
Abstract
A series of moderately conducting and soluble copolymers of poly(aniline-co-o-bromoaniline) (PA-co-o-BrA) having different compositions was obtained by in the situ copolymerization method using different concentrations of monomer units of aniline and o-bromoaniline in the feed. The physio-chemical properties of the copolymers have been studied with sophisticated instrumental techniques. The electrochemical study of the copolymers was conducted by cyclic voltammetry. The band gap of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) of the copolymers was evaluated by UV-vis spectroscopy. The morphological study was conducted by scanning electron microscopy and transmission electron microscopy at high magnification which shows non-uniform tubular to globular morphology of the copolymers. Surface profiles of the polymers were studied by AFM analyses, and it has confirmed the smooth surface of the copolymers while the homopolymers possesses non-uniform surfaces. The particle size distribution curve indicates that the particle sizes vary in the range of 5 to 9000 nm, and a small fraction of particles possess a size in the range of 5–10 nm.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Amongst the conducting polymers, polyaniline and its copolymers possess unique chemical and physical properties which render such materials the most broadly studied materials in the current time due to advancement in technology [1]. The electrochemical polymerization of aniline induces the formation of polyaniline on the electrode by accepted mechanisms [2, 3].The unique ability of the oxidation-reduction reversibility of polyaniline put it in the category of most studied synthetic polymeric material. The different reported oxidized-reduced states of polyaniline are the leucoemeraldine, emeraldine, and perningraniline states [4, 5]. The sulfonation of poly(p-phenylenediamine) by oxidative copolymerization of p-phenylenediamine (pPD) and 3-aminobenzenesulfonic acid is reported [6]. The electrical conductivity of polyaniline-Mn-ZnO.Fe2O4 nano-composites have been reported [7]. Also, the synthesis and characterization of poly(aniline-co-o-toluidine copolymer by chemical and electrochemical methods at low pH environment have been repported [8]. In addition, functionalized aniline derivatives and copolymers are also obtained by electrochemical polymerization methods and possess different chemical and physical properties from the parent polyaniline [9]. Limited studies of the polymerization of aniline derivatives with electron-withdrawing groups such as o-bromoaniline have been reported [10]. Moreover, to improve the solubility of the polyaniline homopolymer and copolymers, –SO3 and –SO4 groups are mainly used as dopants. Incorporating a –SO3 group in the backbone of the polymer modifies the physical properties of parent polymer without altering the conductivity [11,12,13,14]. In addition, halogen substituted polyaniline derivatives substituted with the chloro [15, 16], fluoro [17,18,19], and bromo [20] groups have been reported in the literature. Such mono halogen substituted polyanilines prepared by oxidative polymerization in acidic medium were reported to possess substantially low doping levels with and reduced conductivity than the unsubstituted polyaniline homopolymer. The aim of the current experimental investigations is to prepare a halogenated copolymer with better physical properties such as solubility and electrical conductivity than the mono halogenated polyaniline.
Experimental
Materials
Aniline, o-bromoaniline, ammonium persulphate, hydrochloric acid and sulfuric acid were purchased from the Aldrich Chemical Companyand used as received. Spectroscopic grade N-methyl-2-pyrrolidone (NMP), dimethylsulfoxide (DMSO), and propanol were used as received. Solutions are prepared in double-distilled water. The homopolymers, polyaniline PA and poly(o-bromoaniline) P-o-BrA were synthesized by the oxidative polymerization of aniline and o-bromoaniline, respectively using ammonium persulphate as oxidant in 1 M hydrochloric acid according to procedure outlined in the literature [21,22,23,24] The PA-co-o-BrA copolymers were synthesized [25,26,27] by chemical oxidative coupling of aniline and o-bromoaniline in an aqueous solution of 1 M HCl using ammonium persulphate as an oxidant with molar ratio of monomers to oxidant as unity. The different compositions of the copolymers were obtained by varying the mole fraction of o-bromoaniline to aniline from 0.4 to 0.7. A typical procedure for the copolymerization process is outlined as follows: Aniline (0.93 g) and o-bromoaniline (1.08 g) were dissolved in 200 ml of 1.0 M HCl solution. The solution was cooled to 0–4 °C. Under nitrogen atmosphere, a pre-cooled solution of ammonium persulphate (4.56 g) prepared in 200 ml of 1.0 M HCl was added slowly to the monomer solution with stirring at 0–4 °C. The copolymerization reaction was allowed to continue for 24 h at room temperature, without stirring. The precipitate of the copolymer was filtered and washed with a small volume of 1.0 M HCl and dried in an air oven for few h at 80 °C. The copolymer was obtained as a black powder in the emeraldine salt form. The salt form of the copolymer was converted to the base form by treatment with 1.0 M NH4OH and kept overnight, filtered and dried in an air oven. The general synthesis pathway is given in Scheme 1.

