
Abstract
The complete sequence of CF_YL21, a Potato virus Y (PVY) isolate from Solanum tuberosum in China, was determined to be 9718 nucleotides in length, excluding the 3′-terminal poly(A) tail. The viral genome had a single open reading frame of 9186 nucleotides encoding a polyprotein of 3061 amino acids. The polyprotein was predicted to be cleaved into ten functional proteins by three viral proteases. Sequence analyses indicated that CF_YL21 shared 97% nucleotide identity with Wilga5 (PVYN-Wi), and two putative recombination signals were detected in the P1 and HC-Pro/P3 regions. Phylogenetic analyses, Bayesian Tip-association Significance (BaTS) test, and multiplex RT-PCR assay confirmed that the isolate had the similar molecular and genomic structure with PVYN-Wi, a PVY strain formed by recombination between PVYN and PVYO. To our knowledge, this is the first report of the complete sequence structure of PVYN-Wi strain from potato in China.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Potato virus Y (PVY) is one of the most destructive pathogens affecting potato (Solanum tuberosum) and many other solanaceous crops worldwide. It is a typical member of the genus Potyvirus in the family Potyviridae, with a single-stranded positive-sense genomic RNA of approximately 9.7 kb (King et al. 2012). Infection of field potatoes with PVY could result in 59.31–80.60% yield loss (Rahman and Akanda 2009), depending on the strain and inoculation load of PVY, the time when infection occurs, and the resistance of host genotypes (Gray et al. 2010; Nie et al. 2012). In the past decades, the occurrence and severity of PVY infection increased, posing an increasing threat to the potato industry particularly in underdeveloped regions such as East Asia and Africa. The increased infection may be attributed to emergence of new strains with novel pathogenicity and enhanced aggressiveness, declined effectiveness of chemicals used for vector control, and the use of infected seed potatoes (Perring et al. 1999; Fereres 2000; Robert et al. 2000; Takacs 2000). Increasing aphid numbers due to warmer winters as a result of climatic changes may also contribute to escalating viral infections (Gray et al. 2010).
PVY can induce various symptoms on potato leaves and tubers. Depending on the symptoms induced, PVY is traditionally classified into PVYC, PVYN, and PVYO. PVYC induces hypersensitive responses in a wide range of potato cultivars (Dullemans et al. 2011), forming mild mosaic patterns or stipple streak on leaves. PVYN induces leaf necrosis and mild damage to tubers, and PVYO induces mosaic and leaf drop streaks but does not cause leaf necrosis (Chachulska et al. 1997; Kerlan et al. 1999; Singh et al. 2003).
With the advantage of molecular technology, new and more aggressive PVY strains have been continuously detected around the world (Romancer and Kerlan 1994; Glais et al. 2002; Chikh et al. 2010b). For example, PVYNTN has the ability to induce potato tuber necrotic ringspot disease (Romancer and Kerlan 1994). Tubers infected by PVYNTN become unmarketable, and therefore, infection by PVYNTN results in a larger economic impact than infection by PVYC, PVYN, and PVYO. These new strains are believed to be the recombinants between PVYN and PVYO (Lorenzen et al. 2006) and have dominated in the world populations of PVY (Quenouille et al. 2013). For example, in the 190 complete sequences downloaded from GenBank, we found more than 55.5% have mixing genomic structure of PVYN and PVYO, while less than 38% only has PVYN or PVYO structure (unpublished data).
PVYN-Wi is one of the recombinants formed by PVYN and PVYO. It causes tobacco vein necrosis and has a PVYO serotype. It has two recombinant junctions (RJs) with one each in the P1 and HC-Pro/P3 regions. PVYN-Wi strain was first described in Poland from potato cultivar Wilga in 1991 (Chrzanowska 1991). In 1992, isolates with similar genomic structure were found in North America where they are usually called PVYN:O (Singh et al. 2003; Piche et al. 2004). Subsequently, the strain has been detected in many parts of the world including Canada, Spain, and France (McDonald and Singh 1996; Blanco-Urgoiti et al. 1998; Kerlan et al. 1999) in potato and tobacco. No PVYN-Wi strain has been detected from potato in China yet.
