
Abstract
Tumor growth and progression are critically dependent on the establishment of a vascular support system. This is often accomplished via the expression of pro-angiogenic growth factors, including members of the vascular endothelial growth factor (VEGF) family of ligands. VEGF ligands are overexpressed in a wide variety of solid tumors and therefore have inspired optimism that inhibition of the different axes of the VEGF pathway—alone or in combination—would represent powerful anti-angiogenic therapies for most cancer types. When considering treatments that target VEGF and its receptors, it is difficult to tease out the differential anti-angiogenic and anti-tumor effects of all combinations experimentally because tumor cells and vascular endothelial cells are engaged in a dynamic cross-talk that impacts key aspects of tumorigenesis, independent of angiogenesis. Here we develop a mathematical model that connects intracellular signaling responsible for both endothelial and tumor cell proliferation and death to population-level cancer growth and angiogenesis. We use this model to investigate the effect of bidirectional communication between endothelial cells and tumor cells on treatments targeting VEGF and its receptors both in vitro and in vivo. Our results underscore the fact that in vitro therapeutic outcomes do not always translate to the in vivo situation. For example, our model predicts that certain therapeutic combinations result in antagonism in vivo that is not observed in vitro. Mathematical modeling in this direction can shed light on the mechanisms behind experimental observations that manipulating VEGF and its receptors is successful in some cases but disappointing in others.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The initiation of angiogenesis is a critical bifurcation point in cancer progression, and sustained angiogenesis plays a central role in cancer pathology. The molecular basis of tumor angiogenesis has been of keen interest in the field of cancer research for decades. While numerous pro-angiogenic factors that act on endothelial cells and other vascular-associated cells (e.g., pericytes) have been identified, vascular endothelial growth factor (VEGF) and its receptors are arguably the most well studied (Ferrara 2002; Ferrara et al. 2003; McMahon 2007). VEGF has been implicated across a range of human cancers (Ellis and Hicklin 2008) and is associated with malignancy, metastasis and poor prognosis in a variety of solid tumors, including breast, prostate, lung, brain, ovarian and pancreatic (Botelho et al. 2010; Gasparini 2000; Ellis and Hicklin 2008; Jubb et al. 2004). Preclinical studies have shown that VEGF stimulates survival of existing vessels, promotes new vessel growth and contributes to vascular abnormalities such as tortuousness and hyperpermeability. The production of VEGF by tumor cells is stimulated by a variety of signals, including oncogenes, hypoxia and growth factors (Hanahan and Weinberg 2011). Although there are multiple variants of both the VEGF ligand and its receptor, the angiogenic affects of this pathway are primarily initiated through the interaction of VEGF-A, which binds to two tyrosine kinase receptors, VEGFR1 and VEGFR2 (Ferrara et al. 2003; Yao et al. 2011). Initially, it was thought that these receptors were expressed only on endothelial cells; however, recent studies show that VEGFR1 is also expressed on macrophages, hematopoietic stem cells and even some tumor cell types (Yao et al. 2011).
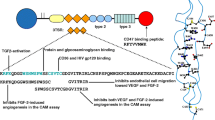
Dimerization and activation of VEGFR2 by tumor-secreted VEGF facilitate autophosphorylation of tyrosine residues in the cytoplasmic portion that either enhance receptor catalytic activity or provide docking sites for downstream signaling proteins resulting in mitogenic, chemotactic and pro-survival signals in endothelial cells (Ferrara 2002; Ferrara et al. 2003; McMahon 2007; Yao et al. 2011). The fundamental understanding of the interaction between the vascular endothelium and cancer cells has long been based on this one-sided concept that endothelial cells are responsive to migration, proliferation and survival signals (VEGF and other angiogenic stimulators) originating from tumor cells. One of the most interesting new discoveries associated with VEGF is that both autocrine and paracrine VEGF signaling occurs in tumor cells, and that this signaling contributes to key aspects of tumorigenesis, independent of angiogenesis. While the effect VEGF on angiogenesis is well established, much less is known about the direct effect of VEGF signaling on tumor cell growth dynamics. Experimental evidence shows that VEGF can play a major role in the proliferation and survival of tumor cells (Kaneko et al. 2007). Specifically, VEGF not only enhances endothelial cell survival by up-regulating Bcl-2 expression through a pathway mediated by VEGFR2, but also mediates tumor cell proliferation and survival via pathways regulated by VEGFR1 (Kaneko et al. 2007). The proposed mechanism by which VEGF initiates a signaling cascade that results in enhanced expression of Bcl-2, CXCL1 and CXCL8 in tumor cells is a signaling pathway initiated by VEGFR1. This bidirectional communication between endothelial cells and tumor cells is centrally regulated by VEGF and its downstream targets (see Fig. 1).

Schematic illustrating the proposed mechanism by which tumor cell expression of VEGF under hypoxia initiates a paracrine signaling cascade that results in enhanced expression of Bcl-2 in endothelial cells and promotes their proliferation in a signaling pathway mediated by VEGFR2. VEGF also initiates autocrine signaling in tumor cells mediated by VEGFR1 that results in Bcl-2 up-regulation and the expression of the proliferative chemokines CXCL1 and CXCL8. An increase in vascular endothelial cell number (and hence tumor vascularity) results in a decrease in hypoxia in the tumor microenvironment
The cross-talk between endothelial cells and tumor cells can thus be thought of as a “two-way street.” Our goal is to develop a mathematical model that will help us better understand—at a qualitative as well as quantitative level—the role of VEGF-mediated cross-talk between endothelial and tumor cells on each cell type’s survival and proliferation. This will help elucidate the implications of VEGF’s biphasic role, both pro-angiogenic and pro-tumorigenic, in cancer pathogenesis.
To accomplish our aim, we will build experimentally based mathematical models that describe VEGF-mediated proliferation and survival of endothelial and tumor cells. We will use the model to show that VEGF-mediated cross-talk between endothelial and tumor cells results in significant and quantifiable modifications in tumor cell phenotype in vitro and that VEGF fulfills a direct pro-tumorigenic role via autocrine–paracrine cellular effects. While numerous models of tumor angiogenesis and related treatments have been proposed, few work across the scales are proposed here, from intracellular signaling to tissue-level cancer growth (for a review of recent models, see Byrne 2010; Peirce 2008; Wang et al. 2015). The approach followed here is based on our previous models (Jain et al. 2008, 2009) that describe tumor angiogenesis, vascular tumor growth and response to treatment focusing on the following levels: (1) intracellular level: regulation of signaling pathways that are critical to cell proliferation, apoptosis and migration; (2) cellular level: cell surface dynamics of receptor–ligand binding and receptor activation that lead to intracellular signal transduction cascades; and (3) tissue level: dynamics of signaling chemicals and anti-cancer agents within the tissue, tumor growth dynamics, and tumor and vascular response to treatment.
The dependency of tumor expansion on angiogenesis and the identification of VEGF as the predominant angiogenic factor has served to propel investigations aimed at suppressing VEGF signaling by targeting either the ligand or its receptors. However, the impact of autocrine and paracrine signaling loops on treatments that interfere with VEGF and receptors is still unclear, making the approach described above that captures these dynamics a potentially important investigative tool. Therefore, our second objective is to use our modeling framework to make predictions on the efficacy of therapies that target VEGF, VEGFR1 and VEGFR2. This type of mathematical study has the potential to yield better strategies for administering therapies that can be antagonistic for endothelial cells, but are beneficial for tumor cells or vice versa.
The rest of this paper is structured as follows. In Sect. 2, we develop a mathematical model to study the effect of VEGF signaling on endothelial cells and tumors cells grown either in isolation or in co-cultures, in vitro. Our model is based on studies in Kaneko et al. (2007) that focus on head and neck squamous cell carcinomas (HNSCC). In these experiments, human dermal microvascular endothelial cells (HDMECs) stably transduced with Bcl-2 (HDMEC-Bcl-2) and empty vector controls HDMEC-LXSN were grown alone or together with the HNSCC cell line, UM-SCC-17B. We use data provided in Kaneko et al. (2007) to test and train our model, and then we use our model to make predictions on the outcome of several relevant treatment scenarios.
In Sect. 3, we propose a preliminary vascular tumor growth model that simulates tumor xenografts grown in mice, to help understand why these highly vascularized HNSCC tumors have only shown modest results when treated with anti-angiogenic strategies. The understanding of the dynamic interplay between tumor and endothelial cells may have important implications in the anti-angiogenic treatment of HNSCC and perhaps other malignancies that are resistant to current anti-angiogenic therapies.
2 Modeling In Vitro Co-cultures of Endothelial and Tumor Cells
We begin with a mathematical description of the interaction between endothelial and tumor cells in an in vitro setting, by tracking the temporal evolution of key cellular, molecular and physical species listed in Table 1.
2.1 Pre-treatment Tumor Cell and Endothelial Cell Growth Model
Our model describes a cell proliferation assay wherein endothelial cells (ECs) are cultured in the lower chamber and tumor cells (TCs) in the upper chamber of a noncontact co-culture system. We model the population dynamics of these two cell types as they grow and secrete VEGF, which mediates and enhances EC and TC proliferation. VEGF-mediated signaling also regulates intracellular Bcl-2 levels that in turn impact both EC and TC survival.
Endothelial Cells: Equation (1) describes the rate of change of endothelial cells. Bound VEGF is vital for EC proliferation, and cells cannot divide without it. Consequently, as in Jain et al. (2008) and Jain et al. (2009) the EC proliferation rate is assumed to be an increasing function of the fraction of activated VEGFR2 per cell (\(\phi _E\)). For simplicity, this rate is taken to be \(\mu _E\phi _E\), where \(\mu _E\) represents the maximum rate of EC proliferation. The limited carrying capacity of the culture environment is accounted for via a logistic term in the EC proliferation rate. Following Jain et al. (2009), since Bcl-2 is a pro-survival protein, EC death rate is assumed to be a decreasing function of Bcl-2 mRNA expression.
Tumor Cells: In contrast to normal cells, one of the hallmarks of cancer is that cell proliferation can occur in the absence of growth factor stimulation. We therefore model tumor growth as exponential in the absence of downstream VEGFR1 signals, with a doubling time of \(\ln {2}/{\mu _T}\). Several research groups have found that VEGF initiates a paracrine signaling cascade that results in enhanced expression of Bcl-2 in tumor cells in a signaling pathway mediated by VEGFR1 (Kaneko et al. 2007). In tumor cells, up-regulation of the anti-apoptotic protein Bcl-2 not only enhances cell survival, but also leads to increased production of CXC chemokines (CXCL1 and CXCL8) which, in turn, stimulate increased cellular proliferation (Kaneko et al. 2007). In several cancers, including HNSCC, VEGFR1 signaling induces activation of the MAPK/ERK and PI3k/Akt pathways, which also enhance proliferation of tumor cells independently of Bcl-2 (Szabo et al. 2016; Wu et al. 2006; Tong et al. 2008). Combining these processes, the rate of TC proliferation is taken to be \((\mu _T + \nu _T\phi _T)\) where \(\phi _T\) represents the fraction of activated VEGFR1 per TC. As with ECs, tumor cell death rate is also assumed to be a decreasing function of Bcl-2 mRNA expression. We remark that when Bcl-2 concentrations are abundant as reflected by a high level of mRNA expression, the death rate parameters will be chosen so that tumor cells are essentially immortal. These assumptions yield the following equation governing tumor dynamics:
We note that in the experiments described in Kaneko et al. (2007), tumor cell cultures are maintained for a period of only 2 days, so that space limitations on their growth may be ignored.
