
Abstract
Purpose
This review presents the current methods used for determining ethyl glucuronide (EtG) and ethyl sulfate (EtS) concentrations in postmortem specimens, including sample preparation, analysis and the role of EtG and EtS in the postmortem assessment of the extent of alcohol abuse.
Methods
Papers pertaining to postmortem investigation were collected from scientific databases and reviewed. The papers were published between January 2006 and October 2020.
Results
Most of the analyses involved postmortem blood and urine samples, with a few reports using other bodily specimens and tissues. The method validation was not conducted for all applications. These reports were mostly intended to present data rather than interpret them, and the lack of effort in relating these ethanol biomarkers with the cause of death and/or determination of the time of deaths due to ethanol intoxication might decrease the applicability of these makers after a promising start between 2006 and 2010. Nevertheless, by the beginning of 2020, papers investigating ethanol biomarkers were still increasing. A considerable number of methods used liquid chromatography coupled with mass spectrometry (LC-MS) techniques that require less sample preparation (e.g., protein precipitation extraction, dilution, filtration, and centrifugation). Although solid-phase extraction can be applied, only three applications were reported.
Conclusions
Matrix effects can be a substantial challenge in analytical methods based on LC–MS because they directly affect the ionization of analytes. However, these problems can be avoided due to the high cutoff values used to identify positive results for these ethanol biomarkers, which are often above 0.1–1 mg/L, and using internal standards. Research on using tissue specimens is recommended as most of the reported results on this type of specimen were promising.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Interpreting the role of alcohol as the cause of death in postmortem specimens is a well-known problem in forensic postmortem toxicology investigations. Postmortem fermentation of alcohol can occur in the body immediately after death [1], during transport of biological specimens, and during storage of the body or specimens before testing [2,3,4,5], and so on. Alcohol in postmortem samples can arise in three main ways: antemortem alcohol ingestion; postmortem alcohol synthesis in the body; and ante- or postmortem alcohol formation by microorganisms. The role of microorganisms is especially important when the time between death and analysis or storage conditions of the body are not appropriate [6, 7]. The presence of bacteria and glucose in the bodily specimens and suitable ambient temperature are known to create conditions conducive to alcohol synthesis in a corpse [8,9,10].
Postmortem ethanol testing uses ethanol biomarkers with the aim of proving that ethanol was ingested antemortem, rather than having been synthesized after death by microorganisms [11, 12]. The use of ethanol biomarkers is not a new practice in postmortem investigations; it is a well-established technique dating back to the first report dealing with postmortem formation of ethanol in corpses in 1936 [10]. The ethanol itself or its primary metabolite, acetaldehyde, can serve as a direct ethanol antemortem biomarker. In addition to ethanol and products from phase I metabolism of ethanol, fatty acid ethyl esters or conjugated metabolites such as ethyl glucuronide (EtG), ethyl sulfate (EtS), and phosphatidyl ethanol represent phase II metabolites that can be directly measured (Fig. 1). These conjugated metabolites can indicate ethanol ingestion before death if specimens are collected at the right time and are at less risk of being contaminated by bacteria that can cause ethanol production after death [13]. Some of the phase I metabolites of ethanol that are produced via oxidative metabolism, such as acetaldehyde and acetic acid, can also be measured, but they are difficult to determine with consistency due to technical issues [14, 15]. Polar metabolites of alcohol (EtG and EtS) are biomarkers of antemortem alcohol consumption and are mostly used to exclude postmortem formation of alcohol because they are easy to detect while also providing reliable information. In addition, plenty of data is already available from the literature [1, 11]. These metabolites are formed from ethanol, are not influenced by liver disease, and are detectable in a variety of biological specimens and tissues [2, 3]. No previous studies found that EtG can be formed after death [16] and EtS is known to be stable even under severe putrefaction; both analytes can be measured in corpses and have been detected even after a period of 27 years had elapsed since death. [17].
Although alcohol biomarkers had been known for a long time, the first application for the simultaneous detection of EtG and EtS was reported in 1995 by Schmitt et al. [18]. Most of these reports, which were reviewed elsewhere [13, 19,20,21,22], pertained to living subjects, and only a few dealt with postmortem cases [23]. To our knowledge, no previous review has characterized forensic postmortem toxicology findings with regard to EtG and EtS in ethanol-related fatalities.
