
Abstract
Morels (Morchella spp.) are highly prized for their culinary qualities and intensively collected worldwide by mycophiles. Morels are divided into three clades by phylogenetic analyses: black morels, yellow morels and the rufobrunnea clade. Morchella importuna, Mel-20 and M. sextelata are included in the black morel clade and are widely distributed in Yunnan province, China. M. importuna and M. sextelata have been artificially cultured in recent years, but their life cycles and reproductive systems are still poorly understood, which delays the progress of morel cultivation. In this study, the genomes of two ascospore isolates of M. importuna with opposite mating-type were sequenced and two idiomorphs, MAT1–1 and MAT1–2, were identified. The MAT1–2 idiomorph was 6.7 kb in length containing a single MAT1–2-1 gene, and the MAT1–1 idiomorph was 10.5 kb containing a MAT1–1-1 gene and two other open reading frames (ORFs), GME3123 and GME3124. These ORFs differed greatly from the homologues of previously published mating-type genes; therefore, we speculate that they are novel mating genes found only in morels. Single-ascospore populations of M. importuna, M. sextelata and Mel-20 were analysed, and the result indicated that the ratios of MAT1–1- and MAT1–2-harbouring idiomorphs were not significantly different from a 1:1 ratio. The results suggest that these three black morels are heterothallic.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
As a group of edible mushrooms with excellent flavour and high medicinal value (Fu et al. 2013; Su et al. 2013; Hu et al. 2013), morels (Pezizomycetes, Ascomycota) are found in most parts of the world and are extensively traded (Pilz et al. 2007; Pildain et al. 2014). In recent years, morels have been commercially cultivated to meet the ever-growing need in China.
Phylogenetic analyses indicate that Morchella consists of three clades: the Elata Clade (black morels), the Esculenta Clade (yellow morels) and the rufobrunnea Clade (O’Donnell et al. 2011). Morchella importuna, Mel-20 and M. sextelata are included in the Elata Clade (Du et al. 2014; Richard et al. 2015). In a previous study, some black morel species were confirmed to form secondary ectomycorrhizal symbioses with spruce (Buscot 1994), while M. importuna and M. sextelata are considered to be saprophytic fungi that can be cultivated artificially (Peng et al. 2016).
Although several species of Morchella have been successfully cultivated (Ower et al. 1986; Miller 2005; Masaphy 2010; Peng et al. 2016), their genetic information, life cycles and reproductive systems remain poorly understood. Based on SNP, SCAR, and AFLP markers, Pagliaccia et al. (2011) consider members of the Morchella sp. Mel-12 phylogenetic lineage to be heterothallic and to outcross in nature. Multiple nuclei are present in a single ascospore or one hyphal segment of Morchella (Hervey et al. 1978; Volk and Leonard 1990). However, whether the ascospores are heterokaryotic or homokaryotic remains unclear.
The sexual reproduction of Ascomycota fungi is controlled by a single mating-type locus (MAT-1) (Coppin et al. 1997) with two highly divergent nonhomologous idiomorphs (Metzenberg and Glass, 1990), designated MAT1–1 and MAT1–2 (Turgeon and Yoder 2000). The MAT1–1 idiomorph is characterized by the MAT1–1-1 gene, which encodes protein with an alpha-box domain, while the gene MAT1–2-1, which encodes a high-mobility group (HMG) domain, is generally located on the MAT1–2 idiomorph (Debuchy and Turgeon 2006). Individual isolates usually contain either the MAT1–1 or MAT1–2 idiomorph in heterothallic species. In contrast, in homothallic species, both mating types are contained in the haploid genome, usually tightly linked, although exceptions exist (Nelson 1996; Debuchy and Turgeon 2006). In Pezizomycetes, edible mushrooms that are as highly praised as morels, Tuber melanosporum and T. indicum, have been identified as heterothallic (Rubini et al. 2011; Belfoori et al. 2013).
In our previous studies, a draft genome database of the Mes-15 strain YAAS2689 included in the Esculenta Clade was obtained (data unpublished), and the sequences of the genes MAT1–1-1 (KP776983) and MAT1–2-1 (KP776984) were verified. In this study, the genomes of two single ascospore isolates from M. importuna (YPL6) with opposite mating-type were sequenced. The aim was (1) to identify the MAT locus structure of M. importuna, (2) to clone and characterise the MAT1–1-1 and MAT1–2-1 mating-type genes of M. importuna, Mel-20 and M. sextelata, and (3) to analyse whether these three black morel species are heterothallic or homothallic.
