
Abstract
Rheological properties of cell wall suspensions strongly depend on particle size and particle-particle interactions. In the present study, an experimental method was developed to study the effect of particle elasticity and electrostatic interactions on the rheological properties of cell suspensions. Enzymes were used to selectively depolymerize the pectin (backbone) and proteins in suspensions. The enzymatic treatments affected the physical properties, thus a hypothesis for the structure-function relationship of these biopolymers was formulated. The enzymatic treatment directly affected particle properties, resulting in looser cell walls as visualized by cryo-SEM. The effect of the enzymatic treatment on the storage modulus was measured as a function of total solid content (below critical packing fraction). Furthermore, experiments were performed in the presence of varying concentrations of sodium chloride in order to change the Debye screening length. Such method assisted in decoupling the electrostatic effects from particle elasticity. In addition, particle properties were measured directly by applying a compressive strain on the particles and measuring the normal force. By fitting the normal stress relaxation with a Maxwell model, particle properties such as time scale of relaxation and elasticity were obtained. It is suggested that for carrot suspensions, pectins on the cell walls could contribute to the particle hardness. The pectins on carrot cell walls are responsible for electrostatic interactions between particles.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Suspensions of plant material, that are rich in polysaccharides, are widely used in products such as paper, biofuels, foods and bioplastic. Engineering these materials requires an in-depth understanding of how the polysaccharides affect bulk properties. These suspensions are chemically heterogeneous with a variety of particle shapes and sizes. This makes a complete understanding of the structure-property relationship of these systems very challenging. It is known that in these suspensions, there are cellulosic particles suspended in a serum phase of solubilized pectin, proteins and other solutes [1]. There is also sufficient evidence to show that there are complex interactions between either the serum-particle or particle-particle phases of the suspensions. The nature of these interactions could be electrostatic, frictional or elastic, depending on the particular system [2]. Electrostatic interactions in the serum phase are common to occur either between protein-pectin [3, 4] or pectin-pectin compounds [5]. It is not yet completely clear what the relative effect of the particle and serum phases to the rheological properties of the whole suspension is [6]. Particles are also deformable, it has been suggested that the elasticity of plant particles can be important to fully understand the rheological properties of plant based suspensions. On shearing, the particles behave like folded elastic sheets that contribute to the storage modulus [2]. It is clear that there are two possible mechanisms of interaction between plant particles, depending on their concentration in the suspensions. They can interact either by an excluded volume effect from electrostatic interactions or by particle elasticity from particle-particle contact. Both types of interactions strongly affect the physical properties such as storage modulus and yield stress. However, a definite link between the polysaccharides and mechanical properties is not clear. Even a qualitative understanding of these relationships would pave way for further quantitative research in the field.
From the world of botany, there has been extensive research on the elasticity and plasticity of cell walls [7, 8]. Viscoelasticity of the cell wall plays an important role during the growth phase of the plant, as it is related to cell wall elongation. This elongation mechanism is driven by polymer rearrangements within the cell wall over long periods of time which affect the “cell wall viscoelasticity” [9]. It has been suggested that the cellulose fibrils are bound by various polysaccharides present within the cell wall [10]. There are various factors suggested to affect the mechanical properties of the cell wall, including: cross linking of matrix polysaccharides, friction between the polysaccharides and/or non-covalent interaction of polysaccharides. The elasticity of the cell wall can be tuned by either breaking such bonds/interactions or by depolymerizing the backbone of major polysaccharides [7].
In this study, a multidisciplinary approach was used to understand the relative importance of particle hardness and electrostatic interactions on the rheology of cell suspensions. It was also aimed to qualitatively connect the function of pectins and proteins to these properties. Enzymes were used to selectively hydrolyze the polysaccharides. These chemical changes were reflected on the measured physical properties. The enzyme treated samples were investigated using rheology, microscopy and multiple angle laser light scattering measurements. Compression experiments were developed to compare the particle hardness between enzyme treated and untreated carrot cell suspensions. This enabled to decouple the effects of particle hardness from electrostatic effects on the rheology of semi-dilute carrot cell suspensions.
Materials and Methods
The following experiments were performed on different fractions obtained from carrot cell suspensions, shown in Fig. 1.