Ethanol biomarkers can be classified into two types: direct and indirect. The change of liver function was noticed after long-term alcohol consumption and liver enzymes, for example, gamma-glutamyl transferase, aspartate aminotransferase, alanine aminotransferase, carbohydrate-deficient transferrin, and mean corpuscular volume, are used as indirect biomarkers for alcohol abuse. Changes in these biomarkers are detected after ongoing overconsumption of alcohol (50–80 g of ethanol daily) for at least 1–2 weeks [24,25,26,27]. While direct biomarkers include EtG, EtS, and phosphatidyl ethanol, which are products of ethyl alcohol metabolism and are thus formed prior to death. As such, they are highly sensitive and selective biomarkers that are superior to indirect biomarkers for determining chronic alcohol consumption [28].
Ethanol, or its metabolite acetaldehyde, has been used as direct biomarkers of ethanol ingestion before death [29, 30]. EtG and EtS are biomarkers of antemortem alcohol consumption and are also commonly used to test postmortem toxicological samples. EtG and EtS are formed by glucuronidation and sulfation of the ethanol and reach a maximum blood concentration 3–5 h post ethanol intake. EtG and EtS are both nonvolatile, stable, and water soluble. Their concentration in urine reaches a maximum at around 22–48 h after alcohol consumption. The formation of EtG after conjugation with glucuronic acid is catalyzed by uridine 5ʹ-diphospho-glucuronosyltransferase.
When postmortem ethanol findings are being interpreted, the most important factor is the distribution of ethanol among blood, other bodily fluids, and tissues. The case history should include information about alcohol ingestion, the postmortem interval (PMI), and the circumstances surrounding the body after death. Furthermore, data on the presence of any other putrefaction products in blood (e.g., n-propanol) are necessary. As mentioned above, the absence of EtG could be due to postmortem degradation, but its presence, as well as that of EtS, provides evidence that ethanol was consumed before death. Some people are not able to form EtG due to reduced activity of the uridine 5ʹ-diphospho-glucuronosyltransferase enzyme, which occurs in Gilbert’s syndrome and also in rare cases of Crigler-Najjar syndrome type 1. No corresponding diseases are known to affect the sulfation process, and a lack of both EtG and EtS formation is less likely to occur [12]. Samples submitted to the laboratory may be in the form of bodily fluids, and/or other tissue specimens. They can be analyzed directly or may need pretreatment. There are many methods used for investigating alcohol biomarkers EtG and EtS. Liquid chromatography–tandem mass spectrometry (LC–MS/MS) is one of the most precise and sensitive methods.
This article is a review of the methods for determining the ethanol biomarkers EtG and EtS in postmortem specimens, including sample preparation, stabilities, and interpretation of the results. The review is aimed at improving the understanding and interpretation of ethanol biomarkers of interest among postmortem forensic toxicologists. The foundation of the review is publications collected from scientific databases, such as ScienceDirect, PubMed, and other databases. These databases were searched using the keywords “ethanol biomarkers,” “ethyl glucuronide,” “ethyl sulfate,” “postmortem samples,” “sample preparation,” and “detection methods” to identify research on ethanol biomarkers used in postmortem investigations. The search focused on publications from the period extending from January 2006 to October 2020.
Analytical methods
The most commonly submitted specimen for analysis in postmortem toxicology is blood, and alcohol is the most frequently encountered substance [31]. However, to detect the origin of ethanol in a dead body, its concentration should be measured in several sample types, such as blood from various sites as well as urine and vitreous humor (VH) [4, 32, 33]. The technical procedures used for measuring ethanol levels in blood from a corpse are the same as those used with the living; however, testing to detect alcohol directly needs to be done within 12 h after death since alcohol breaks down rapidly. Researchers have identified alcohol biomarkers for measurements outside this 12-h window [11]. Table 1 summarizes most of the methods reported for the analysis of EtG and EtS in postmortem specimens.