Better knowledge on the origin and distribution of PVY strain is important for its sustainable control. PVY can be transmitted by aphids, infected seeds, and other mechanisms. Aphids are the primary vector responsible for short to medium distance transmission of PVY while continental dispersal of the pathogen is mainly attributed by international exchanges of potato production (Gray et al. 2010). China has the largest potato production in the world (Li et al. 2013), and potato export in the country has expanded substantially over the past decades. Information on the occurrence and distribution of PVY in China may provide additional knowledge on the efficient control of the pathogen. Here, we study the genomic structure of PVY in China and find that one of the isolates, CF_YL21, has genomic characteristics similar to PVYN-Wi, a N × O recombinant reported in many parts of the world.
Materials and Methods
Viral Sample
CF_YL21 was isolated from a potato (Solanum tuberosum L.) plant showing PVY-alike mosaic symptom (Fig. S1) in August 2011 from a farm field located at Shaanxi province and was confirmed by enzyme-linked immunosorbent assay (ELISA) using a broad-spectrum PVY antibody (Agdia, Elhart, USA), PCR amplification of CP region, and a transmission electron microscope (Gao et al. 2013).
Amplification and Sequence of CF_YL21 PVY Isolate
Total RNA of CF_YL21 was extracted using Easy Pure Plant RNA Kit according to the manufacturer’s instructions (TransGen, Beijing, China). Full complementary DNA (cDNA) was synthesized by reverse transcription polymerase chain reaction (RT-PCR) using a ReverTra Ace qPCR RT Kit (TOYOBO, Shanghai, China). The coding regions of the PVY isolate were amplified from nine overlapping fragments (nucleotides 184–1008, 1009–2403, 2404–3498, 3499–3654, 3655–5556, 5557–5712, 5713–6276, 6277–7008, 7009–8565, and 8566–9366) using the ten degenerate primers described previously (Gao et al. 2014). The 5′- and 3′-terminal ends of untranslated regions (UTR) were determined by 3′ and 5′ Rapid Amplification of cDNA Ends (RACE, respectively (Frohman 1993; Chen and Chen 2002). In 3′ RACE, messenger RNAs (mRNAs) were converted into cDNA using reverse transcriptase and oligo-dT adapter primers. Specific cDNAs were then directly amplified by PCR using gene-specific primers that anneal to the region of known exon sequences and the adapter primers that targeted the poly(A) tail region. This permits the capture of unknown 3′-mRNA sequences that lie between the exon and the poly(A) tail. In 5′ RACE, first-strand cDNA was synthesized from poly(A) + RNA using the gene-specific primers. After synthesis of the first-strand cDNA, the original mRNA template was removed by treatment with the RNase Mix. Unincorporated dNTPs, GSP1, and proteins were separated from cDNA using a S.N.A.P. Column. 5′-RACE and 3′-RACE were performed with the SMARTer RACE cDNA Amplification Kit (Clontech) according to the manufacturer’s instruction.
PCR amplifications of cDNA were conducted on a ABI2710 thermal cycler (Applied Biosystems, USA) in 50 μL volumes, composing of 5.0 μL of TransTaqTM ×10 HiFi Buffer II, 4.0 μL of dNTPs (2.5 mM), 2.0 μL of forward primer (10 μmol/L), 2.0 μL of reverse primer (10 μmol/L), 34.5 μL of ddH2O, 0.5 μL of TransTaqTM HiFi Polymerase (5 U/μL), and 2.0 μL of template cDNA. Thermal cycling conditions involved an initial denaturation step at 94 °C for 5 min followed by 30 cycles of 94 °C for 30 s, 55 °C for 30 s with the exceptions of HC-Pro (52 °C) and P1 cistron (53 °C), and 72 °C for 30–90 s depending on lengths of cistrons (approximate 1 Kb/min). Finally, the products were extended for 10 min at 72 °C.
PCR products were separated by electrophoresis on 1% (w/v) agarose gel at 110 V for 50 min, visualized by a UV transilluminator, and cleaned using a TIANgel Maxi Purification Kit (TianGen, Beijing, China). Nine overlapping fragment amplicons, spanning the entire genome of CF_YL21, were ligated into T-tailed pEASY-T5 Zero vector and transformed into competent E. coli strain Trans1-T1 (TransGen, Beijing, China). Recombinant plasmids were extracted and identified by PCR. Due to high mutation rate in RNA viruses, three to six positive clones randomly selected from each isolate were sequenced in both forward and reverse directions using the M13 primers, and only the sequence identical in at least three clones was used for further analyses to eliminate potential sequence heterogeneity introduced by Taq polymerase. Additionally, DNA segments that are 1- to 2-kbp long were sequenced by primer walking strategy. Sequencing was performed by GenScript Biological Technology Co., Ltd. (Nanjing, China). Consensus sequences were assembled using DNAMAN 8 (Lynnon, Quebec, Canada), and the complete genome sequence of CF_YL21 was deposited in the GenBank database under the Accession Number KJ801915.
