
Abstract
A new DP AdSV method was developed for the determination of the pesticide clothianidin (Clo) based on its nitro group reduction at the in situ renovated bismuth bulk annular band working electrode (BiABE). Crucial point of the proposed procedure is simple and fast regeneration of the BiABE’s surface in the presence of testing solution, by application of the activation potential (E act = −1.5 V) and next short accumulation potential (E acc = −0.6 V). The voltammetric behaviour of clothianidin has been investigated by cyclic voltammetry (CV). The experimental variables such as; potential and time of activation or accumulation, pH, concentration of the supporting electrolyte, DP mode parameters and influence of possible interferences on the Clo signal response, were tested. In the optimized conditions, the peak current was proportional to the concentration of Clo over the range from 0.2 to 23.4 μmol L−1 (0.050 to 5.84 mg L−1) with R = 0.9996. The calculated value of LOD was 0.047 μmol L−1 (0.012 mg L−1) (at S/N = 3), and sensitivity was 0.094 μA/μmol L−1, for 5 s of the accumulation time. The relative standard deviation for 2 μmol L−1 of Clo was 4.2% (n = 5). The presented results were obtained without any pre-concentration time. Finally, the proposed method was successfully applied for determination of Clo in the spiked tap and river waters samples with the recovery test.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Clothianidin (Clo) is a pesticide developed by Takeda Chemical Industries and Bayer AG. This neonicotinoid insecticide controls insects by acting as an agonist at the nicotinic acetylcholine receptor, affecting the synapses in the insect central nervous system [1]. Nowadays, neonicotinoids are one of the most important categories of insecticides being generally regarded as the most powerful for control of insect pests like aphids, leaf- and plant-hoppers, whiteflies, thrips, some micro-Lepidoptera or some other coleopteran pests [2–4]. According to the Environmental Protection Agency (EPA), clothianidin’s major risk concern is to non-target insects. Information from standard tests and field studies, suggest the potential for long-term toxic risk to honey bees and other beneficial insects [5]. High acute risk was also identified from exposure via residues in nectar and pollen. In April 2013, the European Union voted for a two-year restriction on neonicotinoid insecticides. The ban restricted the use of clothianidin for crops that are inviting to bees such as maize, cotton, sunflower and rapeseed. It is moderately toxic in the short-term to mammals that eat it, and long-term ingestion may result in reproductive and developmental effects may cause slight eye irritation, but it does not damage genetic material. It is evidence that it causes cancer in rats or mice; it is unlikely to be a human carcinogen [6].
The method for the determination of clothianidin recommended by the EPA is based on the high performance liquid chromatography (HPLC) [7–11]. However, all of these techniques have some advantages and drawbacks, generally intrinsically related to the sample preparation, which can enhance the selectivity and sensitivity. Thus, additional procedures before measurement based on co-precipitation, solvent extraction and solid-phase extraction are most commonly used. Owing to this, the electroanalytical methods are increasingly used in the clothianidin analysis using the modified and unmodified electrodes.
The hanging mercury drop electrode had been reported as the most sensitive electrode for CLO detection [12, 13]. However, the use of mercury should be avoided because of its toxicity. The problem of the limitation of mercury from the analytical procedure is partially solved by the use of cyclic renewable mercury film silver-based electrode (Hg(Ag)FE) [14, 15]. Great efforts have been taken to develop mercury-free solid electrodes based on the bismuth or carbon materials. One of the promising alternative to mercury electrodes in clothianidin analysis is the bismuth film electrode [16] and the tricresyl-phosphate-based carbon paste electrode [17]. The bismuth film electrode was also used in determination of other neonicotinoid insecticides such as imidacloprid, acetamiprid and thiamethoxam [18, 19].
To the best of our knowledge, only a few reports have concerned application of the bismuth bulk electrodes (BiBEs) in voltammetry. This type of electrodes offers several advantages relative to the bismuth film electrodes. For example, minimization of the amount of generated waste (solution containing bismuth salt is not needed), ease of use and fabrication, cost-efficiency and stability. Until now, the application of BiBEs in trace analysis was mainly focused on the determination of metals (Cd, Pb, Zn, Ni, Co, Pd and Tl) [20–23]. Only several organic compounds have been yet analysed using the BiBE [24–26].