Various steps involved in the copolymerization process
Characterization
The solubility of each polymer and copolymer was measured by dissolving each sample in propanol. In a typical procedure, 10 mg polymer powder was taken into 100 mL of solvent and allowed to disperse thoroughly and kept overnight at room temperature. The dispersion of the polymer was filtered through a sintered glass crucible (porosity 2 μm) and the solubility of the polymer was determined. Cyclic voltammetric measurements were performed on a BAS 100B (Bioanalytical Systems Inc) workstation. The Ag/AgCl electrode acted as the reference electrode, a glassy carbon electrode coated with the respective polymer (5 mg per mL in DMSO) as the working electrode and a platinum wire as the counter electrode. The voltammograms of the homopolymers and copolymers were recorded in 50 mL of a 0.1 M sulphuric acid solution as the electrolyte working solution. Before recording the voltammograms, the sulphuric acid solution was thoroughly degassed with nitrogen for 10 min. The potential sweep operated at a scan rate of 100 mV/s with the potential range of − 400 to 800 mV. UV-Visible absorption spectra of dilute solutions of the polymer samples in DMSO were recorded at room temperature on a Thermo Spectronic Genesys 2 Research Grade Spectrophotometer. Transmission electron microscopy (TEM) images of the copolymers were recorded on a FEI Tecnai TF20 microscope. To investigate the surface topology of the materials, atomic force microscopy (AFM) analyses were conducted at room temperature using a MFP3D Asylum AFM research system (Santa Barbara, CA, USA) using films of the polymers on a silicon wafer. Dilute solution of few μM concentration of the polymers in NMP solvent were prepared by sonication and centrifugation. Particle size distribution curves were measured in NMP solvent using a Malvern Instruments Ltd., Zetasizer Ver. 7.10, UK system. For the morphology studies, scanning electron micrographs of the different polymers were obtained on a JEOL-6700 SEM field emission microscope.