Sequence Analyses
The genome was assembled from overlapping RT-PCR clones after removal of the vector and primer sequence. Nucleotide and protein identities were searched with BLASTN and BLASTP programs implemented in the BLAST software package (http://www.ncbi.nlm.nih.gov/blast), respectively. Cleavage sites in the CF_YL21 genome were identified using online website (http://www.dpvweb.net/potycleavage/). Percentage of sequence identities were calculated using BioEdit software 7.2.0 (Tom Hall, Carlsbad, CA, USA). To determine the strain classification of CF_YL21, we divided its genome (excluding the UTRs) into R1, R2, and R3 regions. R1 (nucleotides 1–5529) starts from P1 and ends at the 3′part of CI cistron; R2 (nucleotides 5630–8382) stretches from VPg to NIb, and R3 (nucleotides 8383–9186) includes most of the CP region.
Recombination Analysis
Recombination events in the CF_YL21 complete genome were determined by similarity plot using SimPlot 3.5 (Lole et al. 1999). PVYN (Mont, AY884983) and PVYO (Oz, EF026074) were chosen as potential parents (Hu et al. 2009), and PVYN-Wi (Wilga5, AJ890350) was chosen as the alternative parent. Recombination breakpoints in the CF_YL21 genome were confirmed by Genetic Algorithm Recombination Detection (GARD) (Kosakovsky Pond et al. 2006) using 23 representative sequences of PVY in Fig. 3 as references (excluding the eight sequences from PVYC, PVYE, and PVYNA-N/NTN strains).
Phylogenetic Classification of CF_YL21
Thirty-one full genomes representing main PVY stains were retrieved from GenBank (Fig. 3), and a Turnip mosaic virus (TuMV) isolate (NC_002509) was used as an outgroup. Multiple sequence alignments were performed with MUSCLE (Codons) implemented in MEGA5 (Tamura et al. 2011), and the conserved regions of the genome were identified with Gblocks program (Talavera and Castresana 2007). Nucleotide substitution model was evaluated by MrModeltest using the Akaike Information Criterion (AIC) (Nylander 2008), and GTR + I + G model was used to reconstruct phylogenetic tree as the model fits best to the data. Maximum-likelihood (ML) tree was reconstructed using MEGA5, and its confidence was evaluated by 1000 bootstrapping.
The phylogenetic relatedness of CF_YL21 to the known PVY strains was further evaluated by Bayesian Tip-association Significance (BaTS) test (Parker et al. 2008) for association index (AI), parsimony score (PS), and maximum monophyletic clade (MC). The three statistics were calculated across all posterior distributions of the tree, and their significances were tested against the null distribution of the tree obtained from 10,000 resamples. Bayesian analysis for PVY sequences was performed with BEAST 1.8.0 (Drummond et al. 2012) using Markov Chain Monte Carlo framework (MCMC). The MCMC was run for 100,000,000 generations, and effective sample size (ESS) of parameters was checked to ensure they were above 200 at the end of running using Tracer 1.6, with the first 10% of sampled trees discarded as burn-in. If the ESS was below 200, additional generations were run for MCMC. Statistical significance of parameters was evaluated via the highest probability density (HPD). In addition, the multiplex RT-PCR assay developed previously (Chikh et al. 2010a) was used to determine the classification of CF_YL21. By mixing six pairs of strain-specific primers together, the multiplex assay was able to separate all known PVY strains, producing two unique fragments with the sizes of 853 and 441 bp for PVYN-Wi strain.