In this paper, the in situ renovated bismuth bulk annular band working electrode (BiABE) for DP AdSV clothianidin determination is presented. The developed method is based on DPV reduction of nitro group at −0.92 V. The main advantage of this electrode is easy regeneration of its surface by electrochemical activation in the presence of the tested solution (E act = −1.5 V, t act = 5 s) and next short accumulation of Clo (E acc = −0.6 V, t acc = 5 s). Moreover, the measurements at the BiABE can be carried out without removing oxygen from the solution. The optimal conditions for the determination of clothianidin in a Britton-Robinson buffer (pH 9.0) at the BiABE were established. The LOD obtained at the BiABE is lower with those achieved for Hg(Ag)FE [15] and BiFE [16]. The recovery of the method was evaluated using natural water samples from the laboratory tap and Rudawa river spiked with clothianidin. Potential interferences from coexisting ions and surface-active substances were determined using commercially available Triton X-100.
Experimental
Apparatus
All voltammetric measurements were performed using the M20 multipurpose electrochemical analyser equipped with the M164 electrode stand (both mtm-anko, Kraków and Poland). Data acquisition and experimental control were processed with the EALab 2.1 software. A quartz cell of 10 mL volume was used with a conventional three-electrode system. The in situ renovated bismuth bulk annular band electrode (BiABE) served as the working electrode (Fig. 1a) and prepared according to the detailed description presented in our previous work [21]. A platinum wire and Ag/AgCl/3 mol L−1 KCl double junction electrode were used as auxiliary and reference electrodes, respectively. A magnetic stirrer (ca. 300 rpm) was used during the activation and accumulation steps. The measurement of pH values during the experiment was performed by means of CPI-505 laboratory pH/ion meter made by ELMETRON, Poland.

Voltammetric measurement system with bismuth bulk annular band electrode a and chemical structure of clothianidin b
Reagents and solutions
All chemicals were of analytical reagent grade. The fresh standard stock solution of 1 mmol L−1 clothianidin (Dr. Ehrenstorfer, Germany) (Fig. 1b) was obtained by dissolution of an appropriate amount of this reagent in 25.0 mL of water-acetone mixture (4:1, v/v) and then was stored in the fridge. Acetone for analysis was purchased from Merck. The 0.04 mol L−1 Britton-Robinson buffer solution (B-R) was prepared by mixing 0.04 mol L−1 phosphoric acid (99.99%, Sigma-Aldrich), 0.04 mol L−1 acetic acid (99.5%, POCh) and 0.04 mol L−1 boric acid (99.97%, Sigma-Aldrich). The required pH value in the range from 5.0 to 11.0 was adjusted by 0.20 mol L−1 sodium hydroxide (99.99%, Sigma-Aldrich). Triton X-100 purchased from Fluka was used. All aqueous solutions were prepared by using quadruple distilled water (two last stages from quartz).
Sample preparation
The presented method was checked by determination of Clo in the spiked tap and Rudawa river water samples. Water collected from the tap at the laboratory originated from the Dobczyce water reservoir. Rudawa river water samples were collected into clean polypropylene bottles from outskirts of Kraków and pre-treatment procedure consisted of successive filtration through a filter paper, Whatman® No. 1 (Sigma-Aldrich) and a 0.45-μm cellulose acetate membrane filter in the Sartorius device. Recovery tests were carried out for the tap and river water samples spiked with standard solutions in amount corresponding to the following concentrations of Clo: 5, 10 and 20 μmol L−1. Clothianidin was added into the water sample about 1 h before filtering.