Results and discussion
Synthesis, yield and solubility
The general schematic molecular structures of the homopolymers and copolymers are shown in Fig. 1. Polyaniline (PA) and poly-o-bromoaniline (P-o-BrA) were synthesized by the chemical oxidative polymerization of aniline and o-bromoaniline monomer, respectively. The oxidant to monomer ratio was maintained as unity for all the polymerization reactions. Similarly, the copolymers of poly(aniline-co-o-bromoaniline)s (PA-co-o-BrA) were prepared by oxidative copolymerization of aniline with o-bromoaniline by slowly adding ammonium persulphate as an oxidant dissolved in hydrochloric acid. Upon varying the ratio of o-bromoaniline and aniline monomer in the feed, copolymers of different compositions were obtained. The physical appearance of different homopolymers as well as the copolymers were observed as black powders. The extent of solubility, percent weight, percent yield, and the bromine content of the homopolymers and copolymers are shown in Table 1. A higher yield is reported for polyaniline than poly-o-bromoaniline and a decrease in the percent yield of copolymers with increasing o-bromoaniline content in the copolymer composition is obtained with a small difference in the percentage yields for the different copolymers, PA-co-o-BrA). The rate determining step in the chemical oxidative polymerization reaction of aniline and o-bromoaniline involves the coupling of oxidized species which results in an overall electrophilic substitution reaction of the respective monomer to the growing chains. The o-bromoaniline is more reactive than aniline because the bromine substituent in the ortho position exerts an electron-withdrawing inductive effect on the benzene ring; The dominant electron-donating resonance effect ultimately influences the reactivity of o-bromoaniline. Thus, with the higher concentration of the more reactive o-bromoaniline in the feed, a statistical copolymer rich in o-bromoaniline segments is formed, especially at low conversion levels. Due to steric hindrance, the size of the bromine group retards the participation of o-bromoaniline in the copolymerization process, thereby reducing the yield of the copolymers even when the concentration of the o-bromoaniline comonomer in the feed is high [28]. The copolymer composition, as indicated by the content of the o-bromoaniline in the copolymers, was determined by elemental analysis for bromine using energy dispersive X-ray emission spectroscopy. The content of bromine varied from zero for polyaniline to 39 wt% for poly-o-bromoaniline. The solubility observed for polyaniline, poly-o-bromoaniline, and the different poly(aniline-co-o-bromoaniline)s in propanol is shown in Table 1. Low solubility of polyaniline in the propanol is observed, whereas poly-o-bromoaniline shows moderate solubility. Moreover, for the copolymers, upon increasing the amount of the o-bromoaniline unit along the polymer backbone, the solubility of bromo functionalized copolymer increases accordingly, and the solubility of the copolymers is relatively better than the corresponding polyaniline, but are less soluble in propanol than poly-o-bromoaniline. The o-bromoaniline unit along the polymer backbone introduces flexibility to the rigid polymer backbone structure and reduces inter-chain interactions, thereby increases the solubility of the copolymers. Also, the increased solubility might be due to the electronegative character of Br atom along the polymer backbone which increases H-bonding with the –OH group of the polar solvent.

General schematic molecular structure of polyaniline, poly-o-bromoaniline and (polyaniline-co-o-bromoaniline)s
Cyclic voltammetry
Figure 2 shows the cyclic voltammograms which depict the electrochemical behavior of polyaniline, poly-o-bromoaniline, and the different (polyaniline-co-o-bromoaniline)s. The polymeric material was cast as a thin film on a titanium plate and used as a thin film electrode in contact with 0.1 M sulfuric acid solution. The poly-o-bromoaniline (P-o-BrA) thin film shows a flat response (constant current over a wide potential range of 2500 mV), which indicates the nonconductive behavior of poly-o-bromoaniline (P-o-BrA). The polyaniline (PA) thin film shows an increased current response over the same potential range. The electrochemical study shows that, upon increasing the amount of o-bromoaniline in the copolymer, a decrease in the conductivity of the copolymer is observed up to 25 % o-bromoaniline content in the copolymer. In fact, the P-o-BrA copolymer (curve blue), shows no conductivity, as per cyclic voltammetry analysis. Furthermore, the poly(aniline-co-o-bromoaniline) copolymer of aniline with 10% of o-bromoaniline content shows higher electrochemical reactivity than the homopolymer P-o-BrA. Generally, introducing the bromine substituent along the polymer backbone, the resultant non-planar conformation restricts the delocalization of electrons along the polymer backbone and thereby reduced electrical conductivity is observed. In addition, the higher conductivity observed for the copolymers relative to the poly-o-bromoaniline homopolymer is due to the presence of larger aniline segments within the copolymer, which facilitates faster charge transport along the polymer chain. The cyclic voltammetry response is purely a qualitative indicator of reactivity of the copolymers and confirms the electro-active nature and order of magnitude of conductivity [29]. The conductivity of the copolymers have been reported in previous work [20].