Schematic of experiments performed for different carrot cell fractions
Sample Preparation
Semi-dilute concentrations of suspensions were prepared as they are relevant to industrial products. Frozen carrots were procured from Ploegmakers Food Ingredients BV (Breda, Netherlands). Carrot cubes of size 1 cm were blanched at 80 ∘C for 2 minutes by the supplier to inactivate endogenous enzyme activity. The cubes were stored and transported at -20 ∘C. Before experiments, the cubes were thawed overnight at room temperature. Thawed carrot cubes were cooked at 90 ∘C for 45 minutes. Cooked carrots were cooled to room temperature in an ice bath. Evaporated water determined by weight loss was replenished with deionized water. The mixture was then blended using an industrial Waring Variable Speed Lab Blender LB20EG 0.40 HP (Waring commercial, Stamford USA) at maximum speed for 5 minutes to obtain a puree of carrot cells. This reference sample was centrifuged at 12000 g and the supernatant was decanted from the pulp. To further remove colloidal particles the supernatant was passed through a 0.45 μm filter. This obtained filtrate (serum phase) was characterized separately.
Enzyme Treatment
Two types of enzymes were used to treat the carrot s namely a protease (Promod 144GL, Biocatalysts Ltd UK) and a pectinase. The pectinase was a mixture of pure endo-polygalacturonase (EC 3.2.1.15, Megazyme International Ireland) and pectin methylesterase (EC 3.1.1.11, Novozyme International Denmark). The mix was made up of ten parts endo-polygalacturonase to one part of pectin methylesterase by volume. The enzyme activity for pure endo-polygalacturonase was 1800 U/ml and pectin methylesterase was 10 U/ml as reported by the suppliers. Both types of enzymes were used at a concentration of 0.1 % w/w in carrot suspensions. All enzymatic treatments were performed at pH 4.5 and a temperature of 45 ∘C for a period of three hours. After enzyme treatment the samples were heated to 90 ∘C for 5 minutes to inactivate the enzymes .
The enzymatically treated samples were centrifuged at 12000 g and the supernatant was decanted from the pulp. To further remove colloidal particles the supernatant was passed through a 0.45 μm filter. The obtained filtrate (serum phase) was characterized separately using MALLS.
Confocal Laser Scanning Microscopy
Confocal laser scanning microscopy (CLSM) was used to visualize the microstructure of the cell wall fragments in the suspensions. The images were taken using a Leica TCS SP5 with a DMI6000 inverted microscope. Congo Red was used for visualization because of its strong affinity with cellulose. The dye was added by gently stirring 1 droplet of Congo Red (0.5 % v/w in water) to 1 ml of sample. A 561 nm laser was used for excitation of the congo red dye. The emitted fluorescence signal was detected by a CCD sensor preceeded by a band pass filter passing light between 565 nm and 680 nm. A 40x objective (NA =1.25; zoom 1) was used to capture images of 1024 x 1024 pixels each. Multiple images were stitched together to give a larger image of approximately 1 mm 2. To enhance clarity the images were converted into grey scale and the contrast was adjusted.
Cryo-Scanning Electron Microscopy
Carrot cells were analyzed using Cryo-Scanning Electron Microscopy (cryo-SEM) to visualize the cellulose structures at a higher resolution, providing information about fibrilar architecture that can not be seen with the confocal microscope. For cryo-SEM a tiny volume of each sample (one droplet) was placed on top of a rivet and plunge-frozen in melting ethane. The sample was cryo-planed using a cryo-ultramicrotome (Leica Ultracut UCT EM FCS), to obtain a freshly prepared cross-section. Cryo-planing was done first with section thickness of 100 nm and a speed of 60 mm/sec using a glass knife. The last sections were made at decreasing thickness, down to 20 nm, with a speed of 2 mm/sec using a diamond knife (Diatome histo cryo 8 mm from DiATOME PA,USA) at -110 ∘C. The rivet was mounted onto a holder and transferred into a Gatan Alto2500 (Gatan Inc, PA,USA) cryo-preparation chamber. To reveal the microstructures under the planed surface, the temperature of the sample was increased for a short while to -90 ∘C in vacuum, to remove a thin layer of water by sublimation. This yielded a semi-3D view on the planed sample. The sample was sputter coated with platinum (120 sec) to improve SEM contrast and to prevent charging by the electron beam. The sample was imaged using a Zeiss Auriga field-emission SEM at -125 ∘C and an accelerating voltage of 3 kV. It is difficult to interpret results from a single electron microscopic image due to heterogeneity and freezing artifacts. Hence, the images shown are representative for the microstructure observed from a set of 20-30 images.
