
Abstract
Oxidant toxicity has been implicated in the pathogenesis of amyotrophic lateral sclerosis (ALS), an insidiously progressive neurodegenerative disorder involving upper and lower motor neurons. Here, we investigated the cellular and molecular mechanisms underlying the neuroprotective effects of an anti-oxidant genistein in SOD1-G93A transgenic mouse model of ALS. Rotarod test, hanging wire test and hindlimb clasping test were used to determined disease onset and assess motor performance. Immunostaining together with neuronal size measurement were used to count viable motor neurons. In addition, immunostaining procedure and ELISA kit were used to assess the inflammatory response in the spinal cord. Our results showed that Genistein administration suppressed the production of pro-inflammatory cytokines and alleviated gliosis in the spinal cord of SOD1-G93A mice. In addition, genistein administration induced autophagic processes and enhanced the viability of spinal motor neurons. As a result, genistein alleviated ALS-related symptoms and slightly prolonged the lifespan of SOD1-G93A mice. Taken together, our results indicate that genistein is neuroprotective in SOD1-G93A mice, suggesting genistein could be a promising treatment for human ALS.

Genistein protects impariments in SOD1-G93A transgenic mouse model.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder that results in death of both upper and lower neurons (Strong 2003). Patients with ALS suffer from progressive loss of function in neuromuscular system, such as difficulty speaking or swallowing and flaccid or spastic paralysis of extremities. Usually, most patients die due to failure of respiratory muscle within 3–5 years after onset of symptoms (Strong 2003).
The molecular mechanisms underlying motor neuron death in ALS patients remain largely unknown. ALS is classified into two forms with 90% being sporadic and 10% being familial (Renton et al. 2014). Previous studies have shown that both genetic and environmental risk factors can be associated with high incidence of ALS. Among 30 reported risk genes, four genes, C9ORF72, SOD1, TARDBP and FUS have been identified in 70% ALS patients with a family history (Chio et al. 2014; Cirulli et al. 2015). SOD1 gene, which encodes superoxide dismutase enzyme, has been widely studied. Furthermore, the transgenic mice expressing mutated SOD1 enzyme (SOD1-G93A mice) presents aggressive neuromuscular defects that mimic the clinical progression and pathological features of ALS in human. Thus, SOD1-G93A mice has provided significant insight into the neuropathology of ALS at molecular, cellular and systemic level. Previous studies have demonstrated that SOD1-G93A mice shows increased oxidative stress (Lev et al. 2009), augmented glutamate signal (Giribaldi et al. 2013; Maragakis and Rothstein 2004), exacerbated inflammation (Debye et al. 2018) and a large number of dead motor neurons in the spinal cord (Gurney 1994; Liang et al. 2017; Rosen 1993).
Genistein is an isoflavone found in soybean that can easily cross blood brain barrier (Du et al. 2018). Numerous studies have shown that genistein is neuroprotective attributed to its anti-oxidant effect and weak estrogen activities (Rahman Mazumder and Hongsprabhas 2016). In addition, a growing amount of evidence has shown that genistein can reduce neuronal death by attenuating the inflammation within the micro-environment around a single neuron (Sadhukhan et al. 2018). The anti-inflammatory effect is attributed to inhibition of kinase activities including extracellular-signal-regulated kinase (ERK) and Mitogen-activated protein kinase (MAPK) in activated microglia (Du et al. 2018; Lu et al. 2018). ERK and MAPK are involved in the production of pro-inflammatory cytokines that contribute to neuronal apoptosis in neurodegenerative disorders (Kim and Choi 2015; Rai et al. 2019). In neurodegenerative disease such as ALS, anti-inflammatory effect of genistein has been shown to be neuroprotective. In a murine model of familial ALS, genistein treatment can protect against neuronal damage caused by oxygen singlet in motor cortex through both estrogen dependent and estrogen non-dependent mechanisms (Trieu and Uckun 1999). Also, genistein can induce autophagic activities in the neurons of mice model with stroke (Trieu and Uckun 1999). Based on the neuropathology related to SOD1 mutation and the pharmacological effects of genistein, we hypothesized that genistein might exert neuroprotective effects on SOD1-G93A mice. Our results indicated that administration of genistein in SOD1-G93A mice reduced loss of motor neurons, inhibited the gliosis and production of pro-inflammatory cytokines and induced the autophagic activities in neurons. As a result, administration of genistein improved the motor performances and prolonged the life span of SOD1-G93A mice.