Measurement procedure
The electroanalytical behaviour of Clo at the BiABE was studied using cyclic voltammetry (CV). The optimization of electrochemical parameters and quantitative determination of Clo were performed using the differential pulse adsorptive stripping voltammetry (DP AdSV) and the standard addition method. Before each series of measurements, the BiABE was polished using 0.05-μm alumina slurries on a Buehler polishing pad and was rinsed with distilled water. Then, 1 mL of 0.04 mol L−1 B-R buffer (pH 9.0), 0.5 mL analysed sample and 3.5 mL distilled water were pipetted into the electrochemical cell to the volume of 5 mL.
Next, determination of Clo procedure was performed in the following steps: (1) short electrochemical activation of the BiABE surface by applying of the activation potential E act = −1.5 V with activation time t act = 5 s, (2) short pre-concentration of Clo on the BiABE at accumulation potential E acc = −0.6 V with accumulation time t acc = 5 s and (3) the rest period of 5 s, the DP voltammograms were recorded in the cathodic direction from −0.6 to −1.2 V. All steps were performed in the same voltammetric cell containing the tested solution. For each measurement, three repetitive scans were recorded. The experiments were carried out for non-deaerated solutions at room temperature.
Results and discussion
Electrochemical behaviour of Clo at the BiABE
The electrochemical behaviour of Clo was studied by cyclic voltammetry in 8 mmol L−1 B-R buffer solution (pH 9.0). The cyclic voltammograms obtained for 20 μmol L−1 Clo using the BiABE were recorded in the range from −0.6 to −1.35 V. As shown in Fig. 2a, one reduction peak is observed at about −0.94 V (solid line), and no peak in the reverse direction was observed (dashed line), indicating that the reduction of Clo at the BiABE electrode is an irreversible process which is in agreement with the literature data using different electrodes such as hanging mercury drop electrode (HMDE) at −0.97 V [12], renewable silver-amalgam film electrode (Hg(Ag)FE) at −0.60 V [15] and bismuth film electrode (BiFE) at −0.95 V [16].

a Cyclic voltammograms obtained for 20 μmol L−1 Clo in 8 mmol L−1 B-R buffer at pH 9.0 at the BiABE for the scan rate of 0.0125, 0.025, 0.050, 0.100, 0.200 and 0.250 V/s−1. b Dependence of the peak current on square root of the scan rate and c log of the peak current on log of the scan rate
The effect of potential scan rate on the voltammetric response Clo reduction on the BiABE was investigated between 0.0125 and 0.250 V s−1. The cathodic peak current (I p) varied linearly with square root of the applied scan rate (v 1/2). The correlation with its equation is shown in Fig. 2b. The linear plot of logarithm of peak current (log I p) versus the logarithm of the scan rate (log v) presented a slope of 0.52 (Fig. 2c); besides this, the obtained value suggests that Clo reduction follows a diffusion-controlled mechanism, and it is in agreement with that reported in the literature [27]. The relationships between the peak current and scan rate for HMDE indicate that the electrode process is controlled by both diffusion and adsorption [12]. In the case of Hg(Ag)FE electrode, reduction of Clo is controlled by adsorption [15].
Activation conditions of the BiABE and effect of accumulation parameters
The bismuth bulk electrode (BiBE) requires renovation of its surface before each measurement, especially in alkaline solutions to ensure reliable results. In particular, purification of the adsorbed surface-active compounds and products of electrochemical reactions is necessary. Moreover, Bi(III) ions are very susceptible to hydrolysis in neutral and alkaline solutions. In highly alkaline media, it has been shown that Bi(III), instead of hydrolysing, forms stable complexes with OH− ions (Bi(OH)2+) that are soluble in aqueous media and can undergo electrochemical reduction on the electrode surface [28]. Hence, for Clo determination, it is recommended to prepare the bismuth film electrode from a plating solution containing 0.02 mol L−1 Bi(NO3)3, 1 mol L−1 HCl and 0.5 mol L−1 KBr in a separate cell (ex situ method) [16]. In addition to these forms, also bismuth oxides such as Bi2O3, especially by contact with atmospheric air can be formed. Above the potential of −1.0 V, Bi2O3 undergo a reduction to metallic bismuth and is deposited on the surface electrode [28]. Another way to have effective pre-treatment of the electrode surface is thorough washing with 0.1 M HCl and later in the buffer supporting electrolyte the electrochemical activation between 15 and 30 cycles from −0.4 to −1.6 V [29]. The studies presented in this work showed that it is possible to determine Clo with high precision and good accuracy after activation of the bismuth bulk electrode carried out by in situ method, i.e. directly in the tested solution. To obtain the most favourable conditions for determination of Clo by means of DPV, the influence of activation potential as well as activation time were investigated.