Sample preparation
Measuring ethanol in any type of postmortem specimen is readily done using traditional methods of gas chromatography with headspace technology that requires little sample preparation [1]. When the polar ethanol metabolites EtG and EtS are measured, some techniques require little to no sample preparation and no derivatizing agents are needed. For example, LC–MS techniques, compared with gas chromatography–mass spectrometry (GC–MS) techniques, do not require the use of derivatizing agents. However, some challenges may be encountered due to the heterogeneity of blood samples and the decomposition of specimens. Under ideal conditions, the sample extract is clean without interference from the matrix, and analyses are repeatable. However, matrix effects are a known problem with LC–MS/MS-related testing, with implications for the precision, accuracy, and reproducibility of the results for the analytes of interest [34]. As previously mentioned, less than 0.1% of the ethanol dose is converted to EtG and EtS [23], and such low concentrations require the use of highly sensitive analytical methods for measurement. Consequently, traditional knowledge of ethanol testing cannot be used for evaluating ethanol biomarkers using LC–MS/MS because the analytes’ concentrations, and even units used for expressing analyte concentrations differ. Hegstad et al. [35] investigated the effects of the quality of blood samples for EtG and EtS testing using LC–MS/MS. In that study, three sample preparation techniques were used to produce acceptable chromatography. First, protein precipitation extraction (PPE) was done, followed by phospholipid removal filtration and, in some cases, dilution of blood samples (10 times) using negative blood. However, EtG peaks were split, which could be explained by matrix components competing with analytes of interest and affecting EtG ionization. This problem was solved by reducing the injection volume from 5 to 0.5 µL. However, lowering the injection volume was at the expense of test sensitivity, given that only a small number of four cases were been examined, and it was uncertain whether the results reflected real-world cases.
Many techniques have been used to limit the effect of endogenous compounds on EtG and EtS ionization in LC–MS/MS as indicated in Table 1. Most previous methods to address matrix effect problems have relied on PPE. PPE is quick, easy, inexpensive, and available in all laboratories, and the second approach to tackle matrix effect phenomena is the use of internal standards deuterium-labeled EtG (EtG-d5) and deuterium-labeled EtS (EtS-d5) this enables compensation for the matrix effects. However, despite the advantages of PPE in routine analysis of ethanol biomarkers, it increases the risk of matrix effects for EtG and EtS extraction and the possibility of low recovery, and it introduces matrix interference on the LC–MS/MS system. PPE is known to be a non-selective method of extraction compared to that of liquid–liquid extraction and solid-phase extraction (SPE), PPE does not remove phospholipids from samples containing endogenous matrix compounds, such as fatty acids and steroids, PPE removes a part of the phospholipid content of serum, plasma or whole blood samples, depending on the organic solvent used. Previous studies found that methanol extracts contain 40% more phospholipids compared to acetonitrile extracts, and are also found to be less clean than ethanol extracts [36,37,38]. According to Chambers et al. significant matrix effects up to 67% to 77% ion suppression, were observed with all PPE regardless of precipitation solvent [39]. Moreover, the degradation products of nontarget components i.e. drugs and metabolites are also introduced in the final sample extract. All of these components lead to the contamination of the LC–MS ion source, and thus contribute to matrix effects and reduced sensitivity of the analysis. More clean-up steps for removing these unwanted degradation products and phospholipids components are required along with PPE such as high-speed centrifugation to reduce the interferent levels in PPE final extracts and to obtain a good response from the analytes of interest [1, 35, 40]. These interferences can cause peak splitting, peak shoulders, broad peaks, and peak tailing, and more importantly, the ion source could be contaminated due to the accumulation of deposits that affect the ionization of the target analytes. The ethanol metabolites are very polar and elute early using reversed-phase liquid chromatography techniques, causing analytes to be eluted in the void volume zone and measurement to be susceptible to interference from degradation products and endogenous components of specimens and column materials and a need exists for improved extraction and analysis methods [41,42,43,44]. Therefore, PPE is often combined with other pretreatment technologies for removing phospholipids. Santunione et al. [2] used sonication before PPE and then precipitated the protein using ice-cold methanol at a ratio of 1:4 (v:v) blood and 1:9 (w:v) for liver specimens. Supernatants were obtained following centrifugation for 10 min, then endogenous phospholipids were removed using Phree™ Phospholipid Removal Tubes (Phenomenex, Torrance, California). In that study, the matrix effect was within an acceptable range for method validation (± 25%) with less than 5% carryover observed when blank samples were injected into the system following the analysis of a control spiked with analytes of interest at concentrations at the upper limit of quantification. However, spiked blood samples may not accurately reflect real case samples. In another study, a postmortem blood clot from a middle-aged man who died 27-years earlier only required two steps for sample preparation, including PPE with acetonitrile and then high-speed centrifugation of the supernatants for 10 min, before quantifying EtG and EtS [17]. In that study, kidney and liver tissues were also extracted and analyzed for EtG and EtS. The results for the analytes of interest in these liver and kidney tissues showed no matrix effects were detected. A different approach has been reported using phospholipid plates and high-speed centrifugation [45]. Samples were first centrifuged at a high speed for 5 min, and then the supernatants were filtered using an Amicon Ultra 3 K filter (Merck Millipore, Tullagreen, Ireland); matrix effects were examined, percentages above 100% indicate the presence of ion enhancement, while those below 100% indicate the presence of ion suppression. In that study, both ion suppressions (94%) and ion enhancement (108%) were observed. It seems that high-speed centrifugation is more effective in a combination involving less sample preparation than using PPE with low-speed centrifugation. In recent work, postmortem blood samples were extracted using ice-cold methanol, centrifugation at 13,200g for 5 min, followed by filtration through a 30-mm nylon syringe filter with 0.2-µm pore size (Thermo Fisher, Rockwood, TN, USA). Matrix effects were detected with ion suppression values of 94% and 98% and ion enhancement values of 101% and 108% in blood and urine with respect to the target concentration, respectively [1].