Materials and methods
Strains and isolations of ascospores
The fruiting body WXLBD7 of Mel-20 was collected from the forests of Weixi (Yunnan province). YPL2 and YPL6 of M. importuna and HL1 of M. sextelata were obtained from morel cultivation fields in Chuxiong (Yunnan province) and Huili (Sichuan province), respectively (Table 1). The corresponding strains were obtained by tissue isolation. Single-ascospore strains were isolated from the fruiting bodies after spore dilution. Every ascospore isolate was numbered with an Arabic numeral suffix corresponding to the ascocarp. For example, YPL6–3 refers to the 3rd ascospore isolate from ascocarp YPL6. All the cultures were identified by phlogenetic analyses (Table S1; Figs. S1, S2) and deposited in Mushroom Center of Yunnan Crops Genebank (in Yunnan Academy of Agricultural Sciences, YAAS), Kunming, China.
DNA extraction and PCR amplification
Each isolate was incubated on PDA at 23 °C for 7 days. Mycelia were scraped and ground in liquid nitrogen. DNA extraction was performed using an EZgene™ Fungal gDNA Kit (BIOMIGA Inc.). All primer pairs were designed by Primer Premier v.5 and synthesized by Beijing Tsingke Biotechnology. The PCR amplifications were performed in a 25-μL mixture containing 12.5 μL of PCR Mix (TSINGKE), 1 μL of primer (10 μM/L) and 10 ng of DNA. Long-range PCR amplification was performed using LA Taq DNA polymerase (TaKaRa Biotechnology (Dalian)). The primer sequences and main PCR cycling parameters are listed in Table 2.
Genome sequencing
According to the conserved amino acid sequences of the MAT1–2-1 gene HMG domain of ascomycetes, primer pair p1 was designed to amplify the corresponding gene fragments from the single-ascospore isolate group which contained 105 isolates of M. importuna YPL6. The isolates that contained the predicted amplification bands were considered to contain the MAT1–2 idiomorph, while the isolates that lacked detectable bands were hypothesised to contain the MAT1–1 idiomorph.
Genomic DNA from the isolates YPL6–1 with the MAT1–2 idiomorph and YPL6–3 with the MAT1–1 idiomorph were submitted to the Beijing Genomics Institute (BGI). Genomic libraries were constructed and paired-End sequences were produced using Illumina HiSeq™ 2000. The clean reads were assembled into contigs by the SOAPdenovo assembly programme (http://soap.genomics.org.cn/soapdenovo.html), and the scaffolds were built by analysing the information of paired reads that covered different contigs. Genes were predicted using GeneMark-ES (v.2.3e)software and corresponding gene annotations were conducted by BLASTx analysis with the KEGG, KOG, SwissProt, NR and GO databases.
Identification and verification of the MAT1–1 and MAT1–2 idiomorphs of M. importuna
The MAT1–2 and MAT1–1 idiomorphs were identified by alignment analyses of two scaffolds containing MAT1–2-1 or MAT1–1-1. Ten PCR primer pairs (p2–1, p2–2, p2–3, p2–4, p2–5, p3–1, p3–2, p3–3, p3–4, and p3–5) (Table 2) were employed to verify the two idiomorphs based on their genome sequences. PCR was performed under standard conditions (Table 2). The products were recovered from agarose gels and purified using a Gel/PCR Extraction Kit (BIOMIGA), then cloned into the pGM-T vector and sequenced. Sequence comparison and dotplot analysis were conducted to verify the two idiomorphs of M. importuna.
Isolation of MAT1–2-1 and MAT1–1-1 from Mel-20 and M. sextelata
Conserved sequences could be found by analysis of the flanking regions of MAT1–2-1 gene in M. importuna (KY782629) and Mes-15 (KY782632). A pair of specific primers (p2–3f and p2–3r) were designed based on the conserved regions to amplify the complete sequences of the MAT1–2-1 gene in Mel-20 and M. sextelata. The PCR amplicons (approximately 5 kb) were cloned and sequenced.
Because no conserved portions could be located in the 10-kb flanking regions from the upstream to the downstream region of the MAT1–1-1 gene in M. importuna (KY782630) and Mes-15 (KY782631), a primer pair (p7f and p7r) was designed to sequences within the MAT1–1-1 gene to amplify the corresponding DNA fragments. To obtain the complete sequence of the MAT1–1-1 gene, the DNA walking method was performed using the Genome Walking Kit (TaKaRa Biotechnology (Dalian)) with SP Primers (p9-1f, p9-2f, p9-3f, p9-1r, p9-2r, and p9-3r) based on MAT1–1-1 fragments of Mel-20 and M. sextelata.