Effect of the activation potential (E act) on the peak current Clo and the baseline level were tested in the range from −1.2 to −2.0 V vs. Ag/AgCl/3 mol L−1 KCl for 20 μmol L−1 of Clo with accumulation potential of −0.6 V. The voltammograms shown in Fig. 3a were recorded without activation potential (dotted line) and after applying the activation potential of −1.5 V (solid line). The results showed that application of the more negative activation potential than −1.2 V causes a fast reduction of bismuth compounds (observed at potential −0.65 V) and effective cleaning of the electrode surface with products of electrode reactions. Initially, the signals slightly increased to −1.6 V (about 20%) and then remained at constant level. Above −1.5 V significant distortions of the voltammograms were observed. Hence, for further experiments the activation potential of −1.5 V was used, which ensures excellent repeatability of the recorded signals and lack of additional peaks of bismuth compounds.

a DP AdSV voltammograms recorded without activation potential (dotted lines) and with activation potential (E act = −1.5 V) (solid lines). b DP AdSV voltammograms obtained without accumulation potential (dashed lines) and after application of accumulation potential (E acc = −0.6 V) (solid lines). For both, concentration of Clo 20 μmol L−1. Other conditions as in Table 1
The effect of the activation time (t act) on Clo peak height was studied in the range from 2 to 15 s. In the whole tested range, the peak current was stable, but under the 5 s registered curves were characterized by worse precision, hence the activation time of 5 s was selected for subsequent experiments.
The influence of accumulation potential (E acc) on the peak current Clo and the baseline level were tested in the range from −0.5 to −0.75 V vs. Ag/AgCl/3 mol L−1 KCl for the same concentration of Clo with activation potential of −1.5 V. The change of accumulation potential had no effect on the peak current. The voltammograms shown in Fig. 3b were recorded without accumulation potential (dashed line) and after applying the accumulation potential of −0.6 V (solid line). As can be seen, the Clo response is about 50% lower for any accumulation potential and presented signals exhibit less repeatability. For further experiments, the accumulation potential of −0.6 V was used.
The effect of the accumulation time (t acc) on Clo peak height was studied in the range from 2 to 30 s. Contrary to expectations, in over the tested range, the peak current remained at a constant level. Based on this results and previous studies for character of electrode process, it can be concluded that clothianidin is partially adsorbed on the electrode surface. For subsequent experiments, the activation time of 5 s was selected.
Effect of pH and Britton-Robinson buffer concentration
The electrochemical behaviour of Clo has been investigated in different buffer solution; acetate (pH 3.5–5.0), phosphate (pH 5.7–8.0), borate (pH 7.0–9.0), Britton-Robinson (pH 5.0–11.0) and ammonium (pH 8.0–11.0). The performed studies and available literature data [12, 15, 16] show that the highest sensitivity and reproducibility of results were obtained in Britton-Robinson buffer (pH 8.1–9.0).