Al-Asmari et al. [11] extracted EtG and EtS from postmortem urine samples using two steps. First, 200 µL of acetonitrile was transferred to a tube containing 100 µL of urine and internal standards, and then the mixture was centrifuged at 3700g for 10 min. In that study, matrix effects remained within a range of 99% to 105%. In fact, urine is thought to be a much cleaner matrix compared with postmortem blood samples, and dilution may be a sufficient pretreatment for urine samples prior to LC–MS/MS.
Direct analytical approaches can be used to analyze postmortem blood and VH samples after collection without any pretreatment other than 1:1 (v:v) dilution with 0.9% NaCl to solve the viscosity issue of VH specimens [46]. Wang et al. [47] developed a method that used dried blood spots on filter paper to quantify EtG and EtS in postmortem specimens. This technique allows stable storage of the collected blood, and it uses methanol for the extraction and n-propanol for reconstitution before injection of the supernatant into the system for analysis.
SPE is believed to be more sensitive and selective in forensic toxicology applications compared with PPE and dilution. It is aimed at preparing a clean extract that can be injected into analyzers with less interference from the matrix. However, it has rarely been reported for the analysis of postmortem specimens for EtG, and no SPE procedure is available for EtS in postmortem specimens yet. SPE has been used for postmortem specimens, with two SPE procedures having been reported for postmortem blood and two others for testing hair [17, 48, 49]. Three of these SPE procedures used aminopropyl SPE cartridges (NH2, 500 mg/3 mL) (Varian, Milan, Italy) and one used Oasis MAX cartridges (30 mg/3 mL) (Waters, Milford, USA). In addition to blood, rib bone marrow, liver, muscle, fat tissue, urine, and bile have been successfully extracted to measure EtG, which was isolated using aminopropyl SPE, with a column conditioned using 2.5 mL of methanol, water, and acetonitrile/hydrochloric acid solution. Following sample-loading, the cartridge was washed using 2 mL of n-hexane, and EtG was then eluted using 1.8 mL of aqueous ammonia solution (1%). Matrix effects were not a concern, with good recoveries ranging from 61 to 97% [48]. In hair samples, EtG was isolated using similar SPE cartridge chemistry. First, the hair shaft was decontaminated using an organic solvent, followed by two steps of washing with dichloromethane and methanol. The hair samples were then cut into small fragments (1–2 mm), spiked with EtG-d5 (100 pg/mg) and incubated overnight with internal standards in deionized water. The mixture was then subjected to ultrasonication for 2 h and centrifuged at 840g for 10 min before the supernatant was loaded into SPE cartridges. In that study, EtG was successfully identified in all hair segments, but it was not possible to quantify EtG due to severe ion suppression issues [17]. Another SPE application was developed by Kharbouche et al. [49] using an Oasis MAX SPE (mixed-mode anion exchange sorbent) to determine EtG in postmortem hair samples, followed by derivatization with perfluoropentanoic anhydride. In that application, EtG was identified and quantified using gas chromatography negative chemical ion mass spectrometry (GC-NCI-MS) at concentrations ranging from 60 to 820 pg/mg.