Methods & Materials
ALS Mouse Model and Genistein Treatment
All animal experiments were approved by Ethics Committee of the Second Hospital of Hebei Medical University. SOD1-G93A mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA) and maintained heterozygous in the C57BL/6 background. All animals were kept in the animal facility in our institute with standard condition of 22 ± 2 °C, an equivalent light-dark cycle and free access to food and water. Eight weeks old animal were evaluated by rotarod test, hanging wire test and hindlimb clasping test to attain baseline value. Animals were then grouped according to their weight and progenitors with a sex ratio close to 1:1 and fed vehicle or genistein at 16 mg/kg twice a day, every day.
Rotarod Test
Motor coordination, strength and balance before and after genistein administration were assessed by the rotarod test. A rod rotating at 14 rpm were used to trained all animals three times a week. Test was performed in animals from 8 to 16 weeks of age. Presence of arbitrary maximum time of maintenance in the rotating rod of 180 s was defined as symptom free and the first day that the animal was unable to complete the 180 s on the rotating was defined as the time of clinical disease onset.
Hanging Wire Test
Conventional cage wire lid was hanged 50 cm above a padding and was turned upside down after each animal was placed on the lid. The overall duration of observation was 60s. The latency time to fall was recorded as the duration that the mouse could hold on to the wire.
Hindlimb Clasping Test
Hindlimb clasping was assessed based on a previously reported method. The base of the tail was used to suspend the animal. Each animal was videotaped for 15 s. A scale of 0 to 3 based on severity was used to rate hindlimb clasping behavior: 0 = hindlimbs abducted away from the abdomen; 1 = one hindlimb adducted for at least 50% of the observation period; 2 = both hindlimbs partially adducted for at least 50% of the observation period; and 3 = both hindlimbs completely adducted for at least 50% of the observation period. Rate of 0.5, 1.5 or 2.5 was used when the performance of the mouse fell between two levels. Rates from three separate trials over three consecutive days was used to abstain an average.
Immunohistochemistry
Avertin was used to anesthetize the animals and ice-cold phosphate-buffered saline containing heparin (10 U/mL) was transcardially perfused before the animals were sacrificed. The lumbar spinal cords were collected and fixed in 4% paraformaldehyde solution at 4 °C for 24 h before being processed for frozen sections every 40 μm. Primary antibodies were anti-Iba-1 antibody (1:100, GeneTex, GTX101495), anti-GFAP antibody (1:100, CST, 3670S) and anti-NeuN antibody (1:100, Abcam, ab190195). Secondary antibodies were horse radish peroxidase (HRP)-labeled. All immunostained sections were then visualized with diaminobenzidine. In the meantime, Nissl detection kit (DK0022, solarbio) were used to detect motor neurons, according to the manufacturer’s protocol. An Olympus IX73 inverted microscope with DP80 camera was to attain all images. Motor neurons (MN) were identified and counted following strict size and morphological criteria: First, it should with diameters larger than 20 μm. Second, it should be with polygonal shape and prominent nucleoli. Only MNswith these characteristics werecounted (Dutta et al. 2018; Mancuso et al. 2011). For each animal, three fields of view per section were imaged and at least three sections were analyzed. All images were then analyzed by personnel who were blind to the genotype and treatment of the mice. Image J (National Institutes of Health, USA) was used to quantify the intensity of antibody labeling.