Determination of Molecular Weight Distribution by SEC MALLS-UV-RI
The molecular weight distribution of the polysaccharides in the serum phase was determined using size exclusion chromatography (SEC) coupled to multiangle laser light scattering (MALLS) (PN3621, Postnova analytics, Germany). Simaltaneously the refractive index (RI) was determined (Shodex RI-101, Showa Denko K.K., Kawazaki, Japan) and UV absorption was measured using a diode array detector (G1316A, Agilent technologies, Diegem, Belgium). The serum phase of the carrot suspensions was filtered (0.45 μm) prior to injection. Using an auto-sampler (G1329A, Agilent technologies, Diegem, Belgium) the serum was injected to a series of three Waters columns (Waters, Milford, MA), namely, Ultrahydrogel 250, 1000, and 2000 with exclusion limits of 8 x 104, 4 x 106, and 1 x 107 g/mol, respectively [4]. An eluent was used based on the starting pH of the sample: 0.1 M acetic acid buffer (pH 4.4) with 0.1 M NaNO 3. The eluent was prepared using demineralized water (organic free, 18 M Ω-cm resistivity), filtered (0.1 mm) and degassed by the on-line degasser of the HPLC system (Agilent technologies 1200 Series, Diegem, Belgium). The flow rate was 0.5 ml/min and the columns were kept at 35 0C. Before injection all samples were allowed to equilibrate overnight after filtration. A dn/dc value of 0.146 mL/g was used for all samples, as this value was found to be accurate for both pectins [11] and for starch [12] in different buffers. The molecular weight was calculated using the Debye fitting method (up to 2nd order) of the software provided by the MALLS detector manufacturer (Nova Mals, version 1.0.0.18, Postnova analytics, Germany). The presented elution profiles are averaged chromatograms of two repetitions.
Particle Size Distribution (PSD)
Particle size distribution was measured by conventional laser light diffraction with a Malvern Mastersizer 2000 (Malvern Instruments Ltd, UK). After calibrating the sensors and lasers, a small volume of the sample was loaded into a Hydro 2000S wet dispersion accessory (Malvern Instruments Ltd, UK). The sample was diluted by the water present in the device. To disperse the suspension, an ultrasonication step was first applied for 10 seconds followed by a stirring step at a speed of 3000 rpm for 30 seconds. This suspension was pumped into the diffraction chamber for measuring the particle size distribution of the bulk solution. Samples were measured at least 3 times. Average values of the volume based diameter (D[4,3]) were reported.
Determination of Galacturonic Acid Content
Galacturonic acid content was determined in the serum phase of the carrot suspensions. First the pectin polymers in the serum were hydrolyzed by the addition of concentrated sulphuric acid. To 2 ml of serum solution 8 ml of concentrated H 2 SO 4 was added dropwise while stirring in an ice bath for 5 minutes. 2 ml of deionized water was added to this solution which was then stirred for a period of 60 minutes for hydrolysis by the method of Ahmed and Labavitch [13]. This solution was then filled up to 50 ml in a volume flask. The method for quantitative determination of the uronide fraction was adapted from Blumenkrantz and Asboe-Hansen [14]. 0.6 ml of the above mentioned solution was mixed with 3.6 ml chilled sodium tetraborate solution and transferred to a test tube. This test tube was vortex mixed and immersed in an oil bath of 100 ∘C for 5 minutes. After placing the test tubes in an ice bath, 60 μl of methahydroxydiphenyl reagent was added. A blank sample with only 60 μl of methahydroxydiphenyl was used as reference. The emission at a wavelength of 520 nm was measured using a spectrophotometer. Acid hydrolysis was performed in duplicate and determination of the uronide component was performed in triplicate. For absolute determination of the galacturonic acid content, a standard curve was prepared using monogalacturonic acid.
Kinematic Viscosity of the Serum
A fully automated Ubbelohde viscometer (iVisc Capillary viscometer, Lauda-Brinkman, New Jersey USA) was used to accurately determine the intrinsic viscosity of the serum phase. The capillary ID was 0a with an internal diameter of 0.53 mm, capillary constant of 0.005 mm 2/s 2 and the calculated Hagenbach correction was approximately 1.57 seconds. The capillary was rinsed with deionized water and subsequently cleaned with ethanol/acetone and dried with air before use. The clean capillary with the sample was immersed into a water bath at 25 ∘C. The sample was allowed to equilibrate for 10 minutes. First the fluid was drawn up the glass to wet the glass surface. Subsequently, kinematic viscosity of the sample was evaluated by measuring the time taken for the fluid to drain between the two marked lines on the capillary. Three measurements were performed and the average viscosity was reported.