Results
Genomic Characterization of CF_YL21
The complete genome of CF_YL21 had 9186 nucleotides in length, excluding the poly(A) tail at its 3′-end. Its 5′ and 3′ UTRs consisted of 183 and 349 nucleotides, respectively. The CF_YL21 genome had a single open reading frame (ORF) from nucleotides 184 to 9369 and encoded a polyprotein of 3061 amino acids with an estimated molecular mass of 347.24 kDa. Like other PVY isolates, the polyprotein had nine cleavage sites generating ten mature proteins (P1, HC-Pro, P3, 6K1, CI, 6K2, VPg, NIa, NIb, and CP). The cleavage site of P1/HC-Pro dipeptide was ARSKVTQ/GVMDSMV cut by P1 serine proteinase, and the cleavage site of HC-Pro/P3 dipeptide was IKHYRVG/GIPNACP cut by HC-Pro cysteine proteinase (Table 1). The cleavage sites of seven other dipeptides (from P3/6K1 in 5′ to NIb/CP in 3′) were EYDVRHQ/RSTPGVK, DHEVRHQ/SLDDVIK, LQFVHHQ/AATSLAK, VETVSHQ/GKNKSKR, AQEVEHE/AKSLMRG, HDEVAEQ/AKHSAWM, and SYEVHHQ/GNDTIDA, respectively.
At the complete genome level, CF_YL21 sequence shared 87% nucleotide identity and 94% amino acid identity with Mont (PVYN, AY884983) whereas 94% nucleotide identity and 96% amino acid identity was shared with Oz (PVYO, EF026074). Comparatively, it shared 97% nucleotide identity and 99% amino acid identity with Wilga5 (PVYN-Wi, AJ890350) whereas 97% nucleotide identity and 98% amino acid identity was shared with PB209 (PVYN:O, EF026076) (Table 1). In pairwise comparisons against the three references, the CF_YL21 genome had a total of 1181, 588, and 238 base differences to the Mont, Oz, and Wilga5 genomes, respectively. The majority of base changes between CF_YL21 and Mont genomes occurred in nucleotides 1–308 and 2250–9366 while the majority of base changes between CF_YL21 and Oz were clustered in nucleotides 309–2224 (Fig. 1). On the other hand, base differences in nucleotides between CF_YL21 and Wilga5 occurred evenly across the entire genome. At the cistron level, CF_YL21 shared more than 96% sequence identities with Oz except for P1 and HC-Pro in which the two genomes only shared 81–90% identities, and 75–96% sequence identities with Mont except for HC-Pro cistron in which the two genomes shared 99% sequence identities (Table 1). The dN/dS between CF_YL21 sequence and Mont, Oz, and Wilga5 sequences was 0.037, 0.072, and 0.057, respectively, suggesting a non-neutral evolution of the pathogen. In pipo, CF_YL21 shared 86% nucleotide and 75% amino acid sequence identity with Mont but more than 97% nucleotide and 94% amino acid sequence identity with the remaining isolates (Table 1). In all cistrons, CF_YL21 shared more than 96% sequence identities with Wilga5, a PVYN-Wi isolate.

The comparison of CF_YL21 sequence with that of Mont (PVYN), Oz (PVYO), and Wilga5 (PVYN-Wi). The x-axis corresponds to nucleotide position along the CF_YL21 genome, and the y-axis is the number of nucleotide differences between the genomes. Bars indicate number of base differences according to individual nucleotide (G, C, T, or A, bottom of each panel) and in the pool of all four nucleotides (top of each panel). a Mont (PVYN strain). b Oz (PVYO strain). c Wigla5 (PVYN-Wi strain). The dN/dS between CF_YL21 sequence and Mont, Oz, and Wilga5 sequences was 0.037, 0.072, and 0.057, respectively
Recombination Analyses in the CF_YL21 Genome
Two recombination signals (nucleotides 309 and 2224) were detected in the CF_YL21 genome when Oz (PVYO) and Mont (PVYN) were used as parental references. The two recombination breakpoints were confirmed by GARD analysis with high confidence (p < 0.01, Table 2) when 13 additional PVYN and PVYO sequences (excluding UTRs, see Fig. 3 for the list of sequences) were used as references. Similar to PVYN-Wi isolates, the first recombination junction (nucleotide 309) within P1 switched CF_YL21 genome from PVYO-like to PVYN-like, and the second recombination junction (nucleotide 2224) in HC-Pro/P3 switched the CF_YL21 genome back from PVYN-like to PVYO-like. No recombination signals were detected when Wilga5 (PVYN-Wi) was assumed to be one of the parental strains (Fig. 2).