Ethanol metabolism diagram in human
Determination of EtG and EtS in biological matrices and tissues using LC–MS techniques
Blood
A procedure of LC coupled with single-stage MS for determining EtG in antemortem cases was adapted for determining EtG in postmortem blood cases [16]. In this application, EtG was separated using a Hypercarb HPLC column (2.1 × 100 mm, 5-µm particles size) and protected using a Hypercarb guard column (2.1× 10 mm) (Thermo Electron Corporation, Cambridge, UK). EtG and the corresponding internal standards were separated using an isocratic elution consisting of 25 mM formic acid with 5% acetonitrile at a flow rate of 0.2 mL/min and a run time of 45 min. This application was proved to be sensitive, with a limit of detection (LOD) and limit of quantification (LOQ) values of 0.02 and 0.09 mg/L, respectively. At the time of that work, the phenomena of matrix effects and the importance of analyzing EtS in parallel with EtG had not yet been taken into consideration. In addition, LC–MS was not the gold standard procedure in forensic toxicology compared with GC–MS, and the literature contained few studies about ethanol biomarkers. Further, EtG stabilities had not yet been studied. Also, matrix effects were not considered important parameters in method validation, as shown by the Society of Forensic Toxicologists (SOFT)/American Academy of Forensic Sciences (AAFS) guidelines, for example [50]. In a study by Hoiseth et al. [16] EtG was detected in blood in 19 out of 39 autopsy cases. The intention in that study was to examine the validity of using EtG as an ethanol biomarker for antemortem consumption in real case samples and in cases that tested positive for EtG, and to study if EtG formed after death in any of the negative cases (20 cases).
In a previous report by the same authors, 93 decedents were reported to have a history of antemortem alcohol ingestion and these reports were supported by positive EtG results in all cases, even with low blood alcohol concentration (BAC) [51]. In addition, samples from 53 decedents with no history of alcohol use all tested negative for EtG. In the positive EtG cases, the range of EtG in blood (93 cases) ranged from 0.11 to 56 mg/L. The median concentration of EtG in blood was 0.48 mg/L (range 1.2–40 mg/L) in cases with a high BAC and a history of alcohol ingestion (41 cases). In the second group, which had a history of alcohol use and the intermediate BAC concentration (40 cases), the median EtG concentration was 3.6 mg/L (range 0.5–56 mg/L). In the last 12 cases, which had low BAC and a history of alcohol use, the median EtG concentration was 0.77 mg/L (range 0.11–4 mg/L) [51]. In these studies, EtG was confirmed to be a reliable biomarker for antemortem alcohol ingestion; however, it was not stable under some circumstances. Therefore, additional alcohol biomarkers similar to EtG, but more stable under severe environmental conditions, were sought for postmortem forensic toxicology investigations. A single-quadrupole LC–MS analyzer was able to provide reasonable data on EtG in a postmortem alcohol investigation, leading to the use of more advanced LC–MS/MS techniques with higher sensitivity and selectivity. However, due to the high cost of LC–MS/MS at that time in 2007, few forensic laboratories were able to use this technique in forensic postmortem investigations.
One of the earliest studies using LC–MS/MS to determine EtG was conducted in 2008 [48]. In this study, 12 blood samples from postmortem cases were analyzed using multiple reaction monitoring (MRM) in negative ion mode to determine EtG. In that method, the LOD and LOQ for EtG were 0.05 mg/L and 0.125 mg/L, respectively, as detailed in a previous report [52]. A linear calibration curve over a dynamic range of 0.05–2 mg/L was established, with a good coefficient of determination (R2 = 0.999). In that procedure, the matrix effects and recovery were not investigated. This procedure was suitable for detecting EtG at a concentration range of 0.2–34.9 mg/mL. The median ratio of blood EtG/BAC was 20 (range 8–79). The correlations between BAC and blood EtG were calculated and found to be poor with R2 = 0.425, y = 0.0108x + 0.136.
In another study, the range of EtG concentration in blood samples was 0.64–5.82 mg/L, which was below the LOD (0.083 mg/L) at the lower end of the range; the median blood EtG level was 1.25 mg/L; and the mean value was 2.0 mg/L with 80% recovery [53]. The median ratio of blood EtG/BAC was 1.1 (range 0.4–4.3). The correlations between BAC and blood EtG were calculated and found to be poor, with R2 = 0.306, y = 0.320x + 0.888.