Small Angle Oscillatory Shear (SAOS)
A stress controlled rheometer (TA Instruments AR-2000ex, USA) with a parallel plate geometry of 40 mm diameter was used for all measurements. Wall slip commonly occurs in two-phase systems due to steric, hydrodynamic, viscoelastic and chemical constraints acting near the boundaries [15]. This wall slip was apparent for the concentrated carrot suspensions using a smooth geometry. To avoid this, the plates were covered with a 800 grit sand paper (3M, USA). In such setup the effects of slip were found to be negligible. For all SAOS measurements the gap between the plates was 2500 μm, being an order of magnitude larger than the mean particle size but small enough to avoid flow effects arising from large gaps. The sample was subjected to a pre-shearing step at a shear rate of 5 1/s for 30 seconds followed by 7 minutes of rest prior to measuring. The storage modulus within the linear viscoelastic region was measured by performing strain sweeps from 0.01-100 % strain at a frequency of 10 rad/sec. Each point in the strain sweep was averaged over 6 cycles of oscillations to ensure good quality data. All measurements were repeated at least three times within each experimental batch and averaged over 2 batches.
Compression Experiments
Compression experiments were performed on the suspensions to qualitatively understand the effect of enzyme treatment on carrot particles. A smooth plate-plate geometry of 40 mm diameter with an initial gap of 1 mm was used for the compression experiments. Due to the slip occurring in such a geometry, the flow is dominated by axial compression whereas shear effects were largely eliminated [16].
After loading the sample into the rheometer, it was pre-conditioned as mentioned in Small Angle Oscillatory Shear (SAOS) section. Subsequently, the top plate was lowered to a gap of 10 μm at a speed of 10 μm/sec, thus compressing the particles. The gap was held constant at 10 μm while measuring the normal force. All measurements were repeated at least three times within each experimental batch and averaged over two batches.
Data Analysis
All experiments were repeated at least three times within each experimental batch and averaged over 2 batches. To determine significant differences in the means between samples, a pairwise Tukey test followed by a Bonferroni correction was used, with a p value of 95 %.
Total Solid Content
The total solid content of the samples were measured by placing a small ammount of sample typically less than 5 grams in an oven at a temperature of 80C for 24 hours. Measuring the weight loss due to evaporation gives the total solid content in the sample.
Results and Discussion
Linear Viscoelastic Properties of Carrot Suspension
Figure 2 showed the particle morphology of the untreated samples which presents mostly single cells due to the hot break treatment [2]. The mean particle size for the reference sample was found to be 83 μm. Mean particle size (Fig. 3), particle morphology (Fig. 2) and total solid content did not change significantly after the enzymatic treatments. Hence, the volume fraction is comparable between the enzyme treated and reference samples with the same solid content. The storage modulus of the various samples was measured as a function of the total solid content, shown in Fig. 4. The storage modulus is known to have a power law dependence on the total solid content (Eq. 1) within the semi-dilute region [2, 6, 17]. After fitting the curves with a power law model the coefficients obtained for the respective samples are shown in Fig. 4. For similar systems, the values of “b” reported earlier are 2.99 [2] and 4 [6]. The reason for the differences in “b” is possibly due to the nature of starting material.
where, G’ is the storage modulus [Pa], a is the prefactor, b is the power law exponent and w is the total solid content.

Confocal microscopy images of the untreated and enzyme treated carrot suspensions

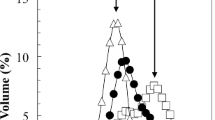
Particle size distribution of the carrot suspensions before and after enzyme incubation. Black squares indicate the untreated sample, green triangles the protease treated sample and red circles indicate the pectinase treated sample

Linear storage modulus as a function of concentration. Black squares indicate the control sample, green triangles the protease treated sample and red circles indicate the pectinase treated sample. a, b are the prefactor and powerlaw exponent in Eq. 1. Error bars are smaller than the symbols. Error bars indicate the standard deviation
The storage modulus of the carrot puree after pectinase treatment is lower throughout the concentration range investigated. This results in the prefactor “a” being lower. However, the coefficient “b” is higher compared to the untreated sample. The changes are significantly different from each other. The storage modulus of the carrot puree after protease treatment appeared similar to the control sample at higher concentrations but is lower at more dilute concentrations. Upon comparing the power law coefficients, it is evident that there are differences between the suspensions. The prefactor “a” for the protease treated sample is lower compared to the untreated sample but higher than the pectinase treated sample, whereas, the coefficient “b” for the protease treated sample is similar to the pectinase treated sample. The lower value of “a” for enzyme treated systems is due to the lower storage modulus. Both pectinase or protease enzyme treatments of the carrot puree resulted in a higher “b” value compared to the untreated sample, indicating that the enzyme treatment induces a more “open” network structure [6, 18]. This could be the result of the enzymes altering the pectin and protein structures present within the serum or particle phase of the suspensions [4, 6]. As a result of enzymatic action, the inter particle interactions are altered either by pectin hydrolysis and demethoxylation (pectinase) or by changing the nature of protein-polysaccharide interactions (protease).