VEGF, VEGFR1 and VEGFR2: Experimental research suggests that VEGF-A binds to and activates its predimerized receptors (Mac Gabhann and Popel 2007). Therefore, we will only consider predimerized VEGF receptors, which we simply refer to as VEGFR1 (\(R_{E1}\) and \(R_{T1}\) on ECs and TCs, respectively) and VEGFR2 (\(R_{E2}\) on ECs only). We denote the VEGF–VEGFR1 complex as \(D_{E1}\) and \(D_{T1}\) on ECs and TCs, respectively. Similarly, VEGF–VEGFR2 complexes that only form on ECs are denoted as \(D_{E2}\). Below is a reaction diagram describing VEGF binding to each of its natural receptors:
where \(k^f_*\) and \(k^r_*\) denote the forward and reverse rates of reactions, respectively, and \(k^p_*\) denote the rates of bound receptor internalization and recycling to cell surface.
This reaction diagram is translated into a system of ordinary differential equations by evoking the law of mass action, which is represented by the first six terms in Eq. (3). We also include the natural decay rate of VEGF, \(\lambda _A\), as well as the following two mechanisms of VEGF synthesis. In an in vitro setting, ECs have been shown to secrete VEGF under Bcl-2-mediated signaling (Kaneko et al. 2007); consequently, a Bcl-2-dependent production of VEGF by ECs is included. Further, tumor cells experience normoxic conditions in vitro and we therefore assume that hypoxia-mediated VEGF production by tumors will not occur in this case; rather, following (Neufeld et al. 1999), we assume a (small) background rate of VEGF secretion by TCs.
The equations for VEGFR1 binding are given below. Note that in addition to binding and releasing VEGF, the rate of change of each receptor species is also affected by the creation of new unbound receptors as a result of cell division and the removal of receptors and complexes due to cell death. This ensures conservation of receptors (see below).
The equations for VEGFR2 binding are given below, and they follow analogously from the equations derived above.
In the above equations, \(R_{Ei}^t\), \(i=1,2\) and \(R_{T1}^t\) represent the VEGFRi and VEGFR1 receptor expression levels on endothelial and tumor cells, respectively. In the absence of treatment, we see from Eqs. (1), (21) and (24) that \(R_{E1}^tE \,=\, R_{E1}\,+\,D_{E1}\), that is, the number of VEGFR1 per endothelial cell is conserved. Likewise, \(R_{E2}^tE \,=\, R_{E2}\,+\,D_{E2}\) and \(R_{T1}^tT \,=\, R_{T1}\,+\,D_{T1} \).
Bcl-2 mRNA Bcl-2 mRNA is constitutively expressed within both cell types and undergoes natural degradation. Activation of its receptors by VEGF leads to additional Bcl-2 synthesis in each cell type. Combining these processes, we obtain the following equations for Bcl-2 mRNA expression in ECs and TCs:
2.2 Parameter Estimation
For cellular growth pre-treatment, a list of parameter values gathered from the literature is provided in Table 2. The remaining parameter values are estimated from data in Kaneko et al. (2007), Nor et al. (1999) and Tubergen et al. (2011) with resulting fits shown in Fig. 2 and estimated values listed in Table 3.

Fits of pre-treatment, in vitro model parameters to data. a Fits to time courses of ECs from cell proliferation assays described in Nor et al. (1999) (black/red squares show experimental data). b Fits to Bcl-2 mRNA expression in ECs as reported in Kaneko et al. (2007). c Fits to VEGF concentration in 24-h EC culture supernatant as reported in Kaneko et al. (2007). d Fits to time courses of TCs from cell co-culture assays described in Kaneko et al. (2007) (black/red squares show experimental data). e Fits to Bcl-2 mRNA expression in TCs as reported in Kaneko et al. (2007). f Fits to VEGF concentration in TC culture supernatant as reported in Tubergen et al. (2011)
In Nor et al. (1999), cell proliferation assays were carried out to demonstrate the effect of Bcl-2 over-expression on HDMEC proliferation and survival. Specifically, control (HDMEC-LXSN) and Bcl-2-over-expressing (HDMEC-Bcl-2) endothelial cells were cultured in vitro and their numbers recorded periodically for 7 days. HDMEC-Bcl-2 showed enhanced proliferation during the course of the experiments, and their numbers were significantly higher compared to HDMEC-LXSN. These data were used to estimate parameters relating to EC proliferation and death (\(\mu _E, \rho _E, \delta _E, \beta _E\)), with best fits shown in Fig. 2a. Bcl-2 mRNA expression levels in HDMEC-LXSN and HDMEC-Bcl-2 and corresponding VEGF concentrations in 24-h supernatant, as reported in Kaneko et al. (2007), were used to estimate the parameters relating to Bcl-2 mRNA production (\(\chi _E, \eta _E, \gamma _E, \omega \)) and EC production of VEGF (\(\mu _A, \beta _A\)). In particular, HDMEC-Bcl-2 constitutively over-expresses Bcl-2 which is reflected in a correspondingly high value for \(\chi _E\), the constitutive expression rate for Bcl-2 mRNA. Best fits to the data are plotted in Fig. 2b, c. We remark that these data show how Bcl-2 expression levels correlate directly with VEGF secretion by endothelial cells.
Figure 2d presents the best fit of the model to data in Kaneko et al. (2007) showing tumor cell (UM-SCC-17B) proliferation when co-cultured with HDMEC-Bcl-2 or HDMEC-LXSN. Notably, tumor cells co-cultured with HDMEC-Bcl-2 showed enhanced proliferation, as compared with tumor cells co-cultured with control endothelial cells. Parameters relating to tumor cell proliferation and death (\(\mu _T,\nu _T,\delta _T\)) were estimated from these data. A value for \(\beta _T\), the sensitivity of tumor cell death rate to Bcl-2 mRNA expression, was fixed to ensure that the maximum death rate of tumor cells (\(\delta _T/\beta _T\)) is greater that their minimum proliferation rate (\(\mu _T\)) since in this situation, Bcl-2 mRNA expression in the cells is negligible and we would expect cell death as a result.
Figure 2e shows the best fit of the model to baseline Bcl-2 mRNA expression levels in tumor cells grown in isolation or in co-culture with both types of endothelial cells, as reported in Kaneko et al. (2007) (Fig. 5a, b). Parameters relating to Bcl-2 mRNA production (\(\chi _T, \eta _T, \gamma _T, \theta \)) were estimated from these data. The data show that tumor cells grown in co-culture with HDMEC-Bcl-2 cells significantly over-express Bcl-2. These in vitro data are supported by in vivo studies in Kaneko et al. (2007) and suggest that endothelial cells secrete factors that influence gene expression levels and proliferation of the tumor cells via a paracrine signaling pathway that is independent of direct cell–cell contact. Finally, \(\nu _A\), the rate of VEGF secretion by tumor cells, is estimated from data in Tubergen et al. (2011), wherein VEGF concentration was measured in HNSCC cell culture supernatant, with best fit shown in Fig. 2f. As expected, in this normoxic, in vitro setting, tumor cells are producing only small amounts of VEGF.
As is clear from the discussion on parameter estimation, our model is able to capture a range of intracellular- and cellular-level data. In the next sections, we use it to predict the impact of a variety of therapeutic approaches targeting VEGF or its receptors on both endothelial and tumor cells.
2.3 In Vitro Therapeutic Interventions
Because of its importance in vascular initiation and remodeling, VEGF has been at the center stage of anti-angiogenic therapy. Inhibitors of the VEGF signaling axis exhibit anti-angiogenic properties with consequential anti-tumor effects; hence, these types of drugs have been eagerly added into clinical protocols for the treatment of several types of tumors (Moserle et al. 2014). Drugs that target various aspects of the VEGF signaling pathway typically fall into three general categories: ligand traps; tyrosine kinase inhibitors; and anti-VEGFR antibodies (Kendrew et al. 2011). Ligand traps, or VEGF-A ligand sequestering agents, bind directly to VEGF. These agents include bevacizumab (Avastin) a humanized monoclonal antibody and aflibercept (Zaltrap), a recombinant fusion protein that has VEGF-binding portions from VEGFR1 and VEGFR2 fused to a portion of the human IgG1 immunoglobulin (Holash et al. 2002; Kim et al. 1993). Alternatively, small molecular weight tyrosine kinase inhibitors (TKI) compete with the ATP binding site of the catalytic domain of several oncogenic tyrosine kinases. Anti-angiogenic drugs in the class include PTK787, sutent, sorafinib, Zactima and recentin (Gotink and Verheul 2010; Wood et al. 2000), which inhibit both VEGFR1 and VEGFR2. The final category consists of antibody or “antibody-like” molecules that block the interaction of VEGF-A with VEGFR-2 such as IMC1121b (ramucirumab) and CDP-791. A number of other antibodies against VEGFR1 and VEGFR2 have been designed and are currently being investigated in multiple clinical trials across a variety of tumor types (Krupitskaya and Wakelee 2009; Lu et al. 2003).
We now use our mathematical model to investigate the effect of treatments that fall into these categories. Specifically, the model equations are amended to include the effects of administering an anti-VEGF antibody, like avastin, an anti-VEGFR1 antibody-like IMC-18F1 and an anti-VEGFR2 antibody, alone and in combination, on HDMEC-LXSN and HDMEC-Bcl-2 grown in culture. We then study the effects of these different therapies on tumor cells grown in the presence of these two types of HDMECs (the full model equations are analogous to those presented in “Appendix” for the in vivo model, and parameters for these treatment strategies are also presented there).
2.3.1 The Effect of Therapy on HDMECs In Vitro
Figure 3a shows the in vitro growth dynamics of control endothelial cells (HDMEC-LXSN) when untreated and when treated with therapies that inhibit VEGF, VEGFR1 and VEGFR2 alone and in combination. The pre-treatment curve (dashed black line) is the same as shown in Fig. 2a. It is clear from the graph that interfering with VEGF and VEGFR2 both lead to reduced numbers of endothelial cells. In fact, there is no significant difference in endothelial cell number over time when treating with a VEGF antibody, a VEGFR2 antibody or a combination of the two treatments alone or together with a VEGFR1 antibody. However, treating with an anti-VEGFR1 antibody alone results in a significant increase in these cells because the anti-VEGFR1 therapies sequester VEGFR1 receptors on endothelial cells, preventing VEGF from binding to them. Thus, a greater amount of free VEGF is available to bind to, and activate, VEGFR2 receptors on ECs. Since in our model, VEGFR2 is assumed to mediate proliferation, survival and autocrine signaling that leads to increased VEGF production by ECs, this results in an increase in EC numbers over time as compared to the untreated control case. This can potentially have a significant impact on the treatment of tumors in vivo, since an anti-VEGFR1 antibody may actually act as an angiogenic stimulus (if there is VEGF available) and thus make the outcome of combination therapy hard to predict with experimental techniques alone.