Thierauf et al. [54] conducted a study using a similar LC–MS/MS procedure to that reported by Weinmann et al. [52] and Schloegl et al. [48] and determined EtG and EtS in postmortem blood samples obtained from 26 cases (BAC range of 346–700 mg/dL). In these cases, no sign of putrefaction was observed due to cold weather and the bodies having been refrigerated immediately; however, some of the corpses had PMI between death and sampling of up to 9 days. Thierauf et al. [54] found positive EtG results in 25 out of 26 cases, with concentrations ranging from 0.35 to19.3 mg/L. Meanwhile, EtS was found to be negative in 2 out of 26 cases, with concentrations in positive cases ranging from 0.13 to 4.9 mg/L. The median urine alcohol concentration (UAC)/BAC ratio was 1.4 (range 0.4–1.8) with a good coefficient of correlation, R2 = 0.895, y = 1.19x + 0.171. The median ratios of blood EtG/blood EtS, blood EtG/BAC, and blood EtS/BAC were 2.3 (range 0.3–6.3), 2.5 (range 0.3–19.3), and 0.9 (range 0.4–4.5), respectively. The correlations between BAC and ethanol biomarkers were calculated and found to be poor, with R2 = 0.209, y = 0.313x + 1.27 and R2 = 0.060, y = 0.0413x + 1.70 for blood EtG/BAC and blood EtS/BAC.
An ultra-performance liquid chromatography coupled with electrospray ionization tandem mass spectrometry (UPLC–ESI-MS/MS) method reported by Hoiseth et al. [55] was adopted by Krabseth et al. [12]. EtG and EtS were analyzed using MRM mode and negative ion mode. Analytes of interest were separated using a Hypercarb HPLC column (2.1 × 100 mm, 5 µm) using a column oven temperature of 40 °C during the analysis, and the isocratic mobile phase consisted of 25 mM formic acid with 5% acetonitrile at 0.2 mL/min, with a run time of 20 min. This application proved to be sensitive, with LOD values of 0.02 and 0.013 mg/L for EtG and EtS; whereas, the LOQ was found to be 0.06 mg/L for EtG and 0.028 mg/L for EtS. This method was applied for 493 corpses positive for ethanol, and 60 cases were found to be negative for EtS (12%). In that study [12], 38% of the total cases that had a BAC ≤ 54 mg/dL were considered as having postmortem alcohol synthesis, while only 2.2% of cases that had a BAC ≥ 55 mg/dL, were considered as having postmortem alcohol synthesis. The median amount of EtG and EtS in the blood was 9.5 μmol/L (range not detected n.d. to 618) and 9.2 μmol/L (range n.d. to 183), respectively, while the median BAC was 120 mg/dL (range 10–490 mg/dL) in the total cases while this was 30 mg/dL (range 10–99 mg/dL) in the 60 cases tested negative for EtS. A positive correlation between BAC and EtG was indicated by Spearman’s rho = 0.671 with Spearman’s rho = 0.670 for BAC and EtS. This is in agreement with a previous study that suggested that the probability of new alcohol formation increased with lower BAC concentrations [4].
Vezzoli et al. [29] reported a validated LC–MS/MS procedure for analyzing EtG, which achieved an LOD and an LOQ as low as 0.001 mg/L and 0.005 mg/L, respectively. Linear responses were obtained over the range of the calibration curve from 0 and 1 mg/L, with a coefficient of determination better than 0.998. Matrix effects were examined using three controls at low, medium, and high concentrations (0.05, 0.1 and 0.2 mg/L). In that study, ion enhancement values of up to 118% were reported. Method accuracy and precision were investigated using similar controls. Acceptable coefficients of variation (%CV) were obtained for accuracy and precision with less than %CV: 15% and %CV: 12%, respectively. However, a matrix effect was not investigated at the LOQ level, and high control of both precision and accuracy was not examined at 80% of the linear dynamic range of method (0–1 mg/L) as recommended by the Scientific Working Group for Forensic Toxicology (SWGTOX) guidelines [56]. In addition, the range of positive EtG values were considered low compared with most published methods that considered 0.1 mg/L as the cutoff due to the probability of false-positive results from matrix effects and other ethanol-containing products such as mouthwash [57]. In a study by Vezzoli et al. [29], the concentration of EtG in femoral blood was used as a marker of recent ethanol use in 63 cases. Nineteen of these cases showed ethanol in the blood, with concentrations ranging from 50 to 300 mg/dL. The median blood EtG concentration was 0.18 mg/L (range 0.02 to 3.27 mg/L). In 13 cases, BAC showed a concentration higher than 50 mg/dL, although the observed blood EtG concentration was lower than 0.01 mg/L. These cases had been classified as putrefied cases, with some putrefaction products detected, such as n-propanol. In contrast, in 14 cases, the BAC was lower than 46 mg/dL and the blood EtG concentration was higher than 0.01 mg/L. The median ratio of blood EtG/BAC was 2.8 (range 0.01–35). The correlations between BAC and EtG were calculated and found to be poor with R2 = 0.376, y = 0.0846x + 0.373.