To investigate the nature of interactions between the particles, NaCl was added to two different packing fractions of particles. Varying the particle concentration enabled to qualitatively control the inter-particle distance. A higher concentration of particles results in more crowding (higher packing fraction) and vice-versa. Logically, the particle properties are more important at higher particle concentrations and the properties of the serum phase are probably more important at lower particle concentrations. The concentration of NaCl was quantified relative to the total solid content. This gives a better indication of how much salt is needed to screen charges on the particles as compared to an absolute measure such as molarity. Figure 5 shows the variation of storage modulus with NaCl addition at two different concentrations. In both concentrations of the particles, the storage modulus of the particles after pectinase treatment is not very sensitive to salt addition. At a higher concentration, the particles are tightly packed bringing the particle closer to each other. This increases the potential for particle-particle interactions and the effect of particles properties become predominant on the rheology. The untreated sample is very sensitive to the addition of salt at a higher concentration of particles. This contrasting behavior of the pectinase treated and untreated samples with addition of salt suggests that the pectins present on the particles could contribute to electrostatic interactions. The combination of PG and PME would cause a reduction in Mw and increase the charge on pectic backbone. The salt insensitivity of the pectinase treated sample suggests that the smaller but more charged pectins have little contribution to particle-particle electrostatic interaction. After protease treatment on the other hand, at the higher particle concentration a similar salt dependent trend to the control sample is observed, while at a lower concentration of particles the storage modulus is insensitive to the salt addition. From these observations, it is suggested that particle-particle interactions in the untreated systems are mainly electrostatic in nature through pectins. The effect of proteins on the rheology is not conclusive from these experiments. From the results it is suggested that the proteins present in these suspensions affect the rheology in more dilute systems. The protease treatment probably alters the protein-polysaccharide interactions which can significantly change the dynamics of such systems. Due to the partially charged pectin backbone and possibly an oppositely charged protein, they are most likely to interact [3, 4].

Effect of NaCl on the storage modulus of the samples at a total solid content of (a) 5.6 % (high) and (b) 3.1 % (low). Black squares indicate the untreated sample, green triangles the protease treated sample and red circles indicate the pectinase treated sample. Error bars indicate the standard deviation
Serum Phase
The serum phase of the suspensions was characterized by measuring the relative viscosity in absence and presence of salt. This allowed investigating the effect of the serum phase on particle-particle interactions by determining the intrinsic viscosity. Relative viscosity of the serum phase decreased in 2M NaCl (Fig. 6). Intrinsic viscosity was determined from fitting a line to the relative viscosity vs galacturonic acid content as shown in Eq. 2. The line was fit to the limit of zero concentration (0,1) and the intrinsic viscosity was measured from the slope. Then the measured intrinsic viscosity of the serum decreases from 0.68 to 0.44 in the presence of 2M NaCl. Hence, the apparent molecular weight of the solubilized polymers in the serum phase also has decreased as given by the Mark-Houwink rule (Eq. 3). The serum phase properties play an important role at a lower packing fraction. However, the decrease in the storage modulus with the addition of salt is not drastic at a lower fraction of particle compared to the higher packing fraction (Fig. 5b). Furthermore, the serum phase polymers result in a lower apparent Mw with the addition of NaCl. Hence, it is unlikely that the serum phase polymers affect particle-particle interactions. This result further strengthens the notion that pectins on the particle surface are responsible for the electrostatic interactions.
where, η is viscosity [Pa.s], η o is solvent viscosity [Pa.s], ϕ is the volume fractions and M w is the molecular weight.