Verification of the coding regions of MAT1–1-1 and MAT1–2-1 genes
Total RNA was extracted from the mycelia of YPL6, YPL6–1, YPL6–3, HL1, HL1–1, HL1–47, WXLBD7, WXLBD7–11 and WXLBD7–23 using a TaKaRa MiniBEST Plant RNA Extraction Kit. The first-strand cDNAs were synthesized using Oligo dT primer (TaKaRa PrimeScript™ II 1st Strand cDNA Synthesis Kit). The primer pairs p8 for the MAT1–1-1 gene and p10 for the MAT1–2-1 gene were designed using predicted sequences adjacent to the start site and end site of the two genes. The design strategy also took advantage of conserved regions, so the primers could be used in all three morel species. Introns were verified manually by comparing the RT-PCR sequences with the corresponding DNA sequences.
Mating type ratios of ascospores from the same ascocarp
Single-ascospore populations of WXLBD7, YPL2, YPL6 and HL1 were obtained to determine the occurrence and frequency of the mating type idiomorphs. Standard PCR tests with p8 and p10 primer pairs were conducted to examine the four single-ascospore populations. A chi-square test was applied to compare the MAT1–1/MAT1–2 ratio at the significance level P = 0.05.
Results
MAT loci structures of M. importuna
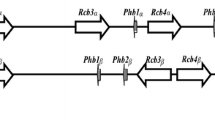
The results of genome assembly and gene annotation of isolations YPL6–1 and YPL6–3 showed that the genome sizes and components of two isolations are similar (Tables S2, S3). The MAT1–1-1 gene was observed only on Scaffold 152 (KY782630) of the M. importuna isolate YPL6–3 genome, which was 80 kb long and contained 18 predicted genes. The MAT1–2-1 gene was found on Scaffold 91 (KY782629) of the YPL6–1 genome, which was 75 kb long and contained 26 predicted genes. Sequence alignment of the two scaffold regions revealed that the MAT1–1 and MAT1–2 idiomorphs were 10.5 and 6.7 kb long, respectively.
The MAT1–2 idiomorph contained only a single MAT1–2-1 gene. However, in addition to the MAT1–1-1 gene, there were two predicted coding sequences GME3123 and GME3124 on the MAT1–1 idiomorph (Fig. 1). They were not homologues to any of the mating genes that have been published; therefore, we speculated that they are novel mating genes unique to morels. GME3123 was found to be 1688 bp containing four introns and encoding 488 amino acids, and GME 3124 was 1825 bp containing five introns and encoding 512 amino acids. BLASTx analysis revealed that GME3123 had 24% and GME3124 21% amino acids similarity to hypothetical protein (AIU38081.1) of Tuber borchii (Belfiori et al. 2016).

Organization of MAT locus in M. importuna. The scale bar indicates sizes of only the MAT idiomorphs, the distance and sizes of other genes are not to scale. Introns in the MAT genes are indicated by black boxes. Thick and thin arrows indicate the orientation of genes and prime pairs respectively. Gene or superfamily names refer to a gene encoding a protein showing the highest percentage identity to the putative gene product following BLASTx analysis. APN2 DNA-(apurinic or apyrimidinic site) lyase 2; cox13 cytochrome c oxidase subunit 6A, mitochondrial; CPSF6: cleavage and polyadenylation specificity factor subunit 6; tfa1 transcription initiation factor IIE subunit alpha; atp-3 ATP synthase subunit 4, mitochondrial; SDH2 succinate dehydrogenase (ubiquinone) iron-sulfur subunit, mitochondrial; Mba1 mitochondrial inner membrane protein; end4 (also known as SLA2) endocytosis protein end4
To compare this analysis with the published morel genome databases, a sequence completely identical to the MAT1–1 idiomorph was found on the Scaffold 48 of the M. conica genome (http://genome.jgi.doe.gov/Morco1/Morco1.home.html) between 239.6 kb and 250.1 kb. Three genes, 10317, 10316 and 10315, were in this area on the Scaffold 48 of the M. conica genome, and they were completely identical with the genes MAT1–1-1, GME3123 and GME3124, respectively. No sequences identical to the MAT1–2 idiomorph were founded in this genome database. Sequences completely identical to the MAT1–2 idiomorph were located on the Scaffold 20 of the M. importuna strain SCYDJ1-A1 genome (http://genome.jgi.doe.gov/Morimp1/Morimp1.home.html) between 230.6 kb and 237.3 kb. Only an ORF known as fgenesh1_pg.20_#_79 was found in this area, and its sequence was the same as the MAT1–2-1 gene. The MAT1–1 idiomorph could not be found in this genome database.