The stability of Clo, its peak current and the peak potentials are strongly dependent upon the pH value of the tested solution. Moreover, the active potential window of the BiABE and the formation of solid bismuth hydroxyl compounds depend too. The effect of solution pH on the reduction of Clo at the BiABE was investigated in a 8 mmol L−1 B-R buffer solution over the pH range from 5.0 to 11.0 (Fig. 4a). It was found that at pH 9.0, the well-shaped, most symmetrical and intense peak of Clo (20 μmol L−1) was obtained. Hence, this pH value was chosen as the optimal for the DP AdSV determination of Clo. Also, the peak potential shifted in negative direction and was linear with increasing pH in the tested range (Fig. 4b). The dependence between Clo peak potential vs. pH value is linear in accordance with equation: Ep = −0.045pH − 0.499 (R = 0.996). The slope of 45.2 mV pH −1 is incompatible as expected for the Nernstian theoretical value of 59.1 mV pH −1 indicating that one electron and one proton are involved in the electrode reaction, which is characteristic for Clo reduction process as it is shown in literature data. Such mechanism is described in literature for Clo at the HMDE as follows: peak I (−0.97 V) relates to 4-electron reduction of NO2 group to NHOH and peak II (−1.34 V) corresponds to further 2-electron reduction of NHOH group to an appropriate NH2 group [12]. Earlier, similar mechanism was well-described in literature in connection with electrode reactions of nitro compounds [30]. In the case of bismuth film electrode [16] and bismuth bulk electrode, the potential window and residual current depend on the amount of electrodeposited bismuth and pH value of the electrolyte. Hence, in this negative potential region for both electrodes only one peak was observed. Such behaviour can be explained by significant role of protons in the complex reduction mechanism, in which the first step involves the reduction of the nitro group to hydroxylamine, observed for BiABE at potential −0.9 V.

a DP AdSV voltammograms of 20 μmol L−1 Clo recorded for the different pH of B-R buffer; (a) 5.0, (b) 6.0, (c) 7.0, (d) 8.0, (e) 9.0, (f) 10.0 and (g) 11.0. b Dependence of the peak current and its potential on pH. c Influence of B-R buffer concentration on the peak current in the range from 1 to 15 mmol L−1. Other conditions as in Table 1. The error bars represent the standard deviation (n = 3)
The influence of the B-R buffer (pH 9.0) concentration on peak current in the solution containing 20 μmol L−1 of Clo was studied in the range from 1 to 15 mmol L−1. As can be seen in Fig. 4c, increasing concentration of the B-R up to 5 mol L−1 caused a sudden rise in peak current. In the range from 5 to 10 mmol L−1, the registered signals were on constant level, at higher concentration than 10 mmol L−1, the negligible decreases of peak current were observed. The highest sensitivity, well-shaped peaks and high ratio of peaks current to the background level were achieved for 8 mmol L−1 of B-R buffer, so this concentration was selected for subsequent experiments.
Influence of DPV parameters on Clo response
The differential pulse voltammetry as the highly sensitive measurement technique was used for the determination of Clo. To optimize the conditions of the DP AdSV determination of Clo, the following instrumental parameters were tested: step potential (E s) in the range 1–7 mV, pulse amplitude (dE) from 10 to 50 mV (both positive and negative modes) and pulse width (t imp) as the sum of waiting time (t w) and current sampling time (t s), both in the range 5–25 ms (t w = t p). Optimal values which were applied in all studies are summarized in Table 1. The step potential of 3 mV, pulse amplitude of 50 mV and pulse width of 40 ms were selected as the best values, which provide good reproducibility, the highest peak current and signal-to-background current ratio.
Analytical performance and stability of the BiABE
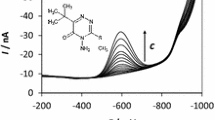
The series of DP AdSV voltammograms for increasing Clo concentration with the corresponding calibration curve were recorded without any pre-concentration time (Fig. 5). The presented voltammograms were obtained after automatic background subtraction. The sensitivity of DP AdSV determination of Clo was 0.094 μA/μmol L−1. The calibration plot was linear from 0.2 to 23.4 μmol L−1 (0.050 to 5.84 mg L−1) and obeyed the equation y = (0.094 ± 0.001)c + (0.011 ± 0.002), where y and c represent peak current (μA) and Clo concentration (μmol L−1), respectively. The correlation coefficient R was 0.9996. LOD was calculated 3 times the standard deviation of the intercept over the slope of the calibration curve and amounts to 0.047 μmol L−1 (0.012 mg L−1) for 5 s of the accumulation time. Obtained results was compared with the results achieved for other electrodes and summarized in Table 2. The estimated LOD is 60 times lower than the value obtained for the BiFE and a few times lower than value obtained for the Hg(Ag)FE. Only for HMDE the lower values of LOD were achieved.

