and stability.
Conclusion
In this work, for the first time, we reported fabrication a modified CPE with graphene nanoparticles and tungsten disulfide nanoparticles (GrNPs/WS2NPs/CPE). By using scanning electron microscope, elemental analysis, and X-ray diffraction, the formation of synthesized nanoparticles graphene nanoparticles and tungsten disulfide nanoparticles was proved and their properties were investigated. Also, chronoamperometry, cyclic voltammetry, impedance, and differential pulse voltammetry techniques were used to investigate the electrochemical behavior of the modified electrode in aqueous solutions. The GrNPs/WS2NPs/CPE was applied as a sensitive sensor for simultaneous determination of NOS and PAP. The GrNPs/WS2NPs/CPE significantly facilitated the electron transport between NOS and PAP and the electrode, improving the oxidation property of NOS and PAP on the electrode. The proposed method displayed suitable characteristics, such as simplicity, low-cost, high sensitivity, rapid analysis procedures, and wide liner range. The modified electrode was successfully applied to determine the concentration of NOS and PAP in real samples.
References
Liu H, Su D, Wang G, Qiao SZ (2012) WS2 anode material with superior electrochemical performance for lithium ion batteries. J Mater Chem 22:17437–17440. https://doi.org/10.1039/C2JM33992G
Yang G, Gong H, Liu T, Sun X, Cheng L, Liu Z (2015) Two-dimensional magnetic WS2@Fe3O4 nanocomposite with mesoporous silica coating for drug delivery and imaging-guided therapy of cancer. Biomaterials 60:62–71. https://doi.org/10.1016/j.biomaterials.2015.04.053
Li S, Jiang Z, Hou X, Xu J, Xu M, Yu X, Ma ZF, Yang J, Yuan X (2019) A relative study on sonochemically synthesized mesoporous WS2 nanorods & hydrothermally synthesized WS2 nanoballs towards electrochemical sensing of psychoactive drug (Clonazepam). Ultrason Sonochem 54:79–89. https://doi.org/10.1016/j.ultsonch.2019.02.012
Li Y (2022) Metal sulfide as catalysts enabling fast polysulfide conversion for high electrochemical performance Li–S batteries. Ionics. https://doi.org/10.1007/s11581-022-04493-8
Wang Y, Chen N, Du X, Zhang X (2021) Hierarchical sulfide nanoarrays as an efficient bifunctional electrocatalyst for overall water splitting. Ionics 27:2591–2602. https://doi.org/10.1007/s11581-021-04037-6
Mayorga-Martinez CC, Ambrosi A, Eng AYS, Sofer Z, Pumera M (2015) Transition metal dichalcogenides (MoS2, MoSe2, WS2 and WSe2) exfoliation technique has strong influence upon their capacitance. Electrochem Commun 56:24–28. https://doi.org/10.1016/j.elecom.2015.03.017
Yang H, Zhao F, Cao X, Liu Q, Zhang X, Zhang X (2020) Enhanced photoelectrocatalytic activity of cobalt sulfide modified with porphyrin as a noble-metal-free photoelectroncatalyst towards methanol oxidation under visible-light. J Taiwan Inst Chem Eng 116:169–177. https://doi.org/10.1016/j.jtice.2020.11.017
Gupta R, Valappil MO, Sakthivel A, Mathur A (2020) Tungsten disulfide quantum dots based disposable paper based lab on genochip for specific meningitis DNA detection. J Electrochem Soc 167:107501. https://doi.org/10.1149/1945-7111/ab8fda
Kozawa D, Carvalho A, Verzhbitskiy I, Giustiniano F, Miyauchi Y, Mouri S, Castro Neto A, Matsuda K, Eda G (2016) Evidence for fast interlayer energy transfer in MoSe2/WS2 heterostructures. Nano Lett 16:4087–4093. https://doi.org/10.1021/acs.nanolett.6b00801
Lei W, Xiao JL, Liu HP, Jia QL, Zhang HJ (2020) Tungsten disulfide: synthesis and applications in electrochemical energy storage and conversion. Tungsten 2:217–239. https://doi.org/10.1007/s42864-020-00054-6
Yang J, Voiry D, Ahn SJ, Kang D, Kim AY, Chhowalla M, Shin HS (2013) Two-dimensional hybrid nanosheets of tungsten disulfide and reduced graphene oxide as catalysts for enhanced hydrogen evolution. Angew Chemie Int Ed 52:13751–13754. https://doi.org/10.1002/anie.201307475