The coding sequences flanking the two idiomorphs were highly conserved. The 5′ flanking region of both idiomorphs contained APN2, which is connected to the cox13 gene, and there were four additional predicted ORFs between the cox13 gene and the MAT idiomorphs (Fig. 1). The APN2 gene displayed 66% amino acid sequence similarity to CCX30259.1 (Pyronema omphalodes), and the cox13 gene displayed 62% similarity to GAP92444.1 (Rosellinia necatrix). The end4 (also known as SLA2) gene was the second gene that located on the 3′ flanking region of two idiomorphs, and it displayed 66% amino acid sequence similarity to CCX33915.1 (Pyronema omphalodes).
Structure of the MAT1–1-1 gene of three morel species
The lengths of the MAT1–1-1 gene sequences of M. importuna, M. sextelata and Mel-20 were 1694, 1730, and 1651 bp, respectively. RT-PCR confirmed that the MAT1–1-1 ORFs contained two introns and encoded proteins of 525, 537 and 511 amino acids, respectively (Table 3). Sequence alignments and dotplot analyses indicated that the MAT1–1-1 gene in M. importuna displayed 92.3 and 87.2% sequence identity to those in M. sextelata and Mel-20, respectively.
The deduced amino acid sequences of these three morel species contained a conserved alpha-box motif of the MAT1–1-1 protein of ascomycetes (Fig. 2a). In addition, the three proteins shared more than 20% amino acid identities to those in Tuber indicum (AHE80942), T. melanosporum (ADU56595) and T. borchii (AIU38080).

Amino acid alignment of conserved regions of MAT proteins of Morchella spp. with those of other ascomycetes. a Amino acid alignment of the conserved alpha-box domain of the following species: (1) Stemphylium callistephi(AAR04468); (2) Alternaria brassicicola(AAK85542); (3) Aspergillus fischeri(XP_001263836); (4) Talaromyces marneffei (ABC68484); (5) Histoplasma capsulatum (ABO26868); (6) Ophiocordyceps sinensis (ALH25059); (7) Cordyceps militaris (AKM95188); (8) Fusarium poae (CAD59610.3); (9) Colletotrichum musae (CAD59611.3); (10) Fusarium avenaceum (CAD59608.4); (11) Morchella sextelata; (12) Morchella importuna; (13) Mel-20; (14) Tuber indicum (AHE80942); (15) Tuber melanosporum (ADU56595); (16) Tuber borchii (AIU38080); (17) Xanthoria polycarpa (CAI59771); (18) Pyrenopeziza brassicae (CAA06844); (19) Sclerotinia homoeocarpa (AJW31369). b Amino acid alignment of the conserved HMG domain of following species: (1) Aspergillus fumigatus (XP_754989.2); (2) Penicillium chrysogenum (CAP17333); (3) Sclerotinia sclerotiorum (AGB05594); (4) Sclerotinia trifoliorum (ANN44262); (5) Sclerotinia homoeocarpa (AJW31335); (6) Sordaria fimicola (CAB63226); (7) Cordyceps militaris (AKM95197); (8) Ophiocordyceps sinensis (AEH27625); (9) Colletotrichum gloeosporioides (AKO22190); (10) Rusavskia elegans (CAI59778.2); (11) Dufourea flammea (CAI59780.2); (12) Passalora fulva (ABG49507); (13) Leptosphaeria maculans (AAO37761); (14) Bipolaris sorokiniana (AAF87724); (15) Morchella importuna; (16) Morchella sextelata; (17) Mel-20; (18) Tuber borchii (AIU38078); (19) Tuber indicum (AHE80950)
Structure of the MAT1–2-1 gene of three morel species
The lengths of MAT1–2-1 gene sequences of M. importuna, M. sextelata and Mel-20 were 1197, 1199 and 1247 bp, respectively. The results of RT-PCR analyses confirmed that the MAT1–2-1 ORFs contained three introns and encoded proteins of 329, 329 and 344 amino acids, respectively (Table 3). Sequence alignments and dotplot analyses indicated that the MAT1–2-1 gene in M. importuna shared 94.3 and 86.3% sequence identity with the corresponding genes in M. sextelata and Mel-20, respectively. The alignment of the MAT1–2-1 protein with corresponding sequences deposited in GenBank indicated that the HMG-box motifs of three morels were conserved (Fig. 2b).