Western Blot
Radioimmunoprecipitation buffer with complete protease inhibitor mixture (Roche Diagnostics, Penzberg, Upper Bavaria, Germany) was used to homogenize the spinal cord tissues. Lysate was centrifuged at 16,000 g for 15 min at 4 °C. The supernatants were collected and the bicinchoninic acid protein assay (Pierce, Waltham, MA) was used to determine the protein concentration. A 4–12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis gel (Invitrogen, Waltham, MA USA) was used to separate proteins that was subsequently transferred onto a nitrocellulose membrane (Merck Millipore). The primary antibodies were anti-TLR2 (1:1000, Abcam, ab9100), anti-TLR4 (1:500, Santa Cruz, SC-13591), anti-p65 (1:500, Santa Cruz, SC-7151), anti-cox2 (1:100, GeneTex, GTX101495), anti-LC3b1 and LC3b2 (1:1000, Thermo Fisher Scientific PA5–32254), anti-GFAP (1:100, CST, 3670S), and anti-GAPDH (1:1000, CST, 2118S). All appropriate secondary antibodies were HRP-conjugated. Bands were attained by enhanced chemiluminescence (Pierce) and quantified by Image J.
Statistical Analysis
All data are presented as the means ± standard deviation (SD) as indicated in graphs. Student’s t test was or two-way ANOVA followed by Bonferroni post hoc test were used for comparison. Mantel-Cox test was used to analyzed onset and survival data. All statistical tests were evaluated at the 5% significance level.
Results
Genistein Administration Alleviated ALS Symptoms and Slightly Prolonged Lifespan in SOD1-G93A Mice
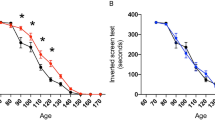
Previous studies have demonstrated that SOD1-G93A mice presents with neurological symptoms similar with the clinic features in human ALS. Here, we evaluated the baseline neuromuscular functions of SOD1-G93A mice at 50 days of age by using rotarod test, hanging wire test and hindlimb clasping test (Fig. 1 a-d). Thereafter, the animals were treated with genistein or vehicle. Neuromuscular functions were evaluated until age 16 weeks. The age of symptom onset was recorded when the animal presented with deficits in any of the three tests described previously. Our results revealed that compared to the vehicle-treated mice, both male and female genistein-treated mice showed a significant delay of symptoms onset (Mantel-Cox test, p < 0.05 (Fig. A-B). In addition, Genistein-treated mice presented with significantly higher motor performance in both rotarod test and hanging wire test than the vehicle-treated mice did (Fig. C). Furthermore, at age of 120 days, Genistein-treated mice displayed much less severe deficit in hindlimb clasping behavior than vehicle-treated mice did, as illustrated by the remarkable decrease in hindlimb severity score (Fig. D). Interestingly, the administration of Genistein significantly prolonged survival in male mice although the survival in female is only slightly prolonged (150 days vs 141 days) (Fig. E). Taken together, our results suggest that genistein might to improve the prognosis and could be a promising treatment for ALS.

Genistein treatment delayed disease onset, increased survival rate and improved motor performance in SOD1-G93A mice. a Genistein treatment delayed 17 days in male (p < 0.01) and 10 days in female SOD1-G93A mice (p < 0.01) regarding the onset of symptoms. Disease onset was assessed by means of rotarod. b Genistein treatment significantly improved the locomotion in SOD1-G93A mice (p < 0.01). Locomotion was evaluated by using rotarod test. c Genistein treated SOD1-G93A mice had significantly longer hanging wire duration than veh-treated mice (**p < 0.01, ***p < 0.001). d Genistein treatment improved the performance in hind-limb clasping behavior (*p < 0.05, ***p < 0.001). e Genistein treatment increased median life span of SOD1-G93A mice (p < 0.05). n = 8. Data was represented as means ± SD
Genistein Administration Improved the Viability of Motor Neurons in the Spinal Cord of SOD1-G93A Mice
ALS is originally featured by death of upper and lower motor neurons. In the present study, mice were sacrificed for motor neuron analysis at the age of 115 days. In SOD1-G93A mice, up to 70% motor neurons died in the end. Here, we combined neuronal nuclear marker NeuN or Nissl staining with neuronal size (diameter > 20 μm) to assess the number of motor neurons in ventral horn of spinal cord. A diameter > 20 μm was used here because neurons with a diameter < 20 μm is more likely to be nonfunctional atrophic motor neurons (Fig. 2a, b). Motor neurons from female and male animals were pooled since no gender difference was observed. Our results indicated that genistein-treated mice had more motor neurons surviving the disease course when compared to the vehicle-treated mice (Fig. 2c). Our results suggest that genistein can protect the motor neurons against the detrimental factors caused by the mutation of SOD1.