Cyclic voltammetry for polyaniline, poly-o-bromoaniline and (polyaniline-co-o-bromoaniline)s
UV-visible spectroscopy
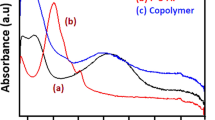
In Fig. 3, the electronic spectra of the homopolymers and copolymers dissolved in DMSO were recorded. Two absorption bands in the electronic spectra of the homopolymers and copolymers are observed. The absorption band around 290–340 nm is due to π→ π* electronic transitions of the phenyl rings along the polymer backbone. The absorption band above 600–620 nm is due to the inter-band charge transfer associated with excitation of benzenoid and quinoid moieties (exciton transitions). For the homopolymer P-o-BrA, the π→ π* transition band shows a hypsochromic shift from 325 to 300 nm, suggesting that there is a decrease in the extent of conjugation and results in an increase in the band gap and thus decrease in the electrical conductivity in the polymer. For the PA-co-o-BrA50 copolymer, no hypsochromic shift is observed, which indicates no decrease in the conjugation system and hence no change in the band gap. A very small change in the λ max values of the copolymer and homopolymer under consideration in the UV-vis spectra is observed. The exciton bands positioned at 620 to 600 nm and the intensity ratio between the exciton and the π→ π* transition bands are also unequal. The exciton band of poly-o-bromoaniline is almost absent in the UV-vis spectrum. The UV-vis results are in agreement with the electrical study of polymers by cyclic voltammetry analyses. Solvent plays an important role in the π→π* transition band of polymers. The hypsochromic shift is observed with the increase in dielectric constant of the solvent. Ghose and Kalpakam [30] explained such a shift on the basis of the dielectric constant of the solvent. The solvotochromism depends on factors such as geometry and electronic structures of the solvent as well as the solute. However, the bathchromic shift in the exciton band is observed with the decrease in dielectric constant of the solvents in the UV-visible absorption spectra. The coil like conformation of the polymer (decrease in conjugation) is favored in a solvent of high dielectric constant and a less polar solvent provides thermodynamically more stable chain conformations and restricts the polymer to a lower energy high planarity state. A bipolaron is created due to exciton (i.e., a negative charge is created on quinoid unit and a positive charge is centered on the adjacent benzenoid units) [31]. The red shift results in a lower energy transition and an increase in the conjugation of system. [32]

UV-vis spectra of (a) poly-o-bromoaniline (b) polyaniline (c) Poly(aniline-co-o-bromoaniline) PA-co-o-BrA50 in DMSO solvent
AFM analysis
The topography of polyaniline PA and the PA-co-o-BrA50 copolymer at nanometer scale were recorded. Figure 4a, b, analyzes the topographic profiles of PA and the poly(aniline-co-o-bromoaniline)s. AFM analysis was conducted because SEM analysis is unable to explain the vertical and surface clarity effectively in the images. The AFM study ensures the no usage of additional material for coating of the polymer sample. In the SEM study, either carbon or gold are required to coat the polymers. A more illustrative 2-D and 3-D perspective and impression of the surface images of polymer are provided by AFM. The dimension of the film used was 3 × 3 μm2 to observe the topographical variation in the roughness of surface of the polymer. The determination of surface roughness of the polyaniline and copolymer was conducted by a quantitative approach. In Fig. 5a, b and c, d, the 3-D surface line profile of PA, PA-co-o-BrA50, respectively, in the AFM images are illustrated. From the line profiles, the degree of roughness of the copolymers coating (SR) are evaluated. The surface line profile of PA indicates a non-uniform surface of the polymer whereas the surface profile of the copolymer PA-co-o-BrA50 shows the smooth and uniform surface. In addition, the degree of roughness is higher in PA sample as compare to PA-co-o-BrA50 copolymer derivative.