Detection and verification of recombination breakpoints in PVY CF_YL21 by Simplot approach using PVYN, PVYO and PVYN-Wi as reference strains
Phylogenetic Analysis
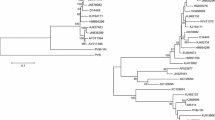
CF_YL21 was clustered together with a tobacco isolate (JN083841) from China with high bootstrap support (Fig. 3). BaTS also supported the phylogenetic structure of the tree (AI and PS; P value < 0.001) (Table 3). Most isolates were strongly associated with their predefined strain group (P MC < 0.05) except in the SYR-I and SYR-II groups. CF_YL21 clustered with PVYN-Wi strains LW, Wilga5, and AQ4 (P MC = 0.02), indicating that they were closely related. Though isolates defined as SYR-I and SYR-II clustered together, these two clades did not have a statistical support (P MC = 1.00).

Phylogenetic relationships of PVY isolates reconstructed by Maximum likelihood approach with a TuMV isolate as outgroup. Numbers above branches indicate bootstrap values of 1000 replicates (only shown >50%). CF_YL21 is marked with a red dot. P potato, T tobacco
Amplification of the CF_YL21 isolate with a multiplex RT-PCR produced two PVYN-Wi-specific bands in the sizes of 853 and 441 bp (Fig. 4).

Multiplex RT-PCR amplification of CF_YL21. The fragments are separated in agarose gel by electrophoresis: 100 bp DNA ladder (lane M), CF_YL21 (lane 1), and healthy potato control (lane 2)
Discussion
It has been widely documented that recombination plays an important role in the evolution of viruses (Gibbs and Ohshima 2010) such as PVY either through the generation of new strains bringing beneficial sequences of two parents together (Quenouille et al. 2013) or removal of deleterious mutations which otherwise may be accumulated quickly in genomes (Chang et al. 2015). Possibly due to this fitness advantage over their parental strains, recombinants have continuously emerged independently and have become prevalent in the PVY populations across various geographical regions over the world (Quenouille et al. 2013). PVYN-Wi is a recombinant strain between PVYN and PVYO. It contains two recombination junctions with one each in P1 and HC-Pro/P3. The strain can infect potato cultivars carrying the Ny resistance gene. Since its first detection in Europe from potato cultivar Wilga in 1991 (Chrzanowska 1991), the strain has been found in many parts of the world. Here, we report the first detection of a PVYN-Wi strain from potato in China, and we have several lines of evidence to support our finding.
First, the CF_YL21 isolate has a similar genomic structure and shared the highest sequence identities with Wilga5, the PVYN-Wi strain reported in the world. When six PVY isolates including Oz (PVYO, EF026074), Mont (PVYN, AY884983), Wilga5 (PVYN-Wi, AJ890350), Mb112 (PVYN:O, AY745491), 34/01 (PVYNTN-b, AJ890342), and SYR-II-2-8 (SYR-II, AB461451) were chosen as references, BLAST search indicated that the R1 region in CF_YL21 shared the highest sequence identity (97–99%) with Wilga5, Mb112, 34/01, and SYR-II-2-8 whereas only 89–91% with Oz and Mont; the R2 region shared the highest sequence identity with Oz, Wilga5, and Mb112 (96–97%) and only 84% with Mont, 34/01, and SYR-II-2-8; and the R3 segment shared the highest sequence identity with Oz, Wilga5, Mb112, and SYR-II-2-8 but only 90–91% with Mont and 34/01. These BLAST results suggest that the CF_YL21 has the typical genome structure of a recombinant between PVYN and PVYO and possesses all molecular characteristics of PVYN-Wi.
Second, CF_YL21 shared phylogenetic clade with all PVYN-Wi isolates (Fig. 3). In phylogenetic analysis, the reliability of trees is usually evaluated by bootstrap confidence (or posterior probability) and can be strongly affected by reference isolates used. In our study, BaTS was employed to further evaluate the robustness of phylogenetic tree and isolate-strain association. This additional evaluation indicated a strong association between the CF_YL21 isolate and PVYN-Wi strain (P MC = 0.02, Table 3).
Third, molecular amplification of CF_YL21 with a multiplex RT-PCR produced two PVYN-Wi-specific bands (Fig. 4). The multiplex RT-PCR amplified with 12 pairs of primers has been successfully used to distinguish main PVY strains including PVYO, PVYN, PVYNTN, and PVYN-Wi (Chikh et al. 2010b). The RT-PCR is able to detect the recombination points in the P1 region of PVY genome, enabling it to differentiate variable recombinant PVY strains.