Mating type ratios of three morel species based on single-ascospore populations
The primer pairs p8 for the MAT1–1-1 gene and p10 for the MAT1–2-1 gene could be used in all three morel species to examine the mating types of single-ascospore populations. Strains that contained only MAT1–1-1 and not MAT1–2-1 were amplified and scored as MAT1–1, and vice versa (Fig. 3). In the four ascospore populations (Table 4), the ratio of MAT1–1 to MAT1–2 ranged from 0.44 to 0.96. Chi-square analysis indicated that the ratio was not significantly different from 1:1 except for the HL1 populations. No sequence difference was detected between the single-ascospore strains and the parent strains in either of the MAT1–1-1 or MAT1–2-1 genes.

MAT gene PCR products of single ascospore populations YPL2, HL1 and WXLBD7. a MAT1–1-1 gene PCR products of YPL2–1 to YPL2–24. b MAT1–2-1 gene PCR products of YPL2–1 to YPL2–24. c MAT1–1-1 gene PCR products of HL1–1 to HL1–24. d MAT1–2-1 gene PCR products of HL1–1 to HL1–24. e MAT1–1-1 gene PCR products of WXLBD7–1 to WXLBD7–24. f MAT1–2-1 gene PCR products of WXLBD7–1 to WXLBD7–24. M DNA marker 2000 (2000,1000, 750, 500, 250 and 100 bp)
Discussion
Heterothallism in M. importuna, M. sextelata and Mel-20
In ascomycetes, the mating type corresponds to two allelic forms of a single locus and, accordingly, heterothallism is bipolar (Whitehouse 1949). This property was first demonstrated in Ascobolus magnificus and A. carbonarius (Dodge 1920) and then in several Neurospora species (Shear and Dodge 1927). Molecular analyses of mating-type in filamentous ascomycetes began with the cloning of A and a mating-types from Neurospora crassa (Glass et al. 1988). It has since been confirmed that the mating-type locus contains one of two highly divergent sequences occupying the same chromosomal locus. Metzenberg and Glass (1990) used the word idiomorph to denote that these large sequences are not obviously related by structural similarity. In heterothallic ascomycetes, ascospore populations can be divided into two groups: those harbouring the MAT1–1 and MAT1–2 idiomorphs. The sexual cycle occurs only between two individuals with opposite mating-type, rather than with self-fertilizing as in homothallic reproduction (Pöggeler 2001; Chilvers et al. 2014).
The genomic data of M. importuna YPL6 confirmed that the MAT locus is divided into two idiomorphs. In the YPL6–3 genome, only the MAT1–1 idiomorph harbouring the MAT1–1-1 gene was found. Similarly, the MAT1–2 idiomorph with the MAT1–2-1 gene was only located in the YPL6–1 genome. Furthermore, the conserved flanking sequences of the two idiomorphs confirmed that they occupy the same chromosomal locus. The characterization of the mating locus is consistent with heterothallic ascomycetes. Mating-type analysis of four ascospore populations from three species, M. importuna, M. sextelata and Mel-20, indicated that two sexual groups were present, and the ratios of MAT1–1- and MAT1–2-harbouring idiomorphs were not significantly different from a 1:1 ratio. These results suggested that these morels are all heterothallic.
Because the lack of visible structures, such as the clamp connection in basidiomycetes precludes visible morphological analysis, these molecular data will facilitate crossbreeding to produce high-quality morel cultivars.
Structure of MAT idiomorphs
In heterothallic ascomycetes, the structure of the MAT idiomorph varies in different species. The mating idiomorphs of Neurospora crassa were the first to be investigated, and the MAT1–1 idiomorph contains three ORFs: MAT1–1-1, MAT1–1-2 and MAT1–1-3 (mat A-1, A-2 and A-3 in N. crassa). The MAT1–2 idiomorph contains the MAT1–2-1 and MAT1–2-2 ORFs (mat a-1 and a-2 in N. crassa) (Pöggeler and Kück 2000). Similar MAT structures have been found in many other species surveyed later (Debuchy and Turgeon 2006; Wilken et al. 2012; Hutchinson et al. 2016). In this study, M. importuna has been identified as possessing a similar MAT idiomorph structure similar to those previously described. The MAT1–1 idiomorph contains three OFRs, and the MAT1–2 idiomorph contains a single MAT1–2-1 gene. However, two ORFs,GME3123 and GME3124,in the MAT1–1 idiomorph were found to be highly divergent from the homologues of mating genes that have been published. At this point, we can only speculate that GME3123 and GME 3124 are new mating-type genes at the MAT1–1 locus of M. importuna.