Intrinsic viscosity of the serum phase of the untreated carrot puree. No salt (black squares) and with 2M NaCl (red circles). The fit linear line is made to pass through (0,1). Error bars indicate the standard deviation
The molecular weight distribution of the serum phase was measured to understand the effect of enzymes on the biopolymers in the serum phase. The results of the RI detector are presented in Fig. 7a. The elution profile of the polymeric compounds shows a clear shift to the right after the treatment with pectinase as expected due to the hydrolysis of the pectic backbone (with minimal changes to the RI profile of the protease treated samples). On the other hand when observing the UV (at 280 nm, Fig. 7b) profile a relatively large increase of the peak around 40 minutes is visible after the protease treatment, most likely suggestive that due to the enzymatic treatment some large Mw UV absorbing material from the particle phase or unfilterable from the serum phase (at 0.45 μm) before the treatment dispersed into serum phase and/or became filterable. As a result the weight average molecular weight (assuming that the dn/dc of the polymers did not change very significantly due to the increased but still small fraction of proteins in the elution profile) of the pectinase treated sample insignificantly decreased from 2.3x10 6 to 1.8x10 6 gr/mol while due to the treatment with protease a significant increase to 4.9x10 6 gr/mol was observed. Changes in the concentration and size of the polymers in the serum phase are likely to affect the rheological properties of the whole system especially in samples where the rheology of the whole systems is dominated by the serum phase (as was suggested for the protease treated samples). A suggested hypothesis is that the protease treatment changes the nature in which pectin binds proteins in the serum phase. However, the precise mechanism is difficult to elucidate within this study due to the limitation of the filter and removal of fully hydrolyzed polymers that result in a superposition of effects in the filterable serum phase. At least, this MALLS study shows that the protease treatment enriches the serum phase with large polymers coming from the particle phase.

Size exclusion elution profile of the serum phase of the enzymatically treated and untreated samples. (a) Refractive index profile, (b) UV signal profile at 280nm. Black squares indicate untreated serum, green triangles indicate protease treated serum and red circles indicate pectinase treated serum phase
Particle Phase
The enzymes affect the cell wall microfibrillar architecture as visualized from cryo-SEM images shown in Fig. 8. Pectins on the cell wall were possibly hydrolyzed by the pectinase enzyme treatment which resulted in significant loosening of the cell wall. Even the protease enzyme treatment resulted in some cell wall loosening. Pectins and proteins are known to crosslink with cellulose fibrils. Upon enzymatic hydrolysis the crosslinks are reduced facilitating the loosening of the cellulose architecture [7].

cryo-SEM images of cell wall architecture for untreated, pectinase treated and protease treated carrot systems
Due to visible changes in the cell wall architecture after enzyme treatment (Fig. 8), the question arises whether the enzymes affect the rheology by changing the physical properties of particles? To answer this, squeeze experiments were performed using a rheometer. The top plate of the rheometer was lowered at a constant rate. This caused the serum phase of the suspensions to percolate through, leaving the particles constrained by the narrow gap. As the gap width tended towards the mean particle size, a direct strain was applied on the particles. The resulting stress response of the particles was measured by the normal force transducer on the bottom plate of the rheometer. Translating the force response to obtain precise rheological material functions is not trivial as the materials are very heterogeneous. Conventional models are applicable only for small deformation typically less than 10 % [19]. Furthermore for anisotropic materials like cell walls, the particle properties depend on the direction of strain applied [20, 21]. Direct visualization of the particle deformation was not possible with the current setup. It is suggested that compressive strain on the originally ellipsoidal particles would cause the cell wall to bend (Fig. 9). The advantage of this method is its robustness and special instrumentation is unnecessary. The downsides are the inability to determine particle properties, the measurement of the average response over many particles and the fact that deformation applied is possibly non-linear. Hence, this method can only be used to qualitatively compare related systems.

Schematic of compression experiment. Inset highlights the possible deformation experienced by the cell wall, dotted lines indicate the compressed cell wall and solid lines indicate the quiescent state of the cell wall
The protocol described here is optimized for the specific rheometer and system. Faster compression rates or higher strains resulted in a normal force larger than specifications of the rheometer. A slower compression rate or lower strain, resulted in poor force resolution. Peak normal force was measured when the plate was stopped and the subsequent normal stress relaxation was fit with a 1-D Maxwell model, shown in Eq. 4.
where, σ is stress (Pa); λ is relaxation time scale (s); σ 0 is peak stress (Pa); t is time (min) and c is residual stress (Pa).
Previous research to evaluate cell hardness has been performed with the use of a micro-manipulator [22] or atomic force microscope [23]. Under a compressive strain the force response has been suggested to be a superposition of two phenomena, the stress response due to particle deformation and water transport through the cell wall [24]. It is difficult to decouple these effects in heterogeneous suspensions. Based on the experiments performed using a micro-manipulator, it is suggested that the peak force is indicative of cell wall elasticity [19]. While the time scale of relaxation is indicative of the water transport that would occur as a result of the applied strain [19].