Genistein treatment decreased the loss of motor neurons in the spinal cord. a Genistein treatment increased the number of motor neurons as indicated by NeuN-staining. Representative pictures from single channel grayscale images are shown. Scale bar =200 μm. For each panel, the region in the black box of the left image was magnified 10x and showed in the right image. b Genistein treatment increased the number of motor neurons as indicated by Nissl staining. Scale = 200 μm. c The quantification of motor neurons in the spinal cord (n = 8, 6 sections per mouse). ImageJ was used to count the numbers of motor neurons. Data was represented as means ± SD. **p < 0.01
Genistein Administration Alleviated Gliosis in the Spinal Cord of SOD1-G93A Mice
A growing number of evidences has suggested that though gliosis can be resulted from motor neuron death, progression of gliosis per se can further exacerbate the degeneration of motor neurons in both animal models and ALS patients. Thus, it is proposed that suppression of gliosis in central nerve system can actually slow down the disease course. Here, we used anti-GFAP antibody to label astroglial cells and anti-Iba-1 antibody to label microglial cells in the spinal cord. Genistein administration substantially reduced the Iba-1 and GFAP immunoactivity when compared to the vehicle treatment. Specifically, the microgliosis and astrogliosis decreased in the spinal cords (Fig. 3a, b).

Genistein treatment alleviated the progression of gliosis in the spinal cord of SOD1-G93A mice. a Genistein treatment remarkably decreased overall signal intensity of GFAP in the spinal cord of SOD1-G93A mice. GFAP is believed to be indicative of astrocytic activation. b Genistein treatment remarkably decreased overall Iba-1 signal intensity in the spinal cord. Iba-1 is believed to be indicative of microglial activation. c, d Genistein treatment decreased expression level of microglial markers including TLR2, TLR4, NF-κB and COX-2. Data was compiled from 8 animals per group with at least 6 sections per mouse and is presented as means ±SD. Scale = 50 μm. *p < 0.05, **p < 0.01, ***p < 0.001
Previous studies have shown that genistein is anti-inflammatory by down-regulating the TLR4/NF-kB signal pathway in microglial cells. First, we tested the activities in the TLR4/NF-kB signal pathway in SOD1G93A mice. Our results showed that activities in this pathway were relatively at high level. Second, we tested the effect of genistein on the increased activities of TLR4/NF-kB signal pathway. Our results revealed that levels of TLR2, TLR4 and NF-kB p65 in spinal cords of Genistein-treated SOD1G93A mice were significantly decreased when compared to that in the vehicle mice (Fig. 3c, d). In addition, the level of cyclooxygenase (COX)-2, an enzyme required for the production of multiple pro-inflammatory cytokines like ILs was also decreased in Genistein-treated SOD1G93A mice, compared to that in the vehicle mice (Fig. 3c, d). To summarize, our results suggest that inflammation in spinal cord is one of the markers for clinical progression of ALS and genistein exerts neuroprotective effect probably via its broad anti-inflammation activity.
Genistein Administration Suppressed the Production of Inflammatory Factors in SOD1G93A Mice
In ALS, activation of microglia and astrocytes leads to increase of inflammatory cytokines including IL-1β, IL-6 and TNF-α, which contribute to degeneration and death of motor neuron. To investigate the effect of genistein on cytokine production, we assessed the levels of IL-1β, IL-6 and TNF-α in the spinal cord lysate of SOD1-G93A mice treated with genistein or vehicle. Compared with the vehicle-treated SOD1-G93A mice, genistein-treated mice showed significantly reduced levels of IL-1β, IL-6 and TNF-α (Fig. 4a, b and c).