Interestingly, AQ4 (JN083841), a PVYN-Wi isolate from tobacco in China (Wang et al. 2012), was phylogenetically closer to CF_YL21 than to other PVYN-Wi isolates from Poland (Fig. 3). There are two possible explanations for this result. It may result from a convergent evolution in response to local environments in China. Alternatively, they may have originated recently from the same ancestor and then have adapted to different hosts.
References
Blanco-Urgoiti B, Tribodet M, Leclere S, Ponz F, Legorburu F, Kerlan C (1998) Characterization of potato potyvirus Y (PVY) isolates from seed potato batches. Situation of the NTN, Wilga and Z isolates. Eur J Plant Pathol 104:811–819
Chachulska AM, Chrzanowska M, Ro-baglia C, Zagorski W (1997) Tobacco veinal necrosis determinants are unlikely to be located within the 5 and 3-terminal sequences of the potato virus Y genome. Arch Virol 142:765–779
Chang F, Zou W, Gao F, Shen J, Zhan J (2015) Comparative analysis of population genetic structure of Potato virus Y from different hosts. Hereditas (Beijing) 37:292–301
Chen J, Chen J (2002) Determination of genome sequence of potyviruses by degenerated PCR and RACE methods. Chin J Virol 18:371–374
Chikh AM, Maoka T, Natsuaki KT, Natsuaki T (2010a) The simultaneous differentiation of Potato virus Y strains including the newly described strain PVYNTN-NW by multiplex PCR assay. J Virol Methods 165:15–20
Chikh AM, Maoka T, Natsuaki T, Natsuaki KT (2010b) PVYNTN-NW, a novel recombinant strain of Potato virus Y predominating in potato fields in Syria. Plant Pathol 59:31–41
Chrzanowska M (1991) New isolates of the necrotic strain of Potato virus Y (PVYN) found recently in Poland. Potato Res 34:179–182
Drummond AJ, Suchard MA, Xie D, Rambaut A (2012) Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29:1969–1973
Dullemans AM, Cuperus C, Verbeek M, van der Vlugt RA (2011) Complete nucleotide sequence of a potato isolate of strain group C of Potato virus Y from 1938. Arch Virol 156:473–477
Fereres A (2000) Barrier crops as a cultural control measure of non-persistently transmitted aphid-borne viruses. Virus Res 71:221–231
Frohman MA (1993) Rapid amplification of complementary DNA ends for generation of full-length complementary DNAs: thermal RACE. Methods Enzymol 218:340–356
Gao F, Shen J, Shi F, Fang Z, Xie L, Zhan J (2013) Detection and molecular variation of Potato virus Y CP gene in China. Sci Agric Sin 46:3125–3133
Gao F, Chang F, Shen J, Shi F, Xie L, Zhan J (2014) Complete genome analysis of a novel recombinant isolate of Potato virus Y from China. Arch Virol 159:3439–3442
Gibbs A, Ohshima K (2010) Potyviruses and the digital revolution. Annu Rev Phytopathol 48:205–223
Glais L, Tribodet M, Kerlan C (2002) Genomic variability in Potato potyvirus Y (PVY): evidence that PVYNW and PVYNTN variants are single to multiple recombinants between PVYO and PVYN isolates. Arch Virol 147:363–378
Gray S, De Boer S, Lorenzen J, Karasev A, Whitworth J, Nolte P, Singh R, Boucher A, Xu H (2010) Potato virus Y: an evolving concern for potato crops in the United States and Canada. Plant Dis 94:1384–1397
Hu X, Karasev AV, Brown CJ, Lorenzen JH (2009) Sequence characteristics of Potato virus Y recombinants. J Gen Virol 90:3033–3041
Kerlan C, Tribodet M, Glais L, Guillet M (1999) Variability of Potato virus Y in potato crops in France. J Phytopathol 147:643–651
King AM, Lefkowitz E, Adams MJ, Carstens EB (2012) Virus taxonomy, ninth report of the international committee on taxonomy of viruses. Elsevier Academic Press, Amsterdam
Kosakovsky Pond SL, Posada D, Gravenor MB, Woelk CH, Frost SD (2006) GARD: a genetic algorithm for recombination detection. Bioinformatics 22:3096–3098
Li YY, Zhang RN, Xiang HY, Abouelnasr H, Li DW, Yu JL, Huang McBeath J, Han CG (2013) Discovery and characterization of a novel carlavirus infecting potatoes in China. PLoS ONE 8, e69255. doi:10.1371/journal.pone.0069255