APN2, SLA2 and APC5 are ancestral companions of the MAT locus in euascomycetes (Debuchy and Turgeon 2006). Similarly, homologues of APN2 and SLA2 were found on each side of the MAT locus in Sordariomycetes, and the ATG3, cox13 and CWF24 genes were also conserved in the up- and downstream regions of MAT (Debuchy and Turgeon 2006; Xu et al. 2016; Lu et al. 2016). For M. importuna, the APN2 gene was connected to a cox13 gene on the 5′ flanking region of both MAT idiomorphs. However, four predicted genes were found between the cox13 gene and the MAT idiomorphs. The SLA2 (end4) gene was located on the 3′ flanking region of two idiomorphs. These results indicated that the MAT locus structure of M. importuna had some conserved sequences but was highly divergent from other species.
Based on the conserved flanking sequences, MAT1–1 and MAT1–2 idiomorphs have been cloned from many species (Martin et al. 2011; Belfiori et al. 2013). In this study, the coding sequences flanking the two idiomorphs of M. importuna were highly conserved, but the cloning of the whole MAT idiomorph of M. sextelata and Mel-20 using long-distance PCR amplification was not successful (data not shown), possibly due to the variability of the sequence in the different species. Unfortunately, we were not able to identify the entire mating idiomorphs of these two morel species.
Expression of mating genes
In Neurospora crassa and Tuber melanosporum, mating genes are constitutively expressed during the vegetative as well as the sexual phases (Ferreira et al. 1996; Rubini et al. 2011). In this study, we did not analyse the expression of mating genes specially in different cultivation periods and conditions. However, we verified the coding region by successfully amplifying the MAT1–1-1 or MAT1–2-1 gene with cDNA from all the tested strains incubated on PDA for 10 days. For the single-ascospore isolates, MAT1–1-1 of YPL6–3, HL1–1 and WXLBD7–11, and MAT1–2-1 of YPL6–1, HL1–47 and WXLBD7–13 were amplified with their cDNA, respectively. In addition, the expression of the two genes was detected in the mycelia from the YPL6, WXLBD7 and HL1 tissues. These results implied that the mating genes in morels are constitutively expressed.
Mating locus and phylogenetic relationship
In ascomycetes, as throughout the eukaryotic kingdom, genes controlling sex determination are evolving more rapidly than other protein-coding genes (Whitfield et al. 1993; Ferris et al. 1997; Pöggeler 1999), which enables the study of the phylogenetic relationship of closely related fungal species. The sequences from the gpd (glyceraldehyde-3-phosphate dehy-drogenase gene), mat A-1 and mat a-1 genes produced phylogenetic trees with a similar topology and strict separation of homothallic and heterothallic species within the genera Neurospora and Sordaria (Pöggeler 1999). Belfiori et al. (2013) revealed the existence of cryptic species of Tuber indicum by comparing the idiomorph structure and sequences of MAT genes.
Morchella importuna and M. sextelata had a closer phylogenetic relationship than did either to Mel-20 (Fig. S1). Based on comparing the DNA sequences of the MAT1–1-1 and MAT1–2-1 genes, M. importuna showed greater identity to M. sextelata than to Mel-20, which was consistent with the molecular evolution relationship based on the analysis of ITS, RPB1, RPB2 and TEF1 (Du et al. 2012; Richard et al. 2015). Acquiring more sequence information on the mating loci will help to investigate the phylogenetic diversity of morel species, thereby producing new insights.