Model predictions of endothelial cell response to anti-VEGF, anti-VEGFR1 and anti-VEGFR2 therapies. a, b Time courses of cell numbers. c Bcl-2 mRNA expression. d VEGF concentration in supernatant
We also investigated the impact of therapy on endothelial cells that over-express Bcl-2 (HDMEC-Bcl-2). Figure 3b shows the temporal changes in HDMEC-Bcl-2 cells, both in the treated and untreated cases as predicted by the model. HDMEC-Bcl-2 cells are essentially immortal, and we see that these cells exhibit greater growth when untreated as compared to the HDMEC-LXSN control cells. Our simulations suggest that while treating with inhibitors of VEGF and VEGFR2 both lead to reduced cell numbers as before, there are now distinct differences between the two therapeutic approaches. HDMEC-Bcl-2 cells continue to grow under anti-VEGF therapy, but are better controlled with anti-VEGFR2 antibodies or with a combination of the two treatments. These simulations also confirm that treatment with an anti-VEGFR1 antibody is antagonistic for ECs, and the therapeutic results are always worse with the addition of this type of treatment. Specifically, anti-VEGFR1 therapy combined with anti-VEGFR2 therapy is less effective (light blue curve) than anti-VEGFR2 alone (yellow curve). These results suggest that in an in vivo setting, if ECs adjacent to the tumor also over-express Bcl-2, then combining therapies has to be done carefully in order to avoid antagonistic effects and achieve desired responses.
Figure 3c shows the model predictions of Bcl-2 mRNA expression in HDMEC-LXSN and HDMEC-Bcl-2 cells under treatment. In control cells, only inhibiting VEGFR1 leads to a remarkable increase in Bcl-2 expression as more VEGF is now available to bind to VEGFR2. This also plays a role in the increased cell numbers shown in Fig. 3a. In HDMEC-Bcl-2 cells, we see that Bcl-2 mRNA expression is not significantly affected by any of the therapeutic interventions. This is not surprising given that these cells naturally over-express Bcl-2 and are essentially immortal; therefore, reducing the levels of bound VEGF does not result in appreciable cell kill, as shown in Fig. 3b.
Finally, Fig. 3d looks at the amount of free VEGF in the culture supernatant after one day. As expected, treating both control and Bcl-2 over-expressing HDMECs with an anti-VEGF antibody results in a substantial reduction in free VEGF. Treatments of both types of ECs with therapies that only block the receptors result in increased amounts of free VEGF, with HDMEC-Bcl-2 producing more VEGF than the control cells in each case. Any combination therapy that includes a VEGF antibody results in reduced VEGF levels as expected. Interestingly, treating both cell types with a combination of VEGFR1 and VEGFR2 antibodies results in the most striking increase in free VEGF; however, these high levels of free VEGF may be taken up by tumor cells and thus have a counter-productive effect in vivo.
2.3.2 The Effect of Therapy on Tumor Cells Alone and in Co-cultures With Endothelial Cells
In HNSCC progression, VEGF has been shown to play a biphasic role: pro-angiogenic and pro-tumorigenic. We now use the model to investigate the impact of anti-VEGF therapy on tumors cells grown alone and in co-culture with control or Bcl-2-over-expressing endothelial cells.

Model predictions of tumor and endothelial cell responses to anti-VEGF, anti-VEGFR1 and anti-VEGFR2 therapies, when tumor cells are cultured alone or in combination with control or Bcl-2-over-expressing endothelial cells for 48 h. a Level of tumor cell growth inhibition. b Level of endothelial cell growth inhibition. c, d Bcl-2 mRNA expression in tumor cells [experimental data taken from Kaneko et al. (2007)]. e Bcl-2 mRNA expression in endothelial cells. f VEGF concentration in supernatant
In Fig. 4a, we show the model predictions for surviving tumor cell fractions versus control as a function of mono-therapy administered for 2 days in three distinct cases: TCs alone, TCs + HDMEC-LXSN and TCs + HDMEC-Bcl-2. The first case of tumor cells growing in isolation is not particularly interesting therapeutically because in this in vitro, normoxic, setting very little VEGF is produced by tumor cells (see Figs. 2f or 4f). Therefore, the effect of VEGF on cell proliferation and survival is minimal, so that the TCs are not sensitive to any of the treatments. When tumor cells are grown in the presence of HDMEC-LXSN, there is more VEGF in the system and most of the therapies reduce the surviving fraction, albeit by less than 20\(\%\). Anti-VEGFR2 therapy, however, aggravates the situation because now ECs are prevented from binding as much VEGF, which leaves more for the tumor cells. Finally, when tumors cells are co-cultured with HDMEC-Bcl-2, we begin to see a significant impact on the surviving fraction. The interesting outcome in this case is that the effect of all therapies, except for anti-VEGFR2, is more pronounced since the very high Bcl-2 levels in ECs result in high levels of VEGF expression and exaggerated VEGF-mediated signaling.
Figure 4b shows the model predictions for surviving endothelial cell fractions when grown in co-culture with tumor cells. The results are similar to those in Fig. 4a. When tumor cells are co-cultured with HDMEC-LXSN, all therapies except for anti-VEGFR1 result in comparable surviving fractions of ECs at about 90\(\%\). In this case, anti-VEGFR1 therapy leads to enhanced survival because VEGFR1 on TCs and ECs is prevented from binding VEGF, which leaves more for VEGFR2 on ECs. When tumor cells are co-cultured with HDMEC-Bcl-2, increased VEGF production, and consequent signaling, results in a more pronounced reduction in surviving cell fraction. These results correlate with those shown in Fig. 4e, where model predictions for Bcl-2 levels in ECs are presented for all possible treatment scenarios. As before, the only therapy that has an impact on the Bcl-2 levels in control cells is blocking VEGFR1, and in this case the outcome is higher levels of Bcl-2, which is undesirable. When compared to Fig. 3b, we see that measurable differences among the other therapies are still not observed. For HDMEC-Bcl-2 cells, we again see a slight increase in Bcl-2 levels when VEGFR1 is blocked. Therefore, treatment with an anti-VEGFR1 antibody is antagonistic in terms of curbing endothelial cell growth in all cases, but is beneficial for controlling tumor cell growth (see Fig. 4a).
Some measure of model validation is provided in Fig. 4c where model fits to Bcl-2 expression data in untreated tumor cells, grown either alone or in co-culture, are presented together with direct comparisons of the model predictions (no data fitting) to the treatment data in Kaneko et al. (2007). The results show that the model predictions of Bcl-2 mRNA levels in tumors after 24-h of treatment correlate very well with the experimental data, even though parameters were only fit to the control data representing untreated tumors. Bcl-2 mRNA levels in tumor cells are further explored in Fig. 4d where model predictions for all possible treatment scenarios are presented. None of the treatments appreciably lower Bcl-2 mRNA levels in the tumor cells grown alone or with control endothelial cells. However, blocking VEGFR2 on ECs results in increased bioavailability of VEGF for TCs and a subsequent increase in Bcl-2 mRNA when tumor cells are grown in co-culture with either type of endothelial cell. As expected, all therapies, except anti-VEGFR2, have the effect of reducing tumor cell Bcl-2 mRNA expression levels back to their untreated value when tumor cells are grown in co-culture with HDMEC-Bcl-2. The interesting outcome here is that in co-culture, anti-VEGFR2 has the most dramatic and least favorable effect on Bcl-2 levels in tumor cells.
Finally, Fig. 4f depicts the amount of free VEGF in the system for each growth scenario. As expected, treating tumor cells grown in isolation or in co-cultures with an anti-VEGF antibody administered alone or in combination with other therapies results in a substantial reduction in free VEGF. Treatments with therapies that only block the receptors result in increased amounts of free VEGF, with TCs grown in co-culture with HDMEC-Bcl-2 consistently leaving more free VEGF than the other cases. While the results in Fig. 4f are fairly intuitive, taken all together model predictions in Fig. 4a, b, d, e imply that we can expect counterintuitive—and hence difficult to predict—tumor growth dynamics in vivo, given that anti-VEGFR2 therapies destroy ECs while boosting TCs, whereas anti-VEGFR1 inhibits TCs while promoting EC growth.
Above, we have discussed a variety of in vitro growth scenarios that were limited to simulating 2 days of treatment following the experimental protocol in Kaneko et al. (2007). The tumor cells grown in co-culture with HDMEC-LXSN are likely the closest to the “real” situation in vivo, where ECs can be induced to over-express Bcl-2 in the presence of VEGF instead of naturally over-expressing it all of the time. In Fig. 5, we take a closer look at this situation and show longer time predictions for cell numbers and activated receptor occupancies under the various treatment combinations.

Model predictions of tumor and endothelial cell response to anti-VEGF/anti-VEGFR1/anti-VEGFR2 treatments given alone or in combination, when SCC-17B cells are co-cultured with HDMEC-LXSN. Time courses of a tumor cell growth inhibition, b fractional occupancy of VEGFR1 on tumor cells, c endothelial cell growth inhibition and d fractional occupancy of VEGFR2 on endothelial cells
Specifically, in Fig. 5a, the surviving fraction of tumor cells versus control is plotted for 10 days. Clearly the most effective strategies for tumor reduction are those that involve blocking the activation of VEGFR1. However, anti-VEGFR2 therapy is undesirable from the point of view of inhibiting tumor cell growth and actually boosts cell numbers, as is also observed in Fig. 4a, d. This effect on tumor cell growth can be explained by looking at fractional occupancies of the VEGFR1 receptors on the tumor cells (\(\phi _T\)), shown in Fig. 5b. As can be seen, anti-VEGFR2 therapy results in a large, transient boost in \(\phi _T\), contributing to TC growth as more VEGF is available for tumor cells since it cannot bind VEGFR2 on ECs. We consider the therapeutic outcomes for the ECs in Fig. 5c. As expected from Fig. 4b, e, anti-VEGFR1 therapy is antagonistic in terms of controlling EC growth and leads to an increase in cell numbers. Interestingly, anti-VEGF + anti-VEGFR1 is also a sub-optimal therapeutic option in the long term even though it inhibited EC growth in the short term (see Fig. 4b). This is because although tumor cell growth is retarded, the cells are still growing at a (slower) exponential rate and the VEGF expressed by them begins to accumulate and eventually overwhelms the anti-VEGF antibody. Since VEGFR1 on ECs and TCs is prevented from binding it, more VEGF is available to activate VEGFR2 on ECs resulting in increased Bcl-2 mRNA expression, and increased proliferation and survival. The fractional occupancies of the VEGFR2 receptors on the endothelial cells (\(\phi _E\)), shown in Fig. 5d, further highlight this.