AFM images of a polyaniline and b (polyaniline-co-o-bromoaniline)s

AFM images of a polyaniline and b poly(aniline-co-o-bromoaniline) PA-co-o-BrA50 and corresponding surface roughness profiles of c polyaniline and d poly(aniline-co-o-bromoaniline) PA-co-o-BrA
Particle size analysis
Figure 6a, b shows the particle size distribution curves of polyaniline PA and the PA-co-o-BrA 50 copolymer in NMP solvent, respectively. For PA, the curve indicates that the particle size varies in the range 5 to 9000 nm and a small fraction of particles possess a size in the range of 5–10 nm. A large fraction of polyaniline possesses the particle size in the range of 100–1000 nm. An average particle size varies in the range of 1000–9000 nm. For the PA-co-o-BrA50 copolymer, a large fraction of the sample, particle sizes in the range of 100–1000 nm are obtained and a small fraction of the particles lies in 8000-9000 nm range only. The particle size varies as result of the degree of bromination of polyaniline. Due to the larger amount of bromine in the copolymer, the particle sizes vary in the range of 100–1000 nm under the observed conditions.

Particle size distribution curves of a polyaniline and b (polyaniline-co-o-bromoaniline)s copolymers
Transmission electron microscopy
TEM images of polyaniline and the copolymer films at high magnification are shown in Fig. 7a, b. For polyaniline PA, the TEM image shows a cluster of non-uniform tubular morphology and particles having diameters in the range of 50–60 nm and the length of the tubes of a few microns in size are observed. At high magnification, the TEM image of the copolymer PA-co-o-BrA50 also shows uniform tubular morphology with particles having smaller diameters and longer lengths when compared to the polyaniline PA derivative.

TEM images of a polyaniline and b (polyaniline-co-o-bromoaniline)s copolymers
Scanning electron micrography(SEM)
Figure 8a–c shows the SEM micrographs of P-o-BrA, PA and the PA-co-o-BrA derivatives, respectively. Non-uniform tubular morphology is observed in the SEM images of polyaniline. The change of tubular morphology of PA to globular morphology for thecopolymers PA-co-o-BrA is observed. Also, corn-flake-like morphology is clearly observed for the P-o-BrA sample. The incorporation of Br along the backbone of the copolymer results in the change in the morphology of homopolymer P-o-BrA and polyaniline from corn-flakes and tubular morphology to non-uniform globular shaped structures for the copolymers.

SEM images of a poly(o-bromoaniline), b polyaniline, and c (polyaniline-co-o-bromoaniline)s copolymers
Conclusion
Polyaniline and poly-o-bromoaniline were prepared by chemical oxidative polymerization of aniline and o-bromoaniline, respectively. A series of copolymers of aniline and o-bromoaniline was synthesized by chemical oxidative copolymerization methods using ammonium persulphate as oxidant in hydrochloric acid medium by varying the ratio of monomers in the feed. The oxidant to monomer ratio was set at 1:1 for all the polymerization reactions. The yields of the polymer products were greater than 50%. The UV-visible spectroscopy supports the characterization of the chemical structures of the polymer samples. Poly-o-bromoaniline and the poly(aniline-co-o-bromoaniline)s exhibit better solubility characteristics than the unsubstituted polyaniline. The solubility of the dispersed copolymer in the solvent such as propanol increases upon increasing the o-bromoaniline concentration in the copolymer, with resultant lower conductivity data for the copolymers. The electrical conductivity of the copolymers decreases with increasing o-bromoaniline comonomer content up to 25% incorporation of the o-bromoaniline comonomer in the copolymer. Copolymers with higher content of the halogenated monomer show little conductivity, as evidenced by cyclic voltammetry data. Surface profile data of the polymers was studied by AFM analyses which suggest that the copolymers has smooth surfaces while the homopolymers possesses non-uniform surfaces. TEM images of polyaniline and the copolymers at high magnification shows a tubular morphology. The particle size distribution curves for the different polymer samples indicate that the particle sizes of the polymer products vary in the range 5 to 9000 nm with a small fraction of particles sizes in the range of 5–10 nm. For the copolymer, poly(aniline-co-o-bromoaniline)50, a large portion of the sample shows particle sizes in the range of 100–1000 nm, with a small portion of particles sizes in the 8000–9000 nm range.