References
Belfiori B, Riccioni C, Paolocci F, Rubini A (2013) Mating type locus of Chinese black truffles reveals heterothallism and the presence of cryptic species within the T. indicum species complex. PLoS ONE 8(12):e82353
Belfiori B, Riccioni C, Paolocci F, Rubini A (2016) Characterization of the reproductive mode and life cycle of the whitish truffle Tuber borchii. Mycorrhiza 26:515–527
Buscot F (1994) Ectomycorrhizal types and endobacteria associated with ectomycorrhizas of Morchella elata (Fr.) Boudier with Picea abies (L.) karst. Mycorrhiza 4:223–232
Chilvers MI, Jones S, Meleca J, Peever TL, Pethybridge SJ, Hay FS (2014) Characterization of mating type genes supports the hypothesis that Stagonosporopsis chrysanthemi is homothallic and provides evidence that Stagonosporopsis tanaceti is heterothallic. Currr Genet 60:295–302
Coppin E, Debuchy R, Arnaise S, Picard M (1997) Mating types and sexual development in filamentous ascomycetes. Microbiol. Mol Biol Rev 61:411–428
Debuchy R, Turgeon BG (2006) Mating-type structure, evolution, and function in Euascomycetes. The Mycota 1:293–323
Dodge BO (1920) The life history of Ascobolus magnificus. Mycologia 12:115–134
Du XH, Zhao Q, O’Donnell K, Rooney AP, Yang ZL (2012) Multigene molecular phylogenetics reveals true morels (Morchella) are especially species-rich in China. Fungal Genet Biol 49:455–469
Du XH, Zhao Q, Yang ZL (2014) Diversity, evolutionary history and cultivation of morels: a review. Mycosystema 33:183–197
Ferris JP, Pavlovic C, Fabry S, Goodenough UW (1997) Rapid evolution of sex-related genes in Chlamydomonas. Proc Natl Acad Sci U S A 94:8634–8639
Ferreira AVB, Saupe S, Glass NL (1996) Transcriptional analysis of the mt A idiomorph of Neurospora crassa identifies two genes in addition to mt A-1. Mol Gen Genet 250:767–774
Fu LH, Wang YP, Wang JJ, Yang YR, Hao LM (2013) Evaluation of the antioxidant activity of extracellular polysaccharides from Morchella esculenta. Food Funct 4:871–879
Glass NL, Vollmer SJ, Staben C, Grotelueschen J, Metzenberg RL, Yanofsky C (1988) DNAs of the two mating-type alleles of Neurospora crassa are highly dissimilar. Science 241:570–573
Hervey A, Bistis G, Leong I (1978) Cultural studies of single ascospore isolates of Morchella esculenta. Mycologia 70:1269–1274
Hu M, Chen Y, Wang C, Cui H, Duan P, Zhai T, Yang Y, Li S (2013) Induction of apoptosis in HepG2 cells by polysaccharide MEP-II from the fermentation broth of Morchella esculenta. Biotechnol Lett 35:1–10
Hutchinson MI, Powell AJ, Tsang A, O’Toole N, Berka RM, Barry K, Grigoriev IV, Natvig DO (2016) Genetics of mating in members of the Chaetomiaceae as revealed by experimental and genomic characterization of reproduction in Myceliophthora heterothallica. Fungal Genet Biol 86:9–19
Lu YZ, Xia YL, Luo FF, Dong CH, Wang CS (2016) Functional convergence and divergence of mating-type genes fulfilling in Cordyceps militaris. Fungal Genet Biol 88:35–43
Masaphy S (2010) Biotechnology of morel mushrooms: successful fruiting body formation and development in a soilless system. Biotechnol Lett 32:1523–1527
Martin SH, Wingfied BD, Wingfield MJ, Steenkamp ET (2011) Structure and evolution of Fusarium mating type locus: new insights from the Gibberella fujikuroi complex. Fungal Genet Biol 48:731–740
Metzenberg RL, Glass NL (1990) Mating type and mating strategies in Neurospora. BioEssays 12:53–59
Miller SC(2005) Cultivation of Morchella US Patent 6,907,691
Nelson MA (1996) Mating systems in ascomycetes: aromp in the sac. Trends Genet 12:53–59
O’Donnell K, Rooney AP, Mills GL, Kuo M, Weber NS, Rehner SA (2011) Phylogeny and historical biogeography of true morels (Morchella) reveals an early cretaceous origin and high continental endemism and provincialism in the Holarctic. Fungal Genet Biol 48:252–265
Ower R, Mills GL, Malachowski JA(1986) Cultivation of Morchella. US patent 4594809
Pagliaccia D, Douhan GW, Douhan LA, Peever TL, Carris LM, Kerrigan JL (2011) Development of molecular markers and preliminary investigation of the population structure and mating system in one lineage of black morel (Morchella elata) in the Pacific northwestern USA. Mycologia 103:969–982
Peng WH, Tang J, He XL, Chen Y, Tang H (2016) Status analysis of artificial cultivation of Morchella in Sichuan(Chinese). Edible Med Mush 24:145–150
Pildain MB, Visnovsky SB, Barroetavena C (2014) Phylogenetic diversity of true morels (Morchella),the main edible non-timber product from native Patagonian forests of Argentina. Fungal Biol 118:755–763