Foroughi MM, Jahani S, Aramesh-Boroujeni Z, Rostaminasab Dolatabad M, Shahbazkhani K (2021) Synthesis of 3D cubic of Eu3+/Cu2O with clover-like faces nanostructures and their application as an electrochemical sensor for determination of antiretroviral drug nevirapine. Ceramics Int 47:19727–19736. https://doi.org/10.1016/j.ceramint.2021.03.311
Setoudeh N, Jahani S, Kazemipour M, Foroughi MM, HassaniNadiki H (2020) Zeolitic imidazolate frameworks and cobalt-tannic acid nanocomposite modified carbon paste electrode for simultaneous determination of dopamine, uric acid, acetaminophen and tryptophan: Investigation of kinetic parameters of surface electrode and its analytical performance. J Electroanal Chem 863:114045. https://doi.org/10.1016/j.jelechem.2020.114045
Sheibani N, Kazemipour M, Jahani S, Foroughi MM (2019) A novel highly sensitive thebaine sensor based on MWCNT and dandelion-like Co3O4 nanoflowers fabricated via solvothermal synthesis. Microchem J 149:103980. https://doi.org/10.1016/j.microc.2019.103980
Arefi Nia N, Foroughi MM, Jahani S (2021) Simultaneous determination of theobromine, theophylline, and caffeine using a modified electrode with petal-like MnO2 nanostructure. Talanta 222:121563. https://doi.org/10.1016/j.talanta.2020.121563
Jijie R, Kahlouche K, Barras A, Yamakawa N, Bouckaert J, Gharbi T, Szunerits S, Boukherroub R (2018) Reduced graphene oxide/polyethylenimine based immunosensor for the selective and sensitive electrochemical detection of uropathogenic Escherichia coli. Sens Actuators B Chem 260:255–263. https://doi.org/10.1016/j.snb.2017.12.169
Wang X, Chen Y, Zheng B, Qi F, He J, Li P (2016) Few-layered WSe2 nanoflowers anchored on graphene nanosheets: a highly efficient and stable electrocatalyst for hydrogen evolution. Electrochim Acta 222:1293–1299. https://doi.org/10.1016/j.electacta.2016.11.104
Foroughi MM, Ranjbar M (2017) Microwave-assisted synthesis and characterization photoluminescence properties: a fast, efficient route to produce ZnO/GrO nanocrystalline. J Mater Sci: Mater Electron 28:1359–1363. https://doi.org/10.1007/s10854-016-5668-x
Lim JY, Mubarak N, Abdullah E, Nizamuddin S, Khalid MJ (2018) Recent trends in the synthesis of graphene and graphene oxide based nanomaterials for removal of heavy metals-A review. J Ind Eng Chem 66:29–44. https://doi.org/10.1016/j.jiec.2018.05.028
Li Y, Niu J, Xue T, Duan X, Tian Q, Wen Y, Lu X, Xu J, Lai L, Chang Y, Li Z, Zhao X, Chen Y (2020) Multifunctional porous nanohybrid based on graphene-like tungsten disulfide on poly(3,4-ethoxylenedioxythiophene) for supercapacitor and electrochemical nanosensing of quercetin. J Electrochem Soc 167:047512. https://doi.org/10.1149/1945-7111/ab721e
Er-Chieh C, Cai-Wan CJ, Jia-Huei Z, Jen-Hsien H, Kuen-Chan L, Bo-Cheng H, Yu-Sheng H (2018) Microwave-assisted synthesis of TiO2/WS2 heterojunctions with enhanced photocatalytic activity. J Taiwan Inst Chem Eng 91:489–498. https://doi.org/10.1016/j.jtice.2018.05.025
Shang X, Yan KL, Liu ZZ, Lu SS, Dong B, Chi JQ, Li X, Liu YR, Chai YM, Liu CG (2017) Oxidized carbon fiber supported vertical WS2 nanosheets arrays as efficient 3D nanostructure electrocatalyts for hydrogen evolution reaction. Appl Surf Sci 402:120–128. https://doi.org/10.1016/j.apsusc.2017.01.059
Cao S, Liu T, Hussain S, Zeng W, Peng X, Pan F (2014) Hydrothermal synthesis of variety low dimensional WS2 nanostructures. Mater Lett 129:205–208. https://doi.org/10.1016/j.matlet.2014.05.013
Latha M, Biswas S, Vatsala Rani J (2020) Application of WS2-G composite as cathode for rechargeable magnesium batteries. Ionics 26:3395–3404. https://doi.org/10.1007/s11581-020-03512-w
Mehew JD, Unal S, Torres Alonso E, Jones GF, Fadhil Ramadhan S, Craciun MF, Russo S (2017) Fast and highly sensitive ionic-polymer-gated WS2-graphene photodetectors. Adv Mater 29:1700222. https://doi.org/10.1002/adma.201700222