References
Duong NN, Hyeonseok Y (2016) Polymers 8(118):1–38
Lapkowski M (1990) Synth Met 35:169–182
Cardova R, del Valle M, Arratia A, Gomez H, Schrelber R (1994) J Electoanal Chem 377:75–83
MacDiarmid A, Chiang J, Richter A, Epstein A (1987) Synth Met 18:285–290
Amer I, Mokrani T, Jewell L, Young DA, Vosloo HCM (2016) Tetrahedron Lett 57:426–430
Gabal MA, Hussein MA, Hermas AA (2016) Int J Electrochem Sci 11:4526–4538
Pron A, Rannou P (2002) Prog Polym Sci 27:135–190
Kumar D (2000) Synth Met 114:369–372
Sahin Y, Percin S, Sahin M, Ozkan G (2003) J App Polym Sci 90:2460–2468
Yue J, Epstein AJ, Zhong Z, Gallopher PZ, Macdiarmid AG (1991) Synth Met 41:765–768
Sahin Y, Perkmez K, Yaldiz A (2002) Synth Met 129:107–115
Sahin Y, Perkmez K, Yaldiz A (2002) J App Sci 85:1227–1235
Sahin Y, Perkmez K, Yaldiz A (2002) Synth Met 131:7–14
Palaniappan S (2000) Polym Int 49:659–662
Rajendra V, Prakash S, Gopalan A, Vasudevan T, Chen W-C, Wen T-C (2001) Matter Chem Phys 69:62–71
Mav I, Zigon M (2001) Synth Met 119:145–146
Sharma AL, Saxena V, Annapoorni S, Malhotra BD (2001) J Appl Polym Sci 81:1460–1466
Sahin Y, Pekmez K, Yildiz A (2002) Synth Metal 129:117–121
Sahin Y, Percin S, Sahin GO, Alsanacak J (2003) Appl Polym Sci 89:1652–1658
Waware US, Mohd Rashid, Summers GJ (2014) J Adv Phys 3:1–6
Gupta MC, Umare SS, Huque MM, Visvanath SG (1997) Ind J Chem 36 B:703–710
Umare SS, Huque MM, Gupta MC, Visvanath SG (1996) Macromol Rep A33:381–389
Waware US, Umare SS, Ingole S, Viswanath SG (2005) Int J Polym Anal Charact 10:1–13
Borkar AD, Gupta MC, Umare SS (2001) Polym Plast Technol Eng 40:225–234
Ye S, Do NT, Dao LH, Vijh AK (1997) Synth Met 88:65–72
Waware US, Umare SS (2005) React Funct Polym 65:343–350
Mahudeswaran A, Vivekanandan J, Vijayanand PS, Kojima T, Kato S (2016) Int J Mod Phy B 30:1650008
Sahin Y et al (2003) J Appl Polym Sci 89:1652–1658
Xu L, Guo P, He H, Zhou N, Ma J, Wang G, Zhang C, Su C (2017) Int J Ionic 23:1375–1382
Ghose S, Kalpagam V (1989) Synth Met 33:11–17
Ginder JM, Epstein AJ (1990) Phys Rev B 41:10674
Zho WS, Cromak K, MacDiarmid AG (1991) J Am Chem Soc 113:2665–2671
Acknowledgments
Funding for the current research from Qatar University, and University of South Africa (UNISA) is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Waware, U.S., Summers, G.J., Rashid, M. et al. Electrochemical, morphological, and spectroscopic study of poly(aniline-co-o-bromoaniline) (PA-co-o-BrA) conducting copolymer. Ionics 24, 1701–1708 (2018). https://doi.org/10.1007/s11581-017-2335-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11581-017-2335-z