DP AdSV voltammograms recorded at the BiABE in the supporting electrolyte containing increasing concentrations of Clo. Concentration of Clo from bottom to top: blank, 0.2, 0.4, 0.8, 1.2, 1.6, 2.0, 3.0, 4.0, 6.0, 8.0, 11.8, 15.7, 19.6 and 23.4 μmol L−1. Inset: the corresponding calibration plot. Other parameters as in Table 1
In order to determine stability of the BiABE, the signals obtained for six different samples with the tested solution containing 2 μmol L−1 of Clo were compared; the calculated RSD was equal to 4.2%. For longer periods of time, when measurements were performed for about 1–2 h per day, fluctuations of ca. 8% in signal sensitivity were noted. The performed tests show very good short-term repeatability. Storage of the BiABE under oxygen, easy access and use of alkaline electrolyte causes the formation of oxides and hydroxides on the surface. Hence, the BiABE required mechanical polishing (0.05 μm Al2O3) before and after each long measurement cycle.
Tolerance to interfering species
The effects of co-existing ionic species and surface-active substances on the stripping peak current of 2 μmol L−1 Clo (0.5 mg L−1) were investigated. A number of metal ions that could potentially interfere was examined: Zn(II), Pb(II), Sn(II), Fe(III), Cu(II), Mg(II), Ni(II), Co(II), Mn(II) and Cr(VI). During the experiments, the mass concentration ratio of potential interfering species and Clo was 1:1. In this case, some interferences were observed only in the presence of Cd(II). The peak potential of Cd(II) (−0.80 V) is very close to the Clo peak (−0.92 V). It was found that the addition of 1.0 and 3.0 mg L−1 of Cd(II) causes decrease of the Clo signal from 80 to 30% of its original value, respectively. However, cadmium concentrations in the environmental samples are many times lower than used in this experiment. It is worth to notice that for Pb(II) and Zn(II), the well-shaped peaks were observed at −0.75 and −1.2 V, respectively.
Voltammetric methods are especially susceptible to organic matter and presence of different kinds of surfactants inevitably present in natural samples. To study the influence of surface-active substances on Clo voltammetric response, the non-ionic surfactant Triton X-100 was used. The effect of Triton X-100 concentration was examined in the range from 0.25 to 5 mg L−1 for solution containing 2 μmol L−1 of Clo. The addition of Triton X-100 at concentration 0.25, 0.50 and 1.0 mg L−1 causes decrease of the stripping peak current up to 25, 40 and 85%, respectively. Addition of 1.5 mg L−1 of Triton X-100 causes complete lack of the signal.
Analytical application of the BiABE
The accuracy of the method was assessed by determination of Clo in the spiked tap and river water samples at various concentrations. The samples were prepared according to procedure described in Section Sample preparation. For each sample, three experiments were performed. Voltammograms obtained in the course of Clo determination in Rudawa water spiked sample (20 μmol L−1) are presented in Fig. 6. Four analytical scans were recorded for each sample as well as after addition of standard solution. The concentrations of Clo in all water samples were calculated by means of four standard additions method. No interferences were observed from the river water components. The obtained results are presented in Table 3. In every measured conditions, recovery was ranged between 95 and 97% which proves good accuracy of the developed method. Thus, analytical usefulness of the clothianidin determination in the examined samples using BiABE was confirmed.