3 Modeling In Vivo Tumor Xenografts
The in vitro model developed in Sect. 2 is able to capture a range of intracellular- and cellular-level data and highlights the potential for expecting counterintuitive and difficult-to-predict dynamics in vivo, when treating vascular tumor growth with therapies targeting VEGF and its receptors. In this section, as a proof of concept, we develop a preliminary model representing tumor growth in vivo and use it to predict the effects of treatment on the growth and degree of oxygenation of the tumor. In order to capture the essential features of an in vivo situation where tumor cells are directly dependent on vascular support for nutrients, several modifications are made to the model equations. These are described in further detail below.
3.1 Pre-treatment Vascular Tumor Growth Model
We begin with a mathematical description of the growth of a tumor without therapeutic intervention. Specifically, our model simulates the growth of HNSCC tumor xenografts and is based on the experiments described in Fujita et al. (2007) wherein HNSCC cell xenografts were established in female BALB/c nu/nu nude mice and tumor volumes measured weekly for a period of 49 days.
Tumor Cells: As in the in vitro case, tumor cells are assumed to proliferate, both independent of, and in response, to VEGF signaling mediated by VEGFR1, and undergo apoptosis at a rate that is a decreasing function of Bcl-2 mRNA expression (\(B_T\)). However, now the rate of TC proliferation dependent on VEGFR1 activation is assumed to be an increasing and saturating function of \(\phi _T\), the fraction of activated VEGFR1 per unit cell number. Further, a tumor growing in vivo has an irregular blood supply and its growth is typically nutrient limited. Consequently, the effect of nutrient (oxygen) availability on TC proliferation and survival must be taken into account. For simplicity, we assume that when oxygen pressure (N) in the tumor falls below a critical threshold \(N_\mathrm{p}\), the cells cease to proliferate and enter quiescence, and a further decline in oxygen pressure below a level \(N_\mathrm{d}\) results in a doubling of the rate of cell death. Together, these assumptions give the following equation governing the growth dynamics of tumor cells in vivo:
Microvascular Endothelial Cells, Vascular Fraction and Oxygen: In normal mature vessels, endothelial cells are physically ensheathed by mural cells (such as pericytes) and chemically maintained in a quiescent state due to the abundance of angiopoietin-1 (Ang1) (Ciric et al. 2012). However, in pathological cases (cancer), this quiescent state is altered by VEGF-mediated signaling which results in the activation of ECs and the sprouting of new blood vessels. For simplicity, we do not explicitly incorporate microvessel density in our model. Instead, we keep track of the temporal dynamics of ECs that are assumed to line intratumoral blood vessels. Microvascular EC proliferation, and hence rate of new microvessel formation, is assumed to be an increasing and saturating function of \(\phi _E\), the fraction of activated VEGFR2 per unit cell number. The rate of endothelial apoptosis that results in microvessel degradation is assumed to be a decreasing function of intracellular Bcl-2 mRNA expression (\(B_E\)). We remark that since they are assumed to line functional blood vessels within the tumor, EC proliferation and death are taken to be independent of nutrient availability. These assumptions result in the following equation governing the dynamics of microvascular ECs:
Finally, the oxygen concentration, N, within the tumor is taken to be proportional to the vascular proportion of the tumor defined as the percentage \(V = 100E/(E+T)\), that is, \(N = \sigma _N V\). An increase in the proportion of ECs compared to the number of TCs reflects an increase in the level of vascularity and hence increased oxygenation in the tumor.
Intratumoral VEGF: The equation representing VEGF dynamics is similar to the in vitro case, with a few modifications. VEGF reacts with VEGFR1 and VEGFR2 on TCs and ECs and is degraded by tissue enzymes such as plasmin and matrix metalloproteinases (Yen et al. 2011). Further, VEGF is produced by tumor cells under conditions of hypoxia (Shweiki et al. 1995), the production being “switched on” when the oxygen concentration falls below a hypoxic threshold \(N_\mathrm{h}\) and “switched off” if \(N > N_\mathrm{h}\). Also, any ECs within the tumor are a part of the microvasculature and therefore do not express VEGF in our model. Combining these assumptions, the rate of change of intratumoral VEGF may be expressed as follows:
VEGFR1, VEGFR2, Bcl-2 mRNA: The equations representing VEGF receptor dynamics and intracellular Bcl-2 mRNA expression in ECs and TCs remain largely unchanged from the in vitro case and are not reproduced here; a full set of model equations can be found in “Appendix.”
3.2 Tumor Xenograft Response to Anti-VEGF Therapies
We now include the action of anti-VEGF therapies in our model of vascular tumor growth. For brevity, we only describe the model equations for anti-VEGF antibody administration. The equations for anti-VEGFR1 antibody and anti-VEGFR2 antibody are similar and are listed in “Appendix.” The experiments described in Fujita et al. (2007) form the basis for our treatment model. Briefly, HNSCC cell xenografts were established in female BALB/c nu/nu nude mice and treated with 2 or 4 mg/kg avastin (anti-VEGF antibody) on days 1 and 5 of each week, for 4 weeks, starting when the tumors reached a size of 50–100 mm\(^3\). The xenografts were then allowed to grow untreated for a further 3 weeks and their volume recorded periodically.
Drug Pharmacokinetics: In the experiments described above, avastin is administered as a series of intraperitoneal injections. Once injected, the drug is assumed to rapidly extravasate into the systemic circulation. Experimental evidence suggests a biphasic plasma concentration–time curve for avastin (Lin et al. 1999). Consequently, the following two-compartment model is proposed to govern its pharmacokinetics. Systemic circulation (together with well-vascularized organs) is taken to be the first compartment from where the drug may be eliminated or enter, in a reversible process, a second compartment which consists of tissues and organs with poor vascular perfusion. Combining these effects, the equations governing avastin pharmacokinetics can be written as follows:
where \(X_{Vs}\) and \(X_{Vp}\) represent the amounts of drug in the systemic circulation and the peripheral compartment, respectively. The dosing function in Eq. (15) represents the periodic administration of avastin and is taken to have the following form:
where \(D_0\) is the amount of drug delivered in each dose; \(t_i\), the timing of each dose; \(t_a\), the length of each injection; and n, the total number of doses.
Intratumoral Drug: Tumor vasculature is characterized by its poor functional quality and is highly disorganized (Siemann 2011). We therefore allow the tumor to reside in a pharmacokinetic compartment of its own. For simplicity, the rates of transfer of drug into and out of the tumor are taken to be the same as those for the peripheral compartment [Eq. (16)]. Further, the tumor is assumed to occupy a negligible volume compared to the volume of the animal; therefore, the extravasation of drug from the tumor into the systemic circulation will not affect the amount of drug in the central compartment. Finally, avastin–VEGF complexes are assumed to undergo the same degradation as free VEGF in tissue. Combined together, these processes yield the following equations governing drug (avastin) dynamics in the tumor space:
where \(X_{V}\) and \(C_{AV}\) represent the amounts of free anti-VEGF antibody and VEGF–anti-VEGF antibody complexes within the tumor.
3.3 Parameter Estimation
The parameter values relating to vascular tumor growth are estimated from data in Carreau et al. (2011), Fujita et al. (2007), Kaneko et al. (2007), Lin et al. (1999), Perfahl et al. (2011) and Yen et al. (2011) as discussed below. Estimated parameter values are listed in Table 6 in “Appendix.”
Parameters relating to the proliferation and death rates of tumor cells (\(\mu _T, \nu _T, \alpha _T, \delta _T, \beta _T\)) and microvascular endothelial cells (\(\mu _E, \alpha _E, \delta _E, \beta _E\)) were estimated by fitting control and treatment time courses of tumor volume as reported in Fujita et al. (2007), with best fits shown in Fig. 6a. These data have already been described earlier. A few simplifying assumptions were made to make parameter estimation easier. For instance, tumor cells were assumed to be highly sensitive to VEGFR1 signaling, which justifies assigning a low value to their VEGF-independent proliferation rate, \(\mu _T\). Further, the exponents \(\rho _1\) and \(\rho _3\) appearing in the TC and EC VEGF-dependent proliferation rates, respectively, where chosen to be equal and high enough so that when the hypoxia switch is turned on, cell proliferation is near its maximum value. Likewise, the exponents \(\rho _2\) and \(\rho _4\) appearing in TC and EC death rates were assumed to be equal and high enough so that at low Bcl-2 mRNA expression levels, cell death is maximized. Once parameters relating to VEGF production by tumor cells are fixed (explained below), the average fractional occupancies of receptors on TCs and ECs under hypoxia can be determined. These provided upper bounds for the half-saturation constants \(\alpha _T\) and \(\alpha _E\) appearing in the TC and EC proliferation rates, respectively. Similarly, the average Bcl-2 mRNA expression in TCs and ECs at the above fractional occupancies of receptors can be numerically estimated. These provided upper bounds for the half-saturation constants \(\beta _T\) and \(\beta _E\) appearing in TC and EC death rates, respectively. We remark that although treatment data are only available for avastin, it is enough to fix the parameters relating to cell death as apoptosis signaling is assumed to occur downstream of VEGF receptor-mediated signaling. Therefore, to simulate treatment with VEGFR1/VEGFR2 inhibitors, it is enough to know the relevant biochemical parameters, which have already been estimated as described in Sect. 2.2 (and listed in Table 2). This highlights an important feature of our approach that includes all of the relevant biochemistry, and without these details it would be necessary to estimate cell death parameters for each drug separately.
Parameters relating to tumor oxygenation and tumor cell response to hypoxia were estimated as follows: Intratumoral oxygen partial pressure typically ranges from 6 to 13 mmHg Carreau et al. (2011). Consequently, a value of 10 mmHg was chosen for \(N_\mathrm{h}\), the threshold below which the angiogenic switch is activated [see Eq. (14)], while the tumor cell quiescence threshold, \(N_\mathrm{p}\), was taken to be 5.5 mmHg [see Eq. (12)]. The threshold \(N_\mathrm{d}\), at which tumor cells begin to undergo high rates of apoptosis due to oxygen deprivation, must have a value lower than \(N_\mathrm{p}\) and was taken to be 1 mmHg. Perfahl et al. (2011) have reported that growing tumors typically comprise \(\sim \)9.2% vessel cells. This allowed us to estimate \(\sigma _N\), the proportionality constant relating the vessel fraction, V, to intramural oxygen levels, N, by requiring that when \(V = 9.2\), \(N = 10\) \(({=}N_\mathrm{h})\) mmHg.