Genistein reduced inflammatory cytokine expression in SOD1-G93A mice. Genistein treatment in SOD1-G93A mice reduced the expression of IL-1β (a), IL-6 (b) and TNF-α (c) in the spinal cord. Cytokine expression was measured by using ELISA kits. Data is represented means ±SD. n = 8 in each group. **p < 0.01. ***p < 0.001
Genistein Administration Induced Autophagic Processes in SOD1-G93A Mice
Aggregated proteins and damaged organelles in cells are usually engulfed and digested by Autophagosome. Autophagic dysfunction can result in accumulation of aggregated protein and damaged organelles that can lead to the major physiopathological changes in numerous neural degenerative disorders. Thus, promoting clearance of aggregated protein and damaged organelles in motor neurons is proposed to be advantageous for ALS patients. Previous evidence has suggested that genistein can modulate the cellular autophagic activities. Here, we test the autophagic states in the motor neurons of spinal cord in SOD1G93A treated with either genistein or vehicle. We immunostained the spinal motor neurons by using anti-p62 antibody and observed prominent p62 positive autophagic granules in genistein treated mice. In contrast, in the motor neurons from vehicle treated mice, the immunostaining signal was sparse and present with small granules. In addition, the p62 protein level significantly increased SOD1G93A mouse (Fig. 5a). It was further found that motor neurons from vehicle treated mice lacked LC3II and Beclin, the markers of autophagy. Interestingly, Genistein administration restored these early markers (Fig. 5b, c), suggesting that genistein induce autophagy. Taken together, our results suggest that genistein induce autophagic flux in the motor neurons from the spinal cord of SOD1G93A mouse.

Genistein treatment induced the autophagic flux in the motor neurons of spinal cord. a Genistein treatment induced large p62 positive autophagosomesin the spinal cord motor neurons. Downregulation of P62 was detected by western blot, which indicated autophagic flux. Genistein treatmentdecreased LC3b2/LC3b1 ratio (b) and Beclin1 expression level (c) in the spinal cord motor neurons. Scale bar =50 μm. Data is represented as means ±SD. n = 8 in each group.*p < 0.05. **p < 0.01
Discussions
ALS is progressive neurodegenerative disorder featured by death of upper and lower motor neurons (Ruffoli et al. 2017; Shibuya 2017). The pathophysiological changes in the nerve system of ALS patient are a complex of cellular events. Progressively increased oxidative stress (Blasco et al. 2017; Bozzo et al. 2017; Islam 2017) during the disease course is believed to be the major contributor to neuronal death. Whereas, the increased oxidative stress can be attributed to activated macroglia and astroglia cells as well as autophagic dysfunction (Monahan et al. 2016; Oakes et al. 2017; Ramesh and Pandey 2017). Here, we demonstrate that SOD1G93A mice, a well-established ALS model had increased inflammatory response in the spinal cord characterized by enhanced microgliosis and gliosis and elevated production of pro-inflammatory cytokines in the spinal cord. Furthermore, the spinal motor neurons from SOD1G93A mice presented significant defect in autophagy. Our results are consistent with the reported pathological findings in SOD1G93A mice (Gomes et al. 2018).
Nowadays, limited treatment options are available for ALS patients. Only two medications, Riluzo and Radicava are approved by FDA (Rothstein 2017). Increasing evidence have demonstrated that oxidant toxicity, inflammation in the CNS, and other defect at cellular and system level might all contribute to the disease course in human ALS as well as SOD1G93A mice (Monahan et al. 2016; Rai et al. 2019; Ramesh and Pandey 2017). During the last decade, SOD1G93A mice has boosted the search for new promising treatments for ALS. Although how mutation of SOD1 leads to such a complex of pathophysiological process in the central nerve system remains mysterious, the resemblance in the pathology and clinical features between the SOD1G93A mice model and human ALS allow scientist to test promising reagents for ALS. Genistein is highlighted recently due to its versatile property including anti-oxidant, anti-inflammation and weak estrogen activities (Ganai and Farooqi 2015; Mukund et al. 2017). Previous studies have shown that genistein inhibits the inflammatory response in multiple neurodegenerative disorders as the pro-inflammatory cytokines such as TNF, ILs are known to trigger the cellular apoptosis pathway and subsequently contribute to the neuronal death (Rai et al. 2019). Most importantly, little toxicity of genistein has been reported. We test genistein first in a small group of wide type healthy mice with no observed adverse events. In SOD1G93A mice, our results indicate that expression of ILs and TNF in the CNS of SOD1G93A mice were elevated. The elevated expression of cytokine expression was significantly down-regulated by administration of genistein (Fig. 4). In addition, the activated microglia and astroglia cells are the major source of pro-inflammatory cytokines in central nerve system (Frakes et al. 2014; Geloso et al. 2017). Our results showed that the spinal cords from SOD1G93A presented with increased micorgliosis and gliosis. As shown in the Fig. 3, the microgliosis and gliosis were alleviated by genistein administration. Activation of microglia and astroglia cells induced by SOD1 mutation can originate from the death of neurons, but can also be due to the loss of function of SOD1 in these cells. Nevertheless, our results suggest that microglia and astroglia cells might be the important targets of genistein in terms of its pharmacological effects on pathophysiological changes in ALS model.