The peak force from the compression test was investigated for a range of concentrations as shown in Fig. 10. It can be observed directly that the control sample shows a lower peak force compared to the enzyme treated samples throughout the concentration range investigated. Statistical analysis shows that the difference observed between the enzyme treated and untreated sample was found to be significantly different with a p-value of 95 %. There was no significant difference found between the peak force values of the pectinase and protease treated samples. From previous studies, enzyme treatment was reported to “soften” the cell wall in the surface plane to facilitate extension [7, 21, 25]. However, it is to be highlighted that this cell wall elasticity is not being measured in the current experimental protocol. The force measured here is the response from a complex cell wall deformation and from pressurized water inside the cells. The deformation of the cell wall results in a combination of bending stress and elastic force measured in the normal direction to the cell wall. A loosened cell wall architecture results in a thicker cell wall. This could cause both the pectinase and protease treated samples to have a significantly higher normal force when compressed. Previous experiments have shown that there is a direct correlation with pectin demethyloxylation and an increased rigidity of the cell wall in growing cells [26, 27]. Hence, the result that the enzyme treated samples shows a higher force is not entirely surprising. The PME induces demethoxylation of the pectic chains which could potentially make the particles stiffer.

Graphs showing (a) peak force (b) relaxation time scales from compression experiments for the untreated, pectinase and protease treated carrot cell suspensions. Error bars indicate the standard deviation
The pectinase treated system had a lower storage modulus (Fig. 10) and a higher peak force (Fig. 10) compared to the untreated system in all the concentrations. Furthermore, the pectinase treated samples were insensitive to the addition of salt (Fig. 5). From these observations, it is suggested that the particle hardness did not affect the bulk rheological properties as significantly as the electrostatic interactions. This is probably due to the low occurrence of particle-particle interactions in concentrations below the packing limit. Contrastingly, the storage modulus of the protease treated sample was lower than the untreated sample only at a lower particle concentration (Fig. 4). Whereas, the peak force of the protease treated sample was consistently higher than the untreated system in all the concentrations investigated (Fig. 10). It could be hypothesized that pectin-protein interactions present in the serum phase affected the rheological properties after protease treatment.
From previous investigations using a micro manipulator, it has been suggested that the force relaxation is a result of water drainage [19]. The relaxation time scale from the Maxwell model is shown in Fig. 10. At higher concentrations, the protease treated sample had the highest relaxation time, followed by the control and pectinase samples. As the concentration decreased the difference between the treatments became insignificant. These results do not indicate a consistent trend. Relaxation time scale for a cell with only turgor pressure was measured to be 10-20 seconds which is longer than what is observed here for systems without turgor pressure [22]. Here the relaxation time scales also varied with the particle concentration. It is possible that in concentrated systems particle-particle interactions affect the stress relaxation. When the concentration of the particles is reduced, the effect of particle-particle interactions also decreases. In that case, the relaxation time scale at the lowest concentration would be the best indicator for each system. However, at the lowest concentration of the particles there is no significant difference in the relaxation time between the different treatments. This suggests that the porosity and torutuosity of the different samples are similar under compression [24].
Conclusions
The effect of protease and pectinase enzymatic treatments on the serum and particle phase properties of carrot cell wall suspensions was investigated. It was shown that both pectinases and proteases altered the network structure present in the serum phase. This was done by both pectin hydrolysis and demethoxylation (pectinase) or changing the protein-polysaccharide interactions by de-polymerization of proteins (protease). The pectinase treated material was insensitive to the addition of NaCl at at both higher and lower concentrations, suggesting that pectins present on the surface of the particles were responsible for the electrostatic particle-particle interactions. This was further verified by measuring the relative and intrinsic viscosity of the untreated serum phase with the addition of NaCl. To investigate the effect of particle properties a series of compression experiments is performed. It was seen that the pectinase and protease treated samples had a higher peak force response compared to the control. However, the time scale for stress relaxation was similar between all the samples investigated. This suggested that particle stiffness had increased due to cell wall loosening caused by the pectinase or protease enzyme treatment. Even though the pectinase treated sample had ’harder’ particles the storage modulus was lower compared to the untreated system. This leads to conclusion that rheological properties of carrot suspensions in the semi-dilute regime were affected by electrostatic interactions rather than particle hardness effects.