Although the tumor xenografts described in Fujita et al. (2007) are supplied by microvessels lined with mouse endothelial cells, for simplicity, these were assumed to have the same properties, such as VEGFR1/VEGFR2 expression levels, as those of human endothelial cells. Further, parameters relating to EC Bcl-2 mRNA production (\(\chi _E, \eta _E, \gamma _E, \omega \)) were estimated as follows: Bcl-2 mRNA expression levels in ECs populating HNSCC cancers (reported in Kaneko et al. 2007) were used to estimate \(\eta _E\) and \(\gamma _E\), with the value for \(\omega \) unchanged from the in vitro case. In the absence of additional data, a value for the baseline rate of Bcl-2 mRNA expression in ECs, \(\chi _E\), was chosen so they express low amounts of mRNA (1 unit) in the absence of any external input. Parameters relating to TC Bcl-2 mRNA production (\(\chi _T, \eta _T, \gamma _T, \theta \)) were estimated as follows: Kaneko et al. (2007) have also estimated the maximum level of Bcl-2 mRNA expression in HNSCC tumors growing in vivo. This was used to estimate \(\eta _T\) and \(\gamma _T\), with the value for \(\theta \) unchanged from the in vitro case. As in the case of ECs, a value for the baseline rate of Bcl-2 mRNA expression in TCs, \(\chi _T\), was chosen so they express low levels of mRNA (1 unit) in the absence of any external input.
As discussed earlier, VEGF is produced by tumor cells in vivo under hypoxia. In the absence of relevant data, such as time course measurements of intratumoral VEGF, it is difficult to estimate \(\nu _A\), the maximum rate of VEGF production by tumor cells, and \(\kappa _A\), the sensitivity of tumor cells to hypoxia. Consequently, values for these parameters were chosen to ensure high receptor occupancies for both cell types when intratumoral oxygen levels are below the hypoxia threshold. We remark that altering these parameter values should not significantly affect the predictions of our model as lower receptor occupancies can be compensated for by lowering the half-saturation constants in TC and EC proliferation and Bcl-2 mRNA expression rates. A value for the decay rate, \(\lambda _A\), of VEGF in tissue was taken from Yen et al. (2011).
Finally, the pharmacokinetic rate constants \(k_{V12}\), \(k_{V21}\) and \(k_{Vel}\) appearing in Eqs. (43) and (44) were fit to data in Lin et al. (1999) wherein mice were treated with 9.3 mg/kg anti-VEGF antibody as a single intravenous injection and drug concentration in serum recorded periodically for 15 days. Best fits to experimental data are shown in Fig. 9 in “Appendix.” Given the large numbers of parameters that were estimated from fitting experimental data for our in vivo model, we also conducted a local and global sensitivity analysis to identify which parameters are most crucial for predicting tumor volumes and degree of oxygenation. Details of this analysis are provided in “Appendix.”

a Model fits to control and treatment time courses of in vivo tumor volume reported in Fujita et al. (2007) (black/blue/red squares show experimental data). The drug is administered on days 1 and 5 of each week for 4 weeks, the treatment period being highlighted with gray shading. b Model predictions of intratumoral oxygen partial pressure corresponding to the tumor volume time courses in panel a. c Model predictions of tumor growth inhibition at the end of 49 days in response to increasing amounts of anti-VEGF or anti-VEGFR1 or anti-VEGFR2 antibodies administered as single therapies following the schedule described in Fujita et al. (2007). d Model predictions of intratumoral oxygen partial pressure averaged over 49 days corresponding to the tumor volume time courses in panel c. Note that the doses of each drug are normalized by the amount of that drug that induces a 50% tumor growth inhibition at the end of 49 days
3.4 The Effect of Anti-VEGF Therapies on Vascular Tumor Growth
A series of numerical simulations were performed to illustrate the effects of anti-VEGF therapies on tumor xenograft growth dynamics. In all treatment cases, the schedule of drug administration was the same as that reported in Fujita et al. (2007), with fixed doses of drug administered on days 1 and 5 for 4 weeks, followed by 3 weeks of untreated growth. Key indicators of tumor development, such as tumor volume, degree of growth inhibition and degree of tumor oxygenation, were studied in particular. The results are summarized below.
Figure 6a shows the model fits to time courses of HNSCC xenograft volume when the tumor is left untreated (control) or is treated with 2 or 4 mg/kg of anti-VEGF antibody (Avastin), and has already been discussed earlier. Figure 6b shows the corresponding time courses of intratumoral oxygen partial pressure. Interestingly, when 2 mg/kg of avastin is administered, the tumor is predicted to exit hypoxia more rapidly than in the control or higher dose (4 mg/kg) cases. This is because, at the lower dose, the anti-VEGF therapy is effective at blocking VEGFR1 signaling on tumor cells leading to reduced proliferation and survival, but is not enough to induce a high degree of vascular disruption, resulting in a better oxygenated tumor. These simulations suggest that a therapeutic window could be created by first treating the tumor with vascular disruption agents to improve blood flow and then administering chemotherapies, which could have a greater impact on tumor growth inhibition due to better delivery across the whole tumor.
Before proceeding with a discussion of the remaining results, we introduce the following notation for ease of comparison between the various treatment scenarios simulated. The model was used to predict the amounts of each drug that induce 50% tumor growth inhibition at the end of 49 days, when given as single agents. All drug doses reported were then scaled by these 50% inhibition amounts.
We now simulate the effects of treating the tumor with increasing doses of anti-VEGF, anti-VEGFR1 and anti-VEGFR2 antibodies, with tumor volume at the end of 49 days (as a fraction of untreated control) shown in Fig. 6c and intratumoral oxygen levels (averaged over 49 days) plotted as a function of drug dose in Fig. 6d. When anti-VEGF or anti-VEGFR1 antibodies are administered as single agents, the results are as expected—tumor volumes decrease monotonically, with anti-VEGFR1 being more effective at higher doses. This is because we have assumed that the tumor cells are highly dependent on VEGF signaling for growth and survival. Tumor oxygen levels only change a little as anti-VEGF antibody dose is increased, indicating that as the tumor shrinks, the degree of vascularity decreases proportionally. In contrast, at high doses of anti-VEGFR1 antibody, the degree of tumor oxygenation increases since the treatment only blocks the action of VEGFR1, and vessel formation and survival are mediated via VEGFR2 signaling in ECs. When anti-VEGFR2 therapy is administered, the results are counterintuitive. At low doses, this treatment actually results in larger tumors as compared to the control, even though there is significant vascular disruption as evidenced by a sharp fall in intratumoral oxygen levels. This is because targeting VEGFR2 has the dual effect of inducing vessel degradation and making more VEGF available to bind and activate VEGFR1 on tumor cells. The situation is further exacerbated since the resulting hypoxia stimulates tumor cells to ramp up the production of VEGF. The therapy is only effective when oxygen levels fall to the quiescent threshold \(N_\mathrm{p}\) for tumor cells, and their proliferation is consequently inhibited. From a clinical view point, these results are undesirable because, not only does the tumor increase in size, it is more hypoxic and therefore probably resistant to alternative therapeutic interventions. The high doses of anti-VEGFR2 required to inhibit tumor growth might be unfeasible due to potential side effects.

Tumor growth response to anti-VEGF/anti-VEGFR1/anti-VEGFR2 treatments given alone or in combination. a Time courses of tumor volume. b Time courses of oxygen partial pressure. c The degree of tumor growth inhibition at the end of the treatment period, and change in tumor volume over the following period off treatment for each possible combination. d The change in tumor oxygenation at the end of the treatment period and at the end of the following period off treatment for each possible combination. The treatment period is highlighted with gray shading
We next investigate the potential of combining the drugs in pairs or giving them all simultaneously, with doses of each drug fixed at their 50% inhibition levels. The predicted time courses of tumor volume and oxygen partial pressure are plotted in Fig. 7a, b, respectively. In terms of inhibiting tumor growth, combining anti-VEGF and anti-VEGFR1 antibodies is the most effective and results in the most well-vascularized tumor. In all cases where anti-VEGFR2 antibody is given, the resulting tumors are more hypoxic. This results in VEGF expression by tumor cells and accelerated growth at the end of the treatment period, even though during treatment these tumors have the slowest growth rate.
Figure 7c, d further underscores these results. The bar graphs in Fig. 7c show the degree of tumor growth inhibition at the end of the treatment period versus control and the subsequent change in tumor volume from day 28 to day 49 (period off treatment). The bar graphs in Fig. 7d show corresponding changes in tumor oxygen levels. As indicated above, combining anti-VEGF and anti-VEGFR1 antibodies is the most effective strategy, resulting in a high level of tumor growth inhibition post-therapy and, crucially, continued growth inhibition even after therapy cessation. This is partly because it is the only combination that results in increased oxygenation of the tumor during both on and off treatment periods. Since the tumor is well supplied with blood vessels, VEGF production is switched off and tumor cells cease proliferation. We remark that if the dependence of tumor cells on VEGFR1 signaling was reduced, tumor growth, rather than growth cessation, might occur in the periods off treatment; however, targeting VEGF and VEGFR1 simultaneously would still be expected to induce maximum growth control. On the other hand, combining anti-VEGFR2 antibody with the other antibodies appears to have an antagonistic effect. Even though tumor growth is most inhibited at the end of treatment in these cases, the resulting tumors are hypoxic and experience accelerated growth in the periods off therapy.

Phase diagrams showing the amounts of: a anti-VEGF and anti-VEGFR1 antibodies. b Anti-VEGF and anti-VEGFR2 antibodies. c Anti-VEGFR1 and anti-VEGFR2 antibodies required to achieve 50% growth inhibition of the tumor after 49 days. For comparison, a curve showing combinations if the drugs were purely additive in action (red dashes) is plotted in each case. d Comparison of tumor response to treatment with increasing doses of anti-VEGF antibody when the tumor cells are assumed to be highly dependent on VEGFR1 signaling (blue curve) versus when they are relatively independent of VEGFR1 signaling (red curve), for survival
To further quantify any synergy or antagonism between the various treatments, an additional set of simulations are conducted, and the results are shown in Fig. 8a–c. These are phase diagrams showing the amounts of antibodies, when given as a combination of two drugs, that need to be co-administered in order to achieve a 50% growth inhibition of tumor xenografts at the end of 49 days. The actions of anti-VEGF and anti-VEGFR1 antibodies appear to be additive (Fig. 8a). However, as indicated in Fig. 7, targeting VEGFR2 is highly antagonistic in combination with either of the other antibodies. Model simulations predict that while anti-VEGF and anti-VEGFR2 antibodies show the most antagonism at low doses of anti-VEGFR2 antibody (Fig. 8b), this trend is reversed when it is combined with anti-VEGFR1 antibody (Fig. 8c).
Finally, we include a simulation of the case when the tumor cells are less dependent on VEGFR1-signaling for survival (Fig. 8d). Specifically, the maximum death rate and the fractional occupancy of VEGFR1 at which Bcl-2 mRNA expression is half its maximum value are reduced by about two-thirds and the effect of treating the tumor with increasing doses of anti-VEGF antibody is recorded. As the drug dose is increased, tumor volumes go down; however, increasing the dosage beyond a certain level has the counterproductive effect of boosting tumor growth. This is because inhibiting VEGF induces endothelial cell death and vessel degradation, and the resulting hypoxia, combined with tumor insensitivity to VEGFR1 signaling, results in reduced tumor growth inhibition. These results, which highlight the importance of tumor cell phenotype (sensitivity to VEGFR1 signaling) on therapeutic outcome, might explain the failure of some anti-VEGF strategies at the clinical level.