Lole KS, Bollinger RC, Paranjape RS, Gadkari D, Kulkarni SS, Novak NG, Ingersoll R, Sheppard HW, Ray SC (1999) Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J Virol 73:152–160
Lorenzen JH, Meacham T, Berger PH, Shiel PJ, Crosslin JM, Hamm PB, Kopp H (2006) Whole genome characterization of Potato virus Y isolates collected in the western USA and their comparison to isolates from Europe and Canada. Arch Virol 151:1055–1074
McDonald J, Singh R (1996) Host range, symptomology, and serology of isolates of Potato virus Y (PVY) that share properties with both the PVYN and PVYO strain groups. Am Potato J 73:309–315
Nie B, Singh M, Murphy A, Sullivan A, Xie C, Nie X (2012) Response of potato cultivars to five isolates belonging to four strains of Potato virus Y. Plant Dis 96:1422–1429
Nylander JAA (2008) MrModeltest v2.3. Program distributed by the author. Evolutionary Biology Centre, Uppsala University
Parker J, Rambaut A, Pybus OG (2008) Correlating viral phenotypes with phylogeny: accounting for phylogenetic uncertainty. Infect Genet Evol 8:239–246
Perring TM, Gruenhagen NM, Farrar CA (1999) Management of plant viral diseases through chemical control of insect vectors. Annu Rev Entomol 44:457–481
Piche LM, Singh RP, Nie X, Gudmestad NC (2004) Diversity among Potato virus Y isolates obtained from potatoes grown in the United States. Phytopathology 94:1368–1375
Quenouille J, Vassilakos N, Moury B (2013) Potato virus Y: a major crop pathogen that has provided major insights into the evolution of viral pathogenicity. Mol Plant Pathol 14:439–452
Rahman MS, Akanda AK (2009) Performance of seed potato produced from sprout cutting, stem cutting and conventional tuber against PVY and PLRV. Bangladesh J Agric Res 4:609–622
Robert Y, Woodford J, Ducray-Bourdin DG (2000) Some epidemiological approaches to the control of aphid-borne virus diseases in seed potato crops in northern Europe. Virus Res 71:33–47
Romancer ML, Kerlan C (1994) Biological characterisation of various geographical isolates of Potato virus Y inducing superficial necrosis on potato tubers. Plant Pathol 43:138–144
Singh RP, McLaren DL, Nie X, Singh M (2003) Possible escape of a recombinant isolate of Potato virus Y by serological indexing and methods of its detection. Plant Dis 87:679–685
Takacs L (2000) Quicksilver from cinnabar: the first documented mechanochemical reaction? JOM 52:12–13
Talavera G, Castresana J (2007) Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol 56:564–577
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Wang B, Jia JL, Wang XQ, Wang ZY, Yang BH, Li XD, Zhu XP (2012) Molecular characterization of two recombinant Potato virus Y isolates from China. Arch Virol 157:401–403
Acknowledgements
This work was supported by the China Agriculture Research System (Grant No. CARS-10-P11) and the Fujian Natural Science Funds for Distinguished Young Scholar (Grant No. 2014J06008), People’s Republic of China. We thank Mr. Wenchao Lin for assistance in manuscript preparation.
Authors’ Contributions
JZ conceived and designed the experiments and wrote the manuscript; FC generated data and wrote the manuscript; WCZ and SZ carried out the experiments; and FLG and JGS analyzed the data. All authors reviewed the manuscript and approved the final manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no competing interests.
Additional information
Fei Chang and Fangluan Gao contributed equally to this work.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Fig. S1
Disease symptoms induced by CF_YL21 on potato (PDF 250 kb)
Rights and permissions
About this article

Cite this article
Chang, F., Gao, F., Shen, J. et al. Complete Genome Analysis of a PVYN-Wi Recombinant Isolate from Solanum tuberosum in China. Potato Res. 58, 377–389 (2015). https://doi.org/10.1007/s11540-015-9307-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11540-015-9307-3