References
B. Yoo, M. Rao, J. Text. Stud. 25, 421 (1994)
P. Lopez-Sanchez, R. Farr, Food Bio. 7, 15 (2011)
N. Takada, P.E. Nelson, J. Food Sci. 48, 1408 (1983)
A.K. Sankaran, J. Nijsse, L. Bialek, A. Shpigelman, Hendrickx, M.E., A.M. Van Loey, Food Hydro. 43, 442 (2015)
K.R.N. Moelants, R. Cardinaels, K. De Greef, E. Daels, S. Van Buggenhout, A.M. Van Loey, P. Moldenaers, M.E. Hendrickx, Food Hydro. 36, 382 (2014)
K.R.N. Moelants, R. Cardinaels, R.P. Jolie, T.A.J. Verrijssen, S. Buggenhout, L.M. Zumalacarregui, A.M. Loey, P. Moldenaers, M.E. Hendrickx, Food and Biopro. Tech (2013)
D.J. Cosgrove, Annu. Rev. Plant Phy. 50, 391 (1999)
D.J. Cosgrove, D.M. Durachko, J. Expt. Bot., 1711–1719 (1994)
B.A. Veytsman, D.J. Cosgrove, Biophy. J. 75, 2240 (1998)
R.D. Preston, Ann. Rev. Plant Physiol. 30, 55 (1979)
M.L. Fishman, H.K. Chau, F. Kolpak, J. Brady, J. Agr. Food Chem. 49, 4494 (2001)
P. Roger, B. Baud, P. Colonna, J. Chromatogr. A. 917, 179 (2001)
A.E.R. Ahmed, J.M. Labavitch, J. Food Biochem. 1, 361 (1978)
N. Blumenkrantz, G. Asboe-Hansen, Anal. Biochem. 54, 484 (1973)
H.A. Barnes, J. Non-Newtonian Fluid. 56, 221 (1995)
C. W. Macosko. Rheology: Principles, Measurements, and Applications (Wiley, 1994)
L. Day, M. Xu, S.K. Oiseth, L. Lundin, Y. Hemar, Colloid Surface B. 81, 461 (2010)
E. Bayod, P. MÃ¥nsson, F. Innings, B. Bergenstahl, E. Tornberg, Food Bio. 2, 146 (2007)
J. Blewett, K. Burrows, C. Thomas, Biotechno. Lett. 22, 1877 (2000)
P. Milani, M. Gholamirad, J. Traas, A. Arneodo, A. Boudaoud, F. Argoul, O. Hamant, Plant J. 67, 1116 (2011)
L. Taiz, Ann. Rev. Plant Physiol. 35, 585 (1984)
L. Wang, D. Hukin, J. Pritchard, C. Thomas, Biotechnol. Lett. 28, 1147 (2006)
M. Gad, A. Itoh, A. Ikai, Cell Biol. Int. 21, 697 (1997)
P. Lopez-Sanchez, M. Rincon, D. Wang, S. Brulhart, J.R. Stokes, M.J. Gidley, Biomacro. 15, 2274 (2014)
A.L. Routier-Kierzkowska, A. Weber, P. Kochova, D. Felekis, B.J. Nelson, C. Kuhlemeier, R.S. Smith, Plant Physiol. 158, 1514 (2012)
E. Parre, A. Geitmann, Planta. 220, 582 (2005)
A. Peaucelle, S.A. Braybrook, L. Le Guillou, E. Bron, C. Kuhlemeier, H. Hofte, Curr. Bio. 21, 1720 (2011)
S. McQueen-Mason, D.J. Cosgrove, Proc. Nat. Acad. Sci. 91, 6574 (1994)
S.J. McQueen-Mason, D.J. Cosgrove, Plant Physiol. 107, 87 (1995)
A. Nussinovitch, M. Peleg, M. Normand, J. Food Sci. 54, 1013 (1989)
M. Peleg, J. Food Sci. 44, 277 (1979)
N. Sakurai. Bot. Maga. Tokyo 104 235 (1991)
J.R. Seth, L. Mohan, C. Locatelli-Champagne, M. Cloitre, R.T. Bonnecaze, Nat. Mat. 10, 838 (2011)
R. Whittenberger, G. Nutting, 8, 420 (1958)
R. Yamamoto, J. Plant Res. 109, 75 (1996)
R. Yamamoto, K. Shinozaki, Y. Masuda, Plant Cell Physiol. 11, 947 (1970)
Acknowledgments
This research was financially supported by the Seventh Framework Programme (FP7) of the European Union under the Marie Curie Initial Training Network ‘HST FoodTrain’ (Grant agreement 264470). R. Cardinaels is indebted to the Research Foundation Flanders for a Postdoctoral Fellowship at KU Leuven.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Sankaran, A.K., Nijsse, J., Cardinaels, R. et al. Effect of Enzymes on Serum and Particle Properties of Carrot Cell Suspensions. Food Biophysics 10, 428–438 (2015). https://doi.org/10.1007/s11483-015-9403-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11483-015-9403-6