4 Discussion
In order to improve patient outcomes for aggressive cancers, an increasing amount of research is now being aimed at understanding the molecular biology of tumor growth, angiogenesis and invasion in an attempt to selectively target pathways involved in tumor progression. Improved understanding of molecular mechanisms associated with tumor angiogenesis and the bidirectional cellular cross-talk between tumor cells and vascular endothelial cells is leading to targeted manipulation of intracellular signaling pathways mediated by VEGF and its receptors. These ligand- and receptor-targeted therapies can, in theory, be administered alone or in combination to achieve maximal anti-tumor effect. Although the significance of VEGF as a conceptual target for cancer treatment is unquestionable, we still do not fully understand how anti-VEGF therapies work in vivo. For example, we know that decreasing VEGF availability to endothelial cells should result in reduced vasculature and tumor burden. However, for certain tumor phenotypes, it is plausible that too much vascular disruption could actually result in more VEGF being produced by, and being made available to, cancer cells, ultimately leading to increased tumor growth as predicted by the numerical simulations in Fig. 6.
Before novel drugs targeted against VEGF and its receptors become uniformly successful as single modality anticancer therapies or are optimized in conjunction with existing chemotherapies, key questions must be answered including: What causes the observed anti-tumor effects and which tumor phenotypes are likely to show the greatest benefit from treatments that disrupt the vasculature? In order to gain insight into these important issues, we developed an adaptable, multilevel modeling framework describing VEGF-mediated cross-talk between tumor cells and vascular endothelial cells both in vitro and in vivo. The model describes the intracellular-, cellular-, and tissue-level response of tumors to combination treatments involving VEGF and its natural receptors. We used the model to rigorously investigate the differential anti-angiogenic and anti-tumor impacts on tumor reduction.
In vitro results for tumor cells co-cultured with control endothelial cells—the closest to the in vivo situation—suggest that the most promising strategies for tumor reduction are those that involve blocking the activation of VEGFR1. Blocking VEGFR2 alone boosted tumor cell numbers because endothelial cell uptake of VEGF was reduced, allowing more of it to be available for binding and activating VEGFR1 on tumor cells. Blocking VEGFR2 in combination with any of the other antibodies had a synergistic effect and inhibited tumor cell growth. Upon developing a version of the model that simulated vascular tumor growth and treatment, we found that in vitro results do not always translate to the in vivo situation. For instance, although targeting VEGFR1 remained the best option therapeutically, combination therapies that include an anti-VEGFR2 antibody were antagonistic and resulted in accelerated tumor growth at the end of treatment cycles due to induction of hypoxia. This antagonism was not present in vitro and highlights the limitations of interpreting in vitro results in a clinical setting. The most effective strategy was the co-inhibition of VEGF and VEGFR1, which induced the maximum level of tumor growth control, coupled with high vascularity within the tumors. This result suggests that such a treatment may be followed up with chemotherapies, which could be more effective due to better drug delivery. Conversely, targeting VEGFR2 in all cases resulted in hypoxic tumors that may be resistant to other forms of chemotherapy. Finally, all of these results must be interpreted while keeping in mind that the tumor cells were assumed to be highly dependent on VEGFR1 signaling for survival. We also included simulations which demonstrated that targeting VEGF may be a counterproductive strategy if this assumption is relaxed, which may partially explain the varying levels of success that anti-VEGF therapies have had in clinical settings.
The in vivo model is presented here as a proof of concept for the overall modeling strategy. Our future work will focus on relaxing many of the simplifying assumptions associated with the current model. For instance, a more biologically realistic ordinary differential equation (ODE) version of the model would be achieved by deriving ODEs for microvascular fraction and oxygen supply to the tumor. This will allow us to simulate drug delivery to the tumor more accurately. We will also explicitly track amounts of Bcl-2 protein (and other members of the Bcl family) rather than approximating its effect on cell death via relative mRNA expression level. We also plan to extend the essential features of this model into a spatiotemporal framework that will be able to mechanistically describe angiogenesis-dependent vascular tumor growth with the level of detail required to accurately predict the therapeutic potential of molecular targets that can affect multiple cell types. The spatial model will allow us to include blood flow, which is critical for blood vessel remodeling and nutrient transfer during tumor progression. We will also be able to capture tumor morphology (e.g., avascular regions) and its effect on oxygenation and drug delivery. This type of model can be used to quantify the influence of the spatially evolving tumor vasculature on treatment outcomes. Although the conclusions and comparisons presented here are based solely on the simplified in vivo model, our future modeling efforts described above will have the potential to make additional therapeutically relevant predictions and further distinguish in vitro versus in vivo tumor response.
Continued modeling efforts in this direction have the potential to shed light on the experimentally observed phenomenon that anti-angiogenic therapy aimed at VEGFR2 initially improves both the structure and function of tumor vessels; however, continued or aggressive regimes result in a vascular supply that is inadequate to support delivery of additional therapeutic agents (Goel et al. 2012; Lin and Sessa 2004). Also, avastin, an agonist for VEGF with no effect on VEGFR2, made the headlines with both positive and negative clinical progress (Honey 2009). It was approved by the FDA for use as a treatment for glioblastoma that has recurred after standard therapies several years ago, even though, around the same time, a clinical trial assessing the ability of avastin to reduce the risk of tumor recurrence in individuals with early-stage colon cancer failed to reach its endpoint (Honey 2009). Considering cancers more broadly, it is clear that anti-angiogenic therapy prolongs survival on the order of months in some settings while failing to induce any survival benefit at all in others (Loges et al. 2010; Moserle et al. 2014). We expect to be able to use our modified model to further the understanding of why manipulating VEGF is successful for some tumor types but disappointing in others and to suggest which cancer cell phenotypes will experience the greatest benefit from therapies targeted against VEGF.
References
Botelho F, Pina F, Lunet N (2010) VEGF and prostatic cancer: a systematic review. Eur J Cancer Prev 19(5):385–392
Byrne HM (2010) Dissecting cancer through mathematics: from the cell to the animal model. Nat Rev Cancer 10(3):221–230
Carreau A, El Hafny-Rahbi B, Matejuk A, Grillon C, Kieda C (2011) Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J Cell Mol Med 15(6):1239–1253
Ciric C, Ciffroy P, Charles S (2012) Use of sensitivity analysis to identify influential and non-influential parameters within an aquatic ecosystem model. Ecol Model 246:119–130
Cunningham SA, Tran TM, Arrate MP, Brock TA (1999) Characterization of vascular endothelial cell growth factor interactions with the kinase insert domain-containing receptor tyrosine kinase: a real time kinetic study. J Biol Chem 274(26):18421–18427
Del Monte U (2009) Does the cell number 10(9) still really fit one gram of tumor tissue? Cell Cycle 8(3):505–506
Ellis LM, Hicklin DJ (2008) VEGF-targeted therapy: mechanisms of anti-tumor activity. Nat Rev Cancer 8:579–591
Ferrara N (2002) VEGF and the quest for tumor angiogenesis factors. Nat Rev Cancer 2:795–803
Ferrara N, Gerber HP, LeCouter J (2003) The biology of VEGF and its receptors. Nat Med 9(6):669–676
Fujita K, Sano D, Kimura M, Yamashita Y, Kawakami M, Ishiguro Y et al (2007) Anti-tumor effects of bevacizumab in combination with paclitaxel on head and neck squamous cell carcinoma. Oncol Rep 18(1):47–51
Gasparini G (2000) Prognostic value of vascular endothelial growth factor in breast cancer. Oncologist 5(Suppl 1):37–44
Goel S, Wong AH-K, Jain RK (2012) Vascular normalization as a therapeutic strategy for malignant and nonmalignant disease. Cold Spring Harb Perspect Med 2(3):a006486
Gotink KJ1, Verheul HM (2010) Anti-angiogenic tyrosine kinase inhibitors: what is their mechanism of action? Angiogenesis 13(1):1–14
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674
Holash J, Davis S, Papadopoulos N, Croll SD, Ho L, Russell M et al (2002) VEGF-Trap: a VEGF blocker with potent antitumor effects. Proc Natl Acad Sci USA 99:11393–11398
Honey K (2009) Good and bad news for an antiangiogenic therapy. J Clin Investig 119(6):1400
Jain HV, Nor JE, Jackson TL (2008) Modeling the VEGF-Bcl-2-CXCL8 pathway in intratumoral angiogenesis. Bull Math Biol 70(1):89–117
Jain HV, Nor JE, Jackson TL (2009) Quantification of endothelial cell-targeted anti-Bcl-2 therapy and its suppression of tumor growth and vascularization. Mol Cancer Ther 8(1):2926–2936
Jubb AM, Pham TQ, Hanby AM, Frantz GD, Peale FV et al (2004) Expression of vascular endothelial growth factor, hypoxia inducible factor 1\(\alpha \), and carbonic anhydrase IX in human tumours. J Clin Pathol 57:504–512
Kaneko T, Zhang Z, Mantellini MG, Karl E, Zeitlin B, Verhaegen M et al (2007) Bcl-2 orchestrates a cross-talk between endothelial and tumor cells that promotes tumor growth. Cancer Res 67(20):9685–9693
Kendrew J, Eberlein C, Hedberg B, Smith NR et al (2011) An antibody targeted to VEGFR-2 Ig domains 4-7 inhibits VEGFR-2 activation and VEGFR-2-dependent angiogenesis without affecting ligand binding. Mol Cancer Ther 10(5):770–783
Kim KJ, Li B, Winer J, Armanini M, Gillett N, Phillips HS et al (1993) Inhibition of vascular endothelial growth factor-induced angiogenesis sup-presses tumour growth in vivo. Nature 362:841–844
King J, Hamil T, Creighton J, Wu S, Bhat P, McDonald F et al (2004) Structural and functional characteristics of lung macro- and microvascular endothelial cell phenotypes. Microvasc Res 67(2):139–151
Kohn-Luque, de Back W, Yamaguchi Y, Yoshimura K, Herrero MA, Miura T (2013) Dynamics of VEGF matrix-retention in vascular network patterning. Phys Biol 10(6):066007
Krupitskaya Y, Wakelee HA (2009) Ramucirumab, a fully human mAb to the transmembrane signaling tyrosine kinase VEGFR-2 for the potential treatment of cancer. Curr Opin Investig Drugs 10:597–605