Chen SM, Umamaheswari R, Mani G, Chen TW, Ali MA, Fahad AH, Elshikh M, Farah MA (2018) Hierarchically structured CuFe2O4 ND@RGO composite for the detection of oxidative stress biomarker in biological fluids. Inorg Chem Front 5:944–950. https://doi.org/10.1039/C7QI00799J
United Nations (1987) Recommended methods for testing opium. United Nations, New York
Gerald MC (1974) Pharmacology: an introduction to drug. Prentice-Hall, Englewood Cliffs, NJ
Yan J, Mi JQ, He JT, Guo ZQ, Zhao M, Chang W (2005) Development of an indirect competitive ELISA for the determination of papaverine. Talanta 66:1005–1011. https://doi.org/10.1016/j.talanta.2005.01.001
Mashkovskii MD (2002) Lekarstvennye sredstva (Pharmaceutical preparations), vol 1. Novaya Volna, Moscow
Han X, Lamshoft M, Grobe N, Ren X, Fist AJ, Kutchan TM, Spiteller M, Zenk MH (2010) The biosynthesis of papaverine proceeds via (S)-reticuline. Phytochemistry 71:1305–1312. https://doi.org/10.1016/j.phytochem.2010.04.022
Huizer H (1987) Analytical studies on illicit heroin. Pharm Weekbl 9:203–211. https://doi.org/10.1007/BF02029331
Gough TA (1991) The examination of drugs in smuggling offences. In: Gough TA (ed) the analysis of drugs of abuse. John Wiley & Sons, Chichester, U.K., pp 511–566
Zhang S, Zhuang Y, Ju H (2004) Flow-injection chemiluminescence determination of papaverine using cerium(IV)-sulfite system. Anal Lett 37:143–155. https://doi.org/10.1081/AL-120027780
Zhuang YF, Cai XL, Yu JS, Ju HX (2004) Flow injection chemiluminescence analysis for highly sensitive determination of noscapine. J Photoch Photobiol A 162:457–462. https://doi.org/10.1016/S1010-6030(03)00391-5
Kasperek R (2008) Determination of diclofenac sodium and papaverine hydrochloride in tablets by HPLC method. Acta Pol Pharm Drug Res 65:403–408
Yin CH, Tang C, Wu XY (2003) HPLC determination of aminophylline, methoxyphenamine hydrochloride, noscapine and chlorphenamine maleate in compound dosage forms with an aqueous-organic mobile phase. J Pharm Biomed Anal 33:39–43. https://doi.org/10.1016/S0731-7085(03)00352-2
Tang Y, Luan J, Wang Q (2002) Determination of papaverine hydrochloride in skin and blood and the drug contents in pig skin. Acta Acad Med Sin 24:413–417
Ghasemi J, Niazi A, Ghorbani R (2006) Determination of trace amounts of lorazepam by adsorptive cathodic differential pulse stripping method in pharmaceutical formulations and biological fluids. Anal Lett 39:1159–1169. https://doi.org/10.1080/00032710600622126
Foroughi MM, Jahani S, Rajaei M (2019) Facile fabrication of 3D dandelion-like cobalt oxide nanoflowers and its functionalization in the first electrochemical sensing of oxymorphone: evaluation of kinetic parameters at the surface electrode. J Electrochem Soc 166:B1300. https://doi.org/10.1149/2.0511914jes
Fathabadi MV, Rafsanjani HH, Foroughi MM, Jahani S, Arefi Nia N (2020) Synthesis of magnetic ordered mesoporous carbons (OMC) as an electrochemical platform for ultrasensitive and simultaneous detection of thebaine and papaverine. J Electrochem Soc 167:027509. https://doi.org/10.1149/1945-7111/ab6446
Tajik S, Taher MA, Beitollahi H (2013) First report for simultaneous determination of methyldopa and hydrochlorothiazide using a nanostructured based electrochemical sensor. J Electroanal Chem 704:137–144. https://doi.org/10.1016/j.jelechem.2013.07.008
Chen TW, Rajaji U, Chen SM, Chinnapaiyan S, Ramalingam RJ (2019) Facile synthesis of mesoporous WS2 nanorods decorated N-doped RGO network modified electrode as portable electrochemical sensing platform for sensitive detection of toxic antibiotic in biological and pharmaceutical samples. Ultrason Sonochemistry 56:430–436. https://doi.org/10.1016/j.ultsonch.2019.04.008
Wang Y, Li Y, Tang T, Lu J, L J, (2009) Application of graphene-modified electrode for selective detection of dopamine. Electrochem Commun 11:889–892. https://doi.org/10.1016/j.elecom.2009.02.013