Little characterizations have been done in terms of the intracellular modification in motor neurons following SOD1 mutation. Most studies focused on the toxicity of excitatory neurotransmitter glutamate. These studies have shown that that the expressions of glutamate metabolic receptors in motor neurons are increased and the subsequent signal transduction after binding of glutamate are substantially enhanced (Giribaldi et al. 2013; Maragakis and Rothstein 2004; Shibuya 2017). A recent study by Laudati G et.al characterized the intracellular modification in vitro. Their findings showed that thimerosal, in SOD1-G93 A cells, but not in SOD1 cells, reduced cell survival. Interestingly, the Sirtuin 1 (SIRT1) activator Resveratrol (RSV) prevented thimerosal-induced cell death by preventing Downstream Regulatory Element Antagonist Modulator (DREAM) reduction and prodynorphin up-regulation. Besides intracellular modification, modified cellular behavior can also contribute to the neuronal death in neurodegenerative disease (Laudati et al. 2019). For example, enhanced neuronal firing rate is well known to be toxic. It can damage multiple cellular process in neurons. Furthermore, the increased calcium level in the firing neurons can lead to paralysis of cellular processes that initially require proper calcium activities. For example, the autophagy in neurons is one of the processes that required proper calcium activities. Here, we checked the autophagic activities in SOD1G93A mice. As expected, the autophagic activities in the motor neurons from the spinal cord of SOD1G93A mice were undetectable by immunostaining the autophagic markers (Fig. 5). Interestingly, genistein restored the autophagic activities in spinal motor neurons. Nevertheless, further studies are needed to determine whether genistein can modulate the motor neurons’ excitability by regulating the glutamate signaling intensity. The relationship between genistein and excitability of motor neurons will be very informative.
Substantial loss of motor neurons ultimately leads to the end stage of ALS. Rome was not built in a day. Chronic pathological processes in motor neurons including chronic inflammation, long-term exposure to oxidative stress, and accumulation of cellular trashes in motor neurons caused by autophagic defect eventually lead to irreversible neuronal damages (Debye et al. 2018; Islam 2017; Kim and Choi 2015; Monahan et al. 2016; Ruffoli et al. 2017; Shibuya 2017). Any reagents that can slow down or suppress these chronic abnormal cellular processes would be able to improve the neuronal viabilities in the long term. Our results in this study and evidence from previous studies have shown that genistein is anti-inflammation, anti-oxidant and promotes autophagy in motor neurons. Based on these findings, we hypothesized that genistein can probably improve motor neurons’ viabilities. As expected, mice treated with genistein had a larger number of motor neurons than the vehicle treated mice did. As a result, mice treated with genistein had a delayed disease onset. More importantly, genistein administration prolonged the life span of SOD1G93A mice (Fig. 2). Thus, our findings suggest that genistein can be a valuable candidate for the phase I clinical trial.
Conclusions
Our results indicate that genistein suppressed the inflammatory response, induced autophagy and improved the viability of motor neurons in an ALS mice model, suggesting genistein could be a promising treatment for human ALS.