Pilz D, McLain R, Alexander S, Villarreal-Ruiz Berch S, Wurtz TL, Parks CG, McFarlane E, Baker B, Molina R, Smith JE (2007) Ecology and Management of Morels Harvested from the Forests of Western North America. Gen Tech Rep PNW-GTR-710. U.S. Department of Agriculture, Forest Service, Pacific Northwest Research Station, Portland
Pöggeler S (1999) Phylogenetic relationships between mating-type sequences from homothallic and heterothallic ascomycetes. Curr Genet 36:222–231
Pöggeler S (2001) Mating-type genes for classical strain improvements of ascomycetes. Appl Microbiol Biotechnol 56:589–601
Pöggeler S, Kück U (2000) Comparative analysis of the mating-type loci from Neurospora crassa and Sordaria macrospore: identification of novel transcribed ORFs. Mol Gen Genet 263:292–301
Richard F, Bellanger JM, Clowez P, Hansen K, O’Donnell K, Urban A, Sauve M, Courtecuisse R, Moreau PA (2015) True morels (Morchella, Pezizales) of Europe and North America: evolutionary relationships inferred from multilocus data and a unified taxonomy. Mycologia 107:359–382
Rubini A, Belfiori B, Riccioni C, Tisserant E, Arcioni S, Martin F, Paolocci F (2011) Isolation and characterization of MAT genes in the symbiotic ascomycete Tuber melanosporum. New Phytol 189:710–722
Shear CL, Dodge BO (1927) Life histories and heterothallism of the red bread mould fungi of the Monillia sitipholia group. J Agric Res 34:1019–1042
Su CA, Xu XY, Liu DY, Wu M, Zeng FQ, Zeng MY, Wei W, Jiang N, Luo X (2013) Isolation and characterization of exopolysaccharide with immunomodulatory activity from fermentation broth of Morchella conica. Daru J Pharmaceuti Sci 21:5
Turgeon BG, Yoder OC (2000) Proposed nomenclature for mating type genes of filamentous ascomycetes. Fungal Genet Biol 31:1–5
Volk TJ, Leonard TJ (1990) Cytology of the life-cycle of Morchella. Mycol Res 94:399–406
Whitfield S, Lovell-Badge R, Goodfellow NP (1993) Rapid sequence evolution of the mammalian sex-determing gene SRY. Nature 364:713–715
Wilken PM, Steenkamp ET, Hall TA, De Beer ZW, Wingfield MJ, Wingfield BD (2012) Both mating types in the heterothallic fungus Ophiostoma quercus contain MAT1-1 and MAT1-2 genes. Fungal Biol 116:427–437
Whiterhouse HLK (1949) Heterothallism and sex in the fungi. Biol Rev 24:411–447
Xu LS, Jardini TM, Chen WD (2016) Direct repeat-mediated DNA deletion of the mating type MAT1-2 genes results in unidirectional mating type switching in Sclerotinia trifoliorum. Sci Rep 6:27083
Acknowledgements
This work was supported by the National Natural Science Foundation Program of PR China (31460014) and the China Agriculture Research System (CARS-24).
Author information
Authors and Affiliations
Corresponding author
Additional information
Section Editor: Eckhard Thines
Electronic supplementary material
Table S1
(DOCX 55 kb)
Table S2
(DOCX 21 kb)
Table S3
(DOCX 24 kb)
Figure S1
Maximum parsimonious phylograms (MP) of Elata Clade (black morels) inferred from combined analysis of ITS, RPB2, and TEF1 sequences from 36 collections. Grey highlight is used to point out that the strain in this study appears to be nested within the corresponding phylogenetically distinct species. The other 31 collections were reported in Du et al.(2012) and O’Donnell et al.(2011) (Supplementary Table S1). Sequences of M. rufobrunnea were used to root the phylogram. The number of each internode (above 60%) represents the MP bootstrap value based on1000 replicates of the data. (JPEG 3571 kb)
Figure S2
Maximum parsimonious phylograms (MP) of Esculenta Clade (yellow morels) inferred from combined analysis of ITS, TEF1 and LSU sequences from 44 collections. Grey highlight is used to point out that strain YAAS2689 appears to be nested within the corresponding phylogenetically distinct species Mes-15. The other 43 collections were reported in Du et al.(2012) and O’Donnell et al.(2011)(Supplementary Table S1). Sequences of M. rufobrunnea were used to root the phylogram. The number of each internode (above 60%) represents the MP bootstrap value based on1000 replicates of the data. (JPEG 1272 kb)
Rights and permissions
About this article

{kind=link}
{kind=link}
Cite this article
Chai, H., Chen, L., Chen, W. et al. Characterization of mating-type idiomorphs suggests that Morchella importuna, Mel-20 and M. sextelata are heterothallic. Mycol Progress 16, 743–752 (2017). https://doi.org/10.1007/s11557-017-1309-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11557-017-1309-x