DP AdSV voltammograms recorded for determination of Clo in dotted Rudawa river water sample using the BiABE in the supporting electrolyte without deaeration. Curves: blank, (a) water sample diluted (1:10), (b) 1, (c) 2, (d) 3 and (e) 4 μmol L−1 of Clo. Other parameters as in Table 1. The inset figure illustrates the corresponding standard addition plot
Conclusions
The presented work is the first to report using the in situ renovated bismuth bulk annular band electrode (BiABE) for clothianidin determination. The proposed DP AdSV method of clothianidin determination in the range of 0.2–23.4 μmol L−1 based on recorded signal of nitro group reduction was developed. As it has been tested, the electrode process is controlled mainly by diffusion of the analyte to the electrode surface, but its partial adsorption is also possible. The main BiABE benefits are ease of cleaning and fast regeneration of the electrode surface in the presence of the alkaline solution by applying activation potential before each measurement, moreover, it has low sensitivity to the presence of oxygen. The presented method shows better sensitivity and lower detection limit comparing to the bismuth film electrode and the renewable silver-amalgam film electrode for Clo detection system. The proposed procedure was also applied for the determination of clothianidin in the spiked real water samples. The analysis was performed with good recovery results. In our opinion, the BiABE with the proposed regeneration method offers increased scope for more widespread use in analysis of nitro compounds.
References
World Health Organization, Food and Agriculture Organization of the United Nations (2011) Pesticide residues in food 2010 – Joint FAO/WHO Meeting on Pesticide Residues, Rome. http://www.fao.org/docrep/013/i1949e/i1949e00.htm. Accessed 27 Feb 2017
Jeschke P, Nauen R, Schindler M, Elbert A (2011) Overview of the status and global strategy for neonicotinoids. J Agric Food Chem 59:2897–2908
Jeschke P, Nauen R (2008) Neonicotinoids-from zero to hero in insecticide chemistry. Pest Manag Sci 64:1084–1098
Elbert A, Haas M, Springer B, Thielert W, Nauen R (2008) Applied aspects of neonicotinoid uses in crop protection. Pest Manag Sci 64:1099–1105
Johnson R (2010) Honey Bee Colony Collapse Disorder, Congressional Research Service Report for Congress. https://fas.org/sgp/crs/misc/RL33938.pdf. Accessed 27 Feb 2017
Australian Pesticides and Veterinary Medicines Authority (2011) Australian evaluation of the new active clothianidin, Canberra. http://apvma.gov.au/. Accessed 27 Feb 2017
Tapparo A, Giorio C, Soldà L, Bogialli S, Marton D, Marzaro M, Girolami V (2013) UHPLC-DAD method for the determination of neonicotinoid insecticides in single bees and its relevance in honeybee colony loss investigations. Anal Bioanal Chem 405:1007–1014
Zhang Y, Xu J, Dong F, Liu X, Li X, Li Y, Wu X, Liang X, Zheng Y (2013) Simultaneous determination of four neonicotinoid insecticides residues in cereals, vegetables and fruits using ultra-performance liquid chromatography/tandem mass spectrometry. Anal Methods 5:1449–1455
Tapparo A, Giorio C, Marzaro M, Marton D, Solda L, Girolami V (2011) Rapid analysis of neonicotinoid insecticides in guttation drops of corn seedlings obtained from coated seeds. J Environ Monit 13:1564–1568
Kamel A (2010) Refined methodology for the determination of neonicotinoid pesticides and their metabolites in honey bees and bee products by liquid chromatography-tandem mass spectrometry (LC-MS/MS). J Agric Food Chem 58:5926–5931
Liu S, Zheng Z, Wei F, Ren Y, Gui W, Wu H, Zhu G (2010) Simultaneous determination of seven neonicotinoid pesticide residues in food by ultraperformance liquid chromatography tandem mass spectrometry. J Agric Food Chem 58:3271–3278
Guziejewski D, Skrzypek S, Łuczak A, Ciesielski W (2011) Cathodic stripping voltammetry of clothianidin: application to environmental studies. Collect Czechoslov Chem Commun 76:131–142