References
Blasco H et al (2017) Panel of oxidative stress and inflammatory biomarkers in ALS: a pilot study. Can J Neurol Sci 44:90–95. https://doi.org/10.1017/cjn.2016.284
Bozzo F, Mirra A, Carri MT (2017) Oxidative stress and mitochondrial damage in the pathogenesis of ALS: new perspectives. Neurosci Lett 636:3–8. https://doi.org/10.1016/j.neulet.2016.04.065
Chio A et al (2014) Genetic counselling in ALS: facts, uncertainties and clinical suggestions. J Neurol Neurosurg Psychiatry 85:478–485. https://doi.org/10.1136/jnnp-2013-305546
Cirulli ET et al (2015) Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 347:1436–1441. https://doi.org/10.1126/science.aaa3650
Debye B, Schmulling L, Zhou L, Rune G, Beyer C, Johann S (2018) Neurodegeneration and NLRP3 inflammasome expression in the anterior thalamus of SOD1(G93A) ALS mice. Brain Pathol 28:14–27. https://doi.org/10.1111/bpa.12467
Du ZR, Feng XQ, Li N, Qu JX, Feng L, Chen L, Chen WF (2018) G protein-coupled estrogen receptor is involved in the anti-inflammatory effects of genistein in microglia. Phytomedicine 43:11–20. https://doi.org/10.1016/j.phymed.2018.03.039
Dutta K, Patel P, Julien JP (2018) Protective effects of Withania somnifera extract in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Exp Neurol 309:193–204. https://doi.org/10.1016/j.expneurol.2018.08.008
Frakes AE et al (2014) Microglia induce motor neuron death via the classical NF-kappaB pathway in amyotrophic lateral sclerosis. Neuron 81:1009–1023. https://doi.org/10.1016/j.neuron.2014.01.013
Ganai AA, Farooqi H (2015) Bioactivity of genistein: a review of in vitro and in vivo studies. Biomed Pharmacother 76:30–38. https://doi.org/10.1016/j.biopha.2015.10.026
Geloso MC, Corvino V, Marchese E, Serrano A, Michetti F, D'Ambrosi N (2017) The dual role of microglia in ALS: mechanisms and therapeutic approaches. Front Aging Neurosci 9:242. https://doi.org/10.3389/fnagi.2017.00242
Giribaldi F et al (2013) Group I metabotropic glutamate autoreceptors induce abnormal glutamate exocytosis in a mouse model of amyotrophic lateral sclerosis. Neuropharmacology 66:253–263. https://doi.org/10.1016/j.neuropharm.2012.05.018
Gomes C, Cunha C, Nascimento F, Ribeiro JA, Vaz AR, Brites D (2018) Cortical neurotoxic astrocytes with early ALS pathology and miR-146a deficit replicate gliosis markers of symptomatic SOD1G93A mouse model. Mol Neurobiol. https://doi.org/10.1007/s12035-018-1220-8
Gurney ME (1994) Transgenic-mouse model of amyotrophic lateral sclerosis. N Engl J Med 331:1721–1722. https://doi.org/10.1056/NEJM199412223312516
Islam MT (2017) Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol Res 39:73–82. https://doi.org/10.1080/01616412.2016.1251711
Kim EK, Choi EJ (2015) Compromised MAPK signaling in human diseases: an update. Arch Toxicol 89:867–882. https://doi.org/10.1007/s00204-015-1472-2
Laudati G et al (2019) Resveratrol treatment reduces the vulnerability of SH-SY5Y cells and cortical neurons overexpressing SOD1-G93A to thimerosal toxicity through SIRT1/DREAM/PDYN pathway. Neurotoxicology 71:6–15. https://doi.org/10.1016/j.neuro.2018.11.009
Lev N, Ickowicz D, Barhum Y, Melamed E, Offen D (2009) DJ-1 changes in G93A-SOD1 transgenic mice: implications for oxidative stress in ALS. J Mol Neurosci 38:94–102. https://doi.org/10.1007/s12031-008-9138-7
Liang H et al (2017) Aldehyde dehydrogenases 1A2 expression and distribution are potentially associated with neuron death in spinal cord of Tg(SOD1*G93A)1Gur mice. Int J Biol Sci 13:574–587. https://doi.org/10.7150/ijbs.19150