Wang Y, Jin Y, Pan E, Jia M (2018) Fe3O4 nanoparticle/grapheme aerogel composite with enhanced lithium storage performance. Appl Surf Sci 458:1035–1042. https://doi.org/10.1016/j.apsusc.2018.07.127
Krishnamoorthy D, Prakasam A (2021) Graphene hybridized with tungsten disulfide (WS2) based heterojunctions photoanode materials for high performance dye sensitized solar Cell device (DSSCs) applications. J Clust Sci 32:621–630. https://doi.org/10.1007/s10876-020-01828-1
Bard AJ, Faulkner LR (2001) Electrochemical methods: fundamentals and applications. Wiley, New York
Rezaei B, Heidarbeigy M, Ensafi AA, Dinari M (2015) Electrochemical determination of papaverine on Mg-Al layered double hydroxide/graphene oxide and CNT modified carbon paste electrode. IEEE Sens 16:3496–3503. https://doi.org/10.1109/JSEN.2016.2533429
Varsha K, Sharma A, Kaur A, Madan J, Pandey RS, Jain UK, Chandra R (2017) Nanostructures for cancer therapy; micro and nano technologies, chapter 28: natural plant-derived anticancer drugs nanotherapeutics: a review on preclinical to clinical success. Elsevier 775
Navaee A, Salimi A, Teymourian H (2012) Graphene nanosheets modified glassy carbon electrode for simultaneous detection of heroine, morphine and noscapine. Biosens Bioelectron 31:205–211. https://doi.org/10.1016/j.bios.2011.10.018
Nicholson RS, Shain I (1964) Theory of stationary electrode polarography. Single scan and cyclic methods applied to reversible, irreversible, and kinetic systems. Anal Chem 36:706–723. https://doi.org/10.1021/ac60210a007
Laviron E (1974) Adsorption, autoinhibition and autocatalysis in polarography and in linear potential sweep voltammetry. J Electroanal Chem 52:355–393
Rezaei B, Zare SZM (2008) Modified glassy carbon electrode with multiwall carbon nanotubes as a voltammetric sensor for determination of noscapine in biological and pharmaceutical samples. Sens Actuators B Chem 134:292–299. https://doi.org/10.1016/j.snb.2008.05.002
Babaei A, Sohrabi M (2016) An electrospun alumina-borate oxide nanofiber and reduced graphene oxide composite modified carbon paste electrode as the electrochemical sensor for simultaneous determination of dopamine and noscapine. Anal Methods 8:6949–6958. https://doi.org/10.1039/C6AY01724J
Ahammad A, Lee JJ, Rahman M (2009) Electrochemical sensors based on carbon nanotubes. Sensors 9:2289–2319. https://doi.org/10.3390/s90402289
Sharifi S, Zarei E, Asghari A (2021) Surfactant assisted electrochemical determination of noscapine and papaverine by TiO2 nanoparticles/multi-walled carbon nanotubes modified carbon paste electrode. Russ J Electrochem 57:183–196. https://doi.org/10.1134/S1023193521020129
Feng LJ, Zhang XH, Zhao DM, Wang SF (2011) Electrochemical studies of bovine serum albumin immobilization onto the poly-o-phenylenediamine and carbon-coated nickel composite film and its interaction with papaverine. Sens Actuators B Chem 152:88–93. https://doi.org/10.1016/j.snb.2010.09.031
Ziyatdinova G, Samigullin A, Budnikov G (2007) Voltammetric determination of papaverine and drotaverine. J Anal Chem 62:773–776. https://doi.org/10.1134/S1061934807080138
Kharitonov SV (2006) Electrochemical response characteristics and analytical application of papaverine ion-selective membrane electrodes. Anal lett 39:259–273. https://doi.org/10.1080/00032710500476912
Eppelsheim C, Aubeck R, Hampp N, Bräuchle C (1991) Determination of ethaverine and papaverine using ion-selective electrodes. Analyst 116:1001–1003. https://doi.org/10.1039/AN9911601001
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ahmadi, E., Zarei, E. & Asghari, A. Electrochemical sensor based on graphene and tungsten disulfide nanoparticles for determination of noscapine and papaverine. Ionics 29, 1579–1591 (2023). https://doi.org/10.1007/s11581-023-04895-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11581-023-04895-2