Guziejewski D, Skrzypek S, Ciesielski W (2012) Application of catalytic hydrogen evolution in the presence of neonicotinoid insecticide clothianidin. Food Anal Methods 5:373–380
Brycht M, Vajdle O, Zbiljić J, Papp Z, Guzsvány V, Skrzypek S (2012) Renewable silver-amalgam film electrode for direct cathodic SWV determination of clothianidin, nitenpyram and thiacloprid neonicotinoid insecticides reducible in a fairly negative potential range. Int J Electrochem Sci 7:10652–10665
Brycht M, Skrzypek S, Guzsvány V, Berenji J (2013) Conditioning of renewable silver amalgam film electrode for the characterization of clothianidin and its determination in selected samples by adsorptive square-wave voltammetry. Talanta 117:242–249
Guzsvány V, Papp Z, Zbiljić J, Vajdle O, Rodić M (2011) Bismuth modified carbon-based electrodes for the determination of selected neonicotinoid insecticides. Molecules 16:4451–4466
Pappl Z, Guzsvány V, Švancara I, Vytřas K (2011) Voltammetric monitoring of photodegradation of clothianidin, nitenpyram and imidacloprid insecticides using a tricresyl phosphate-based carbon paste electrode. Int J Electrochem Sci 6:5161–5171
Guzsvány V, Kádár M, Papp Z, Bjelica L, Gaál F, Tóth K (2008) Monitoring of photocatalytic degradation of selected neonicotinoid insecticides by cathodic voltammetry with a bismuth film electrode. Electroanalysis 20:291–300
Guzsvány V, Kádár M, Gaál F, Bjelica L, Tóth K (2006) Bismuth film electrode for the cathodic electrochemical determination of thiamethoxam. Electroanalysis 18:1363–1371
Armstrong KC, Tatum CE, Dansby-Sparks RN, Chambers JQ, Xue ZL (2010) Individual and simultaneous determination of lead, cadmium, and zinc by anodic stripping voltammetry at a bismuth bulk electrode. Talanta 82:675–680
Baś B, Węgiel K, Jedlińska K (2015a) The renewable bismuth bulk annular band working electrode: Fabrication and application in the adsorptive stripping voltammetric determination of nickel(II) and cobalt(II). Anal Chim Acta 881:44–53
Baś B, Węgiel K, Jedlińska K (2015b) New voltammetric sensor based on the renewable bismuth bulk annular band electrode and its application for the determination of palladium(II). Electrochim Acta 178:665–672
Węgiel K, Jedlińska K, Baś B (2016) Application of bismuth bulk annular band electrode for determination of ultratrace concentrations of thallium(I) using stripping voltammetry. J Hazard Mater 310:199–206
Tall O, Beh D, Jaffrezic-Renault N, Vittori O (2010) Electroanalysis of some nitro-compounds using bulk bismuth electrode. Int J Environ Analyt Chem 90:40–48
Bučková M, Gründler P, Flechsig GU (2005) Adsorptive stripping voltammetric detection of daunomycin at a bismuth bulk electrode. Electroanalysis 17:440–444
Prchal V, Ottenschlagerova A, Vyskocil V, Barek J (2016) Voltammetric determination of 5-nitroindazole using a bismuth bulk electrode. Anal Lett 49:49–55
Grosser DK (1994) Cyclic voltammetry simulation and analysis of reaction mechanisms. VCH Publisher, New York
Economou A (2005) Bismuth-film electrodes: recent developments and potentialities for electroanalysis. Trends Anal Chem 24:334–340
Gaál F, Guzsvány V, Bjelica L (2007) Determination of various insecticides and pharmaceuticals using differently modified glassy carbon electrode. J Serb Chem Soc 72:1465–1475
Squella JA, Bollo S, Nunez-Vergara LJ (2005) Recent developments in the electrochemistry of some nitro compounds of biological significance. Curr Org Chem 9:565–581
Acknowledgements
This work was supported by the Polish National Science Centre (Project No. 2015/19/B/ST5/01380).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Węgiel, K., Baś, B. Voltammetric characteristics and determination of clothianidin using a bismuth bulk annular band electrode regenerated in situ. Ionics 23, 3187–3195 (2017). https://doi.org/10.1007/s11581-017-2105-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11581-017-2105-y