Lu C et al (2018) Genistein ameliorates scopolamine-induced amnesia in mice through the regulation of the cholinergic neurotransmission, antioxidant system and the ERK/CREB/BDNF signaling. Front Pharmacol 9:1153. https://doi.org/10.3389/fphar.2018.01153
Mancuso R, Santos-Nogueira E, Osta R, Navarro X (2011) Electrophysiological analysis of a murine model of motoneuron disease. Clin Neurophysiol 122:1660–1670. https://doi.org/10.1016/j.clinph.2011.01.045
Maragakis NJ, Rothstein JD (2004) Glutamate transporters: animal models to neurologic disease. Neurobiol Dis 15:461–473. https://doi.org/10.1016/j.nbd.2003.12.007
Monahan Z, Shewmaker F, Pandey UB (2016) Stress granules at the intersection of autophagy and ALS. Brain Res 1649:189–200. https://doi.org/10.1016/j.brainres.2016.05.022
Mukund V, Mukund D, Sharma V, Mannarapu M, Alam A (2017) Genistein: its role in metabolic diseases and cancer. Crit Rev Oncol Hematol 119:13–22. https://doi.org/10.1016/j.critrevonc.2017.09.004
Oakes JA, Davies MC, Collins MO (2017) TBK1: a new player in ALS linking autophagy and neuroinflammation. Mol Brain 10(5). https://doi.org/10.1186/s13041-017-0287-x
Rahman Mazumder MA, Hongsprabhas P (2016) Genistein as antioxidant and antibrowning agents in in vivo and in vitro: a review. Biomed Pharmacother 82:379–392. https://doi.org/10.1016/j.biopha.2016.05.023
Rai SN et al (2019) The role of PI3K/Akt and ERK in neurodegenerative disorders. Neurotox Res. https://doi.org/10.1007/s12640-019-0003-y
Ramesh N, Pandey UB (2017) Autophagy dysregulation in ALS: when protein aggregates get out of hand. Front Mol Neurosci 10:263. https://doi.org/10.3389/fnmol.2017.00263
Renton AE, Chio A, Traynor BJ (2014) State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci 17:17–23. https://doi.org/10.1038/nn.3584
Rosen DR (1993) Mutations in cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 364:362. https://doi.org/10.1038/364362c0
Rothstein JD (2017) Edaravone: a new drug approved for ALS. Cell 171:725. https://doi.org/10.1016/j.cell.2017.10.011
Ruffoli R et al (2017) Neurons other than motor neurons in motor neuron disease. Histol Histopathol 32:1115–1123. https://doi.org/10.14670/HH-11-895
Sadhukhan P, Saha S, Dutta S, Mahalanobish S, Sil PC (2018) Nutraceuticals: an emerging therapeutic approach against the pathogenesis of Alzheimer's disease. Pharmacol Res 129:100–114. https://doi.org/10.1016/j.phrs.2017.11.028
Shibuya K (2017) Cortical motor neuron Hyperexcitability and motor neuron death in ALS: dying forward hypothesis. Brain Nerve 69:565–569. https://doi.org/10.11477/mf.1416200783
Strong MJ (2003) The basic aspects of therapeutics in amyotrophic lateral sclerosis. Pharmacol Ther 98:379–414
Trieu VN, Uckun FM (1999) Genistein is neuroprotective in murine models of familial amyotrophic lateral sclerosis and stroke. Biochem Biophys Res Commun 258:685–688. https://doi.org/10.1006/bbrc.1999.0577
Funding
This study was funded by the internal funding resource in our institute.
Author information
Authors and Affiliations
Contributions
Z.C.Z, J.S.F and S.P.L conceived the study; Z.C.Z and J.S.F analyzed the data; Z.Z.L wrote the paper.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that there is no conflict of interests in this paper.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhao, Z., Fu, J., Li, S. et al. Neuroprotective Effects of Genistein in a SOD1-G93A Transgenic Mouse Model of Amyotrophic Lateral Sclerosis. J Neuroimmune Pharmacol 14, 688–696 (2019). https://doi.org/10.1007/s11481-019-09866-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11481-019-09866-x