Lin YS, Nguyen C, Mendoza JL, Escandon E, Fei D, Meng YG et al (1999) Preclinical pharmacokinetics, interspecies scaling, and tissue distribution of a humanized monoclonal antibody against vascular endothelial growth factor. J Pharmacol Exp Ther 288(1):371–378
Lin MI, Sessa WC (2004) Antiangiogenic therapy: creating a unique “window” of opportunity. Cancer Cell 6(6):529–531
Loges S, Schmidt T, Carmeliet P (2010) Mechanisms of resistance to anti-angiogenic therapy and development of third-generation anti-angiogenic drug candidates. Genes Cancer 1(1):12–25
Lu D, Shen J, Vil MD, Zhang H, Jimenez X, Bohlen P et al (2003) Tailoring in vitro selection for a picomolar affinity human antibody directed against vascular endothelial growth factor receptor 2 for enhanced neutraliz- ing activity. J Biol Chem 278:43496–43507
Mac Gabhann F, Yang MT, Popel AS (2005) Monte Carlo simulations of VEGF binding to cell surface receptors in vitro. Biochim Biophys Acta 1746(2):95–107
Mac Gabhann F, Popel AS (2007) Dimerization of VEGF receptors and implications for signal transduction: a computational study. Biophys Chem 128:125–139
Mac Gabhann F, Popel AS (2007) Interactions of VEGF isoforms with VEGFR-1, VEGFR-2, and neuropilin in vivo: a computational model of human skeletal muscle. Am J Physiol Heart Circ Physiol 292(1):H459–74
McMahon G (2000) VEGF receptor signaling in tumor angiogenesis. Oncologist 5(Suppl 1):3–10
Morris MD (1991) Factorial sampling plans for preliminary computational experiments. Technometrics 33:161–174
Moserle L, Jimenez-Valerio G, Casanovas O (2014) Antiangiogenic therapies: going beyond their limits. Cancer Discov 4(1):31–41
Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z (1999) Vascular endothelial growth factor (VEGF) and its receptors. FASEB J 13(1):9–22
Nor JE, Christensen J, Mooney DJ, Polverini PJ (1999) Vascular endothelial growth factor (VEGF)-mediated angiogenesis is associated with enhanced endothelial cell survival and induction of Bcl-2 expression. Am J Pathol 154(2):375–384
Papadopoulos N, Martin J, Ruan Q, Rafique A, Rosconi MP, Shi E et al (2012) Binding and neutralization of vascular endothelial growth factor (VEGF) and related ligands by VEGF Trap, ranibizumab and bevacizumab. Angiogenesis 15(2):171–185
Peirce SM (2008) Computational and mathematical modeling of angiogenesis. Microcirculation 15(8):739–751
Perfahl H, Byrne HM, Chen T, Estrella V, Alarcon T, Lapin A et al (2011) Multiscale modelling of vascular tumour growth in 3D: the roles of domain size and boundary conditions. PLoS One 6(4):e14790
Pianosi F, Sarrazin F, Wagener T (2015) A matlab toolbox for global sensitivity analysis. Environ Model Softw 70:80–85
Reed JC, Tsujimoto Y, Epstein SF, Cuddy M, Slabiak T, Nowell PC et al (1989) Regulation of bcl-2 gene expression in lymphoid cell lines containing normal #18 or t(14;18) chromosomes. Oncogene Res 4(4):271–282
Shweiki D, Neeman M, Itin A, Keshet E (1995) Induction of vascular endothelial growth factor expression by hypoxia and by glucose deficiency in multicell spheroids: Implications for tumor angiogenesis. Proc Natl Acad Sci USA 92(3):768–772
Siemann DW (2011) The unique characteristics of tumor vasculature and preclinical evidence for its selective disruption by tumor vascular disrupting agents. Cancer Treat Rev 37:63–74
Spratlin JL, Cohen RB, Eadens M, Gore L, Camidge DR, Diab S et al (2010) Phase I pharmacologic and biologic study of ramucirumab (IMC-1121B), a fully human immunoglobulin G1 monoclonal antibody targeting the vascular endothelial growth factor receptor-2. J Clin Oncol 28(5):780–787
Szabo E, Schneider H, Seystahl K, Rushing EJ, Herting F, Weidner KM, Weller M (2016) Autocrine VEGFR1 and VEGFR2 signaling promotes survival in human glioblastoma models in vitro and in vivo. Neuro Oncol 18(9):1242–1252
Tong M, Lloyd B, Pei P, Mallery SR (2008) Human head and neck squamous cell carcinoma cells are both targets and effectors for the angiogenic cytokine. VEGF J Cell Biochem 105(5):1202–1210
Van Tubergen E, Vander Broek R, Lee J, Wolf G, Carey T, Bradford C et al (2011) Tristetraprolin regulates interleukin-6, which is correlated with tumor progression in patients with head and neck squamous cell carcinoma. Cancer 117(12):2677–2689
von Tiedemann B, Bilitewski U (2002) Characterization of the vascular endothelial growth factor-receptor interaction and determination of the recombinant protein by an optical receptor sensor. Biosens Bioelectron 17(11–12):983–991
Wang D, Lehman RE, Donner DB, Matli MR, Warren RS, Welton ML (2002) Expression and endocytosis of VEGF and its receptors in human colonic vascular endothelial cells. Am J Physiol Gastrointest Liver Physiol 282(6):G1088–1096
Wang Z, Butner JD, Kerketta R, Cristini V, Deisboeck TS (2015) Simulating cancer growth with multiscale agent-based modeling. Semin Cancer Biol 30:70–78
Wood JM, Bold G, Buchdunger E, Cozens R, Ferrari S, Frei J et al (2000) PTK787/ZK 222584, a novel and potent inhibitor of vascular endothelial growth factor receptor tyrosine kinases, impairs vascular endothelial growth factor-induced responses and tumor growth after oral administration. Cancer Res 60:2178–2189
Wu Y, Hooper AT, Zhong Z, Witte L, Bohlen P, Rafii S, Hicklin DJ (2006) The vascular endothelial growth factor receptor (VEGFR-1) supports growth and survival of human breast carcinoma. Int J Cancer 1:119(7):1519–1529
Yao J, Wu X, Zhuang G et al (2011) Expression of a functional VEGFR-1 in tumor cells is a major determinant of anti-PlGF antibodies efficacy. PNAS 108(28):11590–11595
Yen P, Finley SD, Engel-Stefanini MO, Popel AS (2011) A two-compartment model of VEGF distribution in the mouse. PLoS One 6(11):e27514
Author information
Authors and Affiliations
Corresponding author
Appendix
Appendix
1.1 In Vitro Treatment Equations
The complete set of model equations used for simulating in vitro, pre-treatment tumor growth is described in the main text. For the targeted therapies we are considering, the tumor cell, endothelial cell and Bcl-2 equations do not change. Below we describe the modifications to the model that arise due to treatment.
VEGF-Binding Equations: The equations below describe VEGF binding, and they are identical to those presented in the main text, except for the addition of the last two terms in Eqs. (20)–(23), which represent the binding dynamics of anti-VEGF, VEGFR1 or VEGFR2 antibodies.
VEGF, VEGFR1, VEGFR2 Therapy Equations: The binding dynamics for an anti-VEGF antibody (\(X_V\)), an anti-VEGFR1 antibody (\(X_{R1}\)) and an anti-VEGFR2 antibody (\(X_{R2}\)) are described below:
1.2 In Vivo Treatment Equations
The model equations used for simulating tumor cells, endothelial cells and drug pharmacokinetics for anti-VEGF therapy in vivo are described in detail in the main text. Below we present the remaining model equations associated with treatment for the in vivo situation.
Intratumoral VEGF: Intratumoral VEGF (A) is governed by Eq. (34). The first eight terms of the equation, representing binding, decay and production, are described in detail in the main text. The last terms in the VEGF equation represent the response to therapies that target the VEGF.
VEGFR1 and VEGFR2: The equations representing VEGF receptor dynamics on ECs and TCs remain also largely unchanged from the in vitro case described in the main text. The only addition is the therapeutic binding of VEGFR1 and VEGFR2 antibodies, where appropriate. The first three terms in each of the equations below represent the response to VEGF of each variable (\(R_i\) for \(i = 1, 2 \equiv \) free VEGFR1/VEGFR2 receptors on ECs and/or TCs, and \(D_i\) for \(i = 1,2 \equiv \) VEGF–VEGFR\(_i\) complexes on ECs and TCs). Note that in addition to binding and releasing VEGF, the rate of change of each receptor species is also affected by the creation of new unbound receptors as a result of cell division and the removal of receptors and complexes due to cell death. This ensures conservation of receptors and is described by the fourth term in the \(D_i\) equations and the fourth and fifth terms in the \(R_i\) equations. The last terms in each of the \(R_i\) equations represent the response to therapies that target the VEGF receptors.
1.2.1 Intracellular Bcl-2
Bcl-2 mRNA is constitutively expressed within both cell types and undergoes natural degradation. Activation of its receptors by VEGF leads to additional Bcl-2 synthesis in each cell type. Combining these processes, we obtain the following equations for Bcl-2 mRNA expression in ECs and TCs, as described in the main text:
1.2.2 Molecular-Level Variables Associated with Treatment
Below, we present the model equations for the effects of an anti-VEGF antibody, like avastin, an anti-VEGFR1 antibody-like IMC-18F1, and an anti-VEGFR2 antibody.
Drug Pharmacokinetics: Experimental evidence suggests a biphasic plasma concentration–time curve for avastin Lin et al. (1999). Consequently, a two-compartment model is proposed to govern the pharmacokinetics of all of the drugs we consider. Systemic circulation (together with well-vascularized organs) is taken to be the first compartment from where the drug may be eliminated or enter, in a reversible process, a second compartment which consists of tissues and organs with poor vascular perfusion. Combining these effects, the equations governing drug pharmacokinetics can be written as follows:
where \(X_{is}\) and \(X_{ip}\) (\(i = V, R1, R2\) for anti-VEGF, anti-VEGFR1, and anti-VEGFR2 therapy) represent the amount of drug in systemic circulation and the peripheral compartment, respectively. The dosing function in Eq. (43) represents the periodic administration of avastin and is taken to have the following form:
where \(D_0\) is the amount of drug delivered in each dose; \(t_i\), the timing of each dose; \(t_a\), the length of each injection; and n, the total number of doses.
Intratumoral Drug Dynamics: The tumor is assumed to reside in a pharmacokinetic compartment of its own. For simplicity, the rates of transfer of drug into and out of the tumor are taken to be the same as those for the peripheral compartment [Eq. (44)]. Further, the tumor is assumed to occupy a negligible volume compared to the volume of the animal; therefore, the extravasation of drug from the tumor into systemic circulation will not affect the amount of drug in the central compartment. Finally, avastin–VEGF complexes are assumed to undergo the same degradation as free VEGF in tissue. Combined together, these processes yield the following equations governing drug dynamics in the tumor space. Recall, \(X_{i}\) (\(i = V, R1, R2\) for the anti-VEGF, anti-VEGFR1 and anti-VEGFR2 therapy) represent free forms of each drug and \(C_{j}\) (\(j = AV, E1R1, T1R1, E2R2\)) represent the VEGFR1–anti-VEGFR1 antibody complexes on ECs/TCs and the VEGFR2–anti-VEGFR2 antibody complexes on ECs.
1.2.3 Receptor Conservation
In the above equations, \(R_{Ei}^t\), \(i=1,2\) and \(R_{T1}^t\) represent the VEGFRi and VEGFR1 receptor expression levels on endothelial and tumor cells, respectively. From equations (13), (35), (38) and (49), it follows that \(R_{E1}^tE \,=\, R_{E1}\,+\,D_{E1} \,+\, C_{E1R1}\), that is, the number of VEGFR1 per endothelial cell is conserved. Likewise, \(R_{E2}^tE \,=\, R_{E2}\,+\,D_{E2} \,+\, C_{E2R2}\) and \(R_{T1}^tT \,=\, R_{T1}\,+\,D_{T1} \,+\, C_{T1R1}\).
1.3 Parameters
A list of parameter values estimated from the literature for the in vitro and in vivo treatment models is provided in Table 5. We remark that the rates of forward reaction are typically expressed in units of “per time, per concentration.” However, in our model, all chemical species are expressed in terms of total quantity (fmol in the in vitro case and pmol in the in vivo case). Therefore, to preserve dimensional accuracy, the rates of forward reaction listed in Table 4 (and Table 1 of the main text) have been scaled by the volume of the experiment being simulated. For instance, the rate of forward reaction between avastin and VEGF has been determined experimentally to be 2.9794 (fmol/ml)\(^{-1}\) day\(^{-1}\) (or (pmol/\(\upmu \)l)\(^{-1}\) day\(^{-1}\)). Consequently, \(k_{VAb}^f\) is taken to be 2.9794/Vol\(_C\) fmol\(^{-1}\) day\(^{-1}\) when simulating in vitro experiments and 2.9794/Vol\(_T\) pmol\(^{-1}\) day\(^{-1}\) in the in vivo case. Here, \(Vol_C\) is the volume of the cell culture experiments in ml and \(\hbox {Vol}_T\), the volume of the tumor in \(\mu \)l. Note that \(Vol_T\) \(=\) (volume of 1 million ECs)\(\times E\) \(+\) (volume of 1 million TCs)\(\times T\) and the volume of 1 EC is 2.2\(\times 10^{-6}\) \(\mu \)l King et al. (2004) and that of 1 TC is 1\(\times 10^{-6}\) \(\mu \)l Monte (2009).
A list of parameter values for the in vivo model is provided in Table 6.
The best fit of the in vivo treatment model to avastin pharmacokinetic data reported in Lin et al. (1999) is shown in Fig. 9.

Model fit to avastin pharmacokinetic data reported in Lin et al. (1999)
1.4 Parameter Sensitivity
A local sensitivity analysis is carried out by varying the parameters associated with the vascular growth and response to treatment of in vivo tumor xenografts. These include: the VEGF-dependent rate of tumor cell proliferation, \(\nu _T\); the sensitivity of tumor cell proliferation to VEGFR1 receptor occupancy, \(\alpha _T\); the Bcl-2-modulated rate of tumor cell death, \(\delta _T\); the sensitivity of tumor cell death to Bcl-2 expression, \(\beta _T\); the VEGF-dependent rate of endothelial cell proliferation, \(\mu _E\); the sensitivity of endothelial cell proliferation to VEGFR2 receptor occupancy, \(\alpha _E\); the Bcl-2-modulated rate of endothelial cell death, \(\delta _E\); the sensitivity of endothelial cell death to Bcl-2 expression, \(\beta _E\); the VEGF expression rate of tumor cells, \(\nu _A\); and the sensitivity of tumor cells to hypoxia, \(\kappa _A\) [see equations (12)–(14) in the main text]. The two cases—control (no treatment) and treatment—are considered separately. In each case, the residual between simulated and experimental data (tumor volume time course) is computed as each parameter is varied from its baseline estimate, and the resulting residual is plotted as a function of the percent change in the value of parameter being varied. Given that vascular tumor growth is driven by hypoxia in our model, we also test the sensitivity of tumor oxygenation to the various parameters by plotting the maximum partial pressure of oxygen as a function of the percent change in the value of parameter being varied.
Local Sensitivity Analysis–Control
Figure 10a, c, e reveal that the model predictions of tumor xenograft size are sensitive to endothelial and tumor cell growth parameters, tumor cell death parameters, as well as tumor cell VEGF expression and sensitivity to hypoxia, with low values of \(\alpha _T\) and high values of \(\nu _A\) and \(\nu _T\) resulting in the most change. Since these simulations are without treatment, tumor xenograft size is predictably insensitive to parameters relating to endothelial cell death and tumor cell death. Figure 10b, d, f reveal that tumor oxygenation is most sensitive to high values of \(\mu _E\) and \(\alpha _T\) and low values of \(\alpha _E\), \(\nu _T\) and \(\nu _A\). Once again, cell death has little impact on tumor oxygenation.

Local sensitivity analysis for parameters relating to tumor xenograft growth in the absence of treatment. a, c, e The residual or Euclidean norm between simulated and experimental data (tumor volume time course, see Fig. 6a in main text) is plotted on the y-axis, and percentage variation of the parameters from their baseline values is plotted on the x-axis. b, d, f The predicted maximum partial pressure in the tumor over the time course of the simulation is plotted on the y-axis, and percentage variation of the parameters from their baseline values is plotted on the x-axis
Local Sensitivity Analysis–Treatment
Figure 11a, c, e reveal that the model predictions of tumor xenograft size under avastin therapy are sensitive to endothelial and tumor cell growth parameters, as well as tumor cell VEGF expression and sensitivity to hypoxia, with low values of \(\alpha _T\) and high values of \(\nu _A\) and \(\nu _T\) resulting in the most change. VEGF expression level per tumor cell (\(\nu _A\)) now emerges as the most influential parameter. Interestingly, although the simulations are sensitive to tumor cell death parameters, cellular proliferation rates are still more influential in comparison, indicating that the inhibition of cell proliferation—as opposed to inducing cell death—might be what drives the observed changes in xenograft volume in response to treatment with avastin. Figure 11b, d, f reveal that tumor oxygenation is sensitive to high values of \(\mu _E\), \(\alpha _T\), \(\delta _T\) and \(\beta _T\) and low values of \(\alpha _E\), \(\nu _T\) and \(\nu _A\). In contrast to the earlier case, the tumor cell death rate \(\delta _T\) emerges as the most influential parameter since increased tumor cell death could have the overall effect of increasing tumor oxygen levels.

Local sensitivity analysis for parameters relating to tumor xenograft growth under treatment with avastin. a, c, e The residual or Euclidean norm between simulated and experimental data (tumor volume time course, see Fig. 6a in main text) is plotted on the y-axis, and percentage variation of the parameters from their baseline values is plotted on the x-axis. b, d, f The predicted maximum partial pressure in the tumor over the time course of the simulation is plotted on the y-axis, and percentage variation of the parameters from their baseline values is plotted on the x-axis
Global Sensitivity Analysis
Although the above analysis provides some information on which parameters are more influential in our model, this analysis is local, with each parameter varied individually, keeping all others fixed. This approach suffers from the limitation that only a small fraction of parameter space is explored and does not take into account the simultaneous variation, and thus possible interactions, of model parameters. We therefore also conduct a global sensitivity analysis on the same set of parameters using the elementary effects method developed by Morris (1991) and implemented in MATLAB by Pianosi et al. (2015). An elementary effect is computed as follows. Consider a model with n independent input parameters \(x_i\), \(i=1,\ldots ,n\) with each parameter uncertainty interval divided into p equal intervals resulting in an n-dimensional p-level hyperspace. For any given set of parameter values, the elementary effect of the ith input factor on the output y is defined as:

Global sensitivity analysis for parameters relating to tumor xenograft growth in the absence of treatment. a Phase plane showing the mean versus standard deviation of elementary effects (EEs). b Bar graphs showing the corresponding global sensitivity indices for the various parameters when the output function is taken to be the residual or Euclidean norm between simulated and experimental data (tumor volume time course, see Fig. 6a in main text). c Phase plane showing the mean versus standard deviation of elementary effects (EEs). d Bar graphs showing the corresponding global sensitivity indices for the various parameters when the output function is taken to be the maximum partial pressure in the tumor over the time course of the simulation
where \(\Delta = p/(2(p-1))\). The distribution of elementary effects associated with \(x_i\) is then obtained by randomly sampling r different sets of parameters from the prescribed hyperspace. Thus, the Morris method depends not only on p, but also on the sampling number r. The sensitivity measures proposed by Morris are \(\mu _i\) and \(\sigma _i\)—the mean and the standard deviation, respectively, of the distribution of elementary effects for each \(x_i\). The mean \(\mu _i\) assesses the overall influence of one parameter on the output while the standard deviation \(\sigma _i\) assesses the parameter’s higher-order effects, such as interactions with other parameters or nonlinear effects on the output. For instance, a low value of \(\sigma _i\) indicates that the effect of \(x_i\) is almost independent of the values taken by the other factors. Elementary effects are typically represented graphically in the \((\mu _i,\sigma _i)\) plane, and the most influential parameters appear on the upper right corner.
An alternative and more quantitative measure to determine a parameter \(x_i\)’s sensitivity is its global index (Ciric et al. 2012), defined as:
Figure 12a, b reveal that in the control case, the output y—defined as the residual between simulated and experimental data (tumor volume time course)—is most sensitive to \(\alpha _T\) (tumor cell proliferation rate sensitivity to activated VEGFR1) and \(\mu _E\) (activated VEGFR2-mediated endothelial cell proliferation rate), followed by \(\nu _T\) (activated VEGFR1-mediated tumor cell proliferation rate), \(\nu _A\) (tumor cell VEGF expression level) and \(\alpha _E\) (endothelial cell proliferation rate sensitivity to activated VEGFR2). However, as shown in Fig. 12c, d, when the output is chosen as the maximum level of tumor oxygenation, the most influential parameters emerge as \(\alpha _T\) and \(\nu _A\), followed by \(\delta _T\) (tumor cell death rate), \(\alpha _E\) and \(\mu _E\). We remark that \(\delta _T\) has a high global sensitivity index even though locally the model is insensitive to it.
In the treatment case, Fig. 13a, b reveal that when the output is taken as the residual between simulated and experimental data, the most influential parameters remain \(\alpha _T\), \(\mu _E\) and \(\nu _T\) followed by \(\nu _A\) and \(\alpha _E\). However, as shown in Fig. 13c, d, when the output is chosen as the maximum level of tumor oxygenation, the most influential parameters emerge as \(\alpha _T\), \(\delta _T\) and \(\alpha _E\), followed by \(\nu _A\), \(\mu _E\), \(\nu _T\) and \(\delta _E\).

Global sensitivity analysis for parameters relating to tumor xenograft growth under treatment with avastin. a Phase plane showing the mean versus standard deviation of elementary effects (EEs). b Bar graphs showing the corresponding global sensitivity indices for the various parameters when the output function is taken to be the residual or Euclidean norm between simulated and experimental data (tumor volume time course, see Fig. 6a in main text). c Phase plane showing the mean versus standard deviation of elementary effects (EEs). d Bar graphs showing the corresponding global sensitivity indices for the various parameters when the output function is taken to be the maximum partial pressure in the tumor over the time course of the simulation
Rights and permissions
About this article

Cite this article
Jain, H., Jackson, T. Mathematical Modeling of Cellular Cross-Talk Between Endothelial and Tumor Cells Highlights Counterintuitive Effects of VEGF-Targeted Therapies. Bull Math Biol 80, 971–1016 (2018). https://doi.org/10.1007/s11538-017-0273-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11538-017-0273-6