An LC–MS/MS with a triple-quadrupole mass spectrometer operated with MRM and negative ion mode was used for EtG and EtS analysis [58]. In that method, EtG and EtS were separated using a Hydro-RP 100A column (2 × 100 mm, 2.5 μm) and a gradient mobile phase consisted of 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B) at a flow rate of 0.25 mL/min; the total run time was 6 min. This procedure was sensitive for ethanol metabolites, with an LOD of 0.001 mg/L and an LOQ of 0.005 mg/L. Ion suppression was observed in the range of 76–80%, 88–94% and analytical recovery of 78 and 90% was obtained for EtG and EtS, respectively.
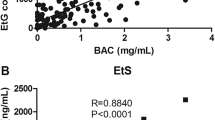
Wang et al. [58], investigated the role of ethanol biomarkers for determining antemortem alcohol consumption, with n-propanol used as a putrefaction marker. In their study, 42 cases were tested for n-propanol, EtG, and EtS. BACs in these cases ranged from 10 to 500 mg/dL. The authors found that n-propanol was not a reliable putrefaction marker, and up to 17% of the cases in which EtG and EtS were present or absent would be considered false results if only n-propanol results were used. In addition, the concentration of EtG in blood was always higher than that of EtS, which was in agreement with previous studies [3, 29, 59]. In these cases, good correlations were observed between BAC and ethanol biomarkers with R = 0.792 for EtG and R = 0.884 for EtS.
A UPLC–MS/MS method for EtG and EtS was developed and validated for the testing of blood specimens obtained at autopsy with putrefaction and without putrefaction of the decedents’ corpse [35]. The authors used a hydrophilic interaction LC column (HSS T3 column, 2.1 × 100 mm 1.8 μm; Waters) to separate analytes of interest, which was maintained at a 50 °C oven temperature. Separation of analytes of interest was conducted using a gradient mobile phase consisting of 0.1% formic acid in water (A) and 100% methanol (B); the run time was 2.5 min which is the shortest run time reported yet for the analysis of EtG and EtS. A Xevo TQ-S tandem MS (Waters instrument) equipped with a Z-spray electrospray interface was used for the quantification and identification of analytes of interest using negative ESI mode. This procedure provided a good coefficient of correlation (0.999) over the ranges of the calibration curves from 0.08 to 2 mg/L EtG and 0.025 to 2 mg/L EtS. Method accuracy and precision were examined using three controls at 0.12, 0.6, and 1.5 mg/L for EtG and 0.04, 0.6, and 1.5 mg/L for EtS. Accuracy was expressed as the relative error (%E), and values were in the range of − 2.4% to 1.8% with a within-assay %CV value in the range of 2.7–4.6% and a between-assay %CV value ranging from 1.7 to 7%. An ion suppression matrix value of less than 80% was observed, while extraction recoveries were approximately 51 and 55% for EtG and EtS, respectively. Nine autopsy blood samples were tested for EtG and EtS, the median EtG and EtS concentrations were 1 mg/L (range 0.2–7.5 mg/L) and 1.1 mg/L (range 0.1–1.9 mg/L), respectively. In these cases, the ratio value of the median blood EtG/blood EtS was 1.5 (range 0.2–3.9), and a good correlation relationship was observed between EtG and EtS with R = 0.643, y = 3.26x − 1.24.
Liu et al. [60] studied the stabilities of EtG and EtS in six real postmortem blood samples. They used their previous sensitive method of analysis [44], which was developed to achieve sensitive detection of lower LODs which ranged from 0.0001 to 0.01 mg/L for EtG and EtS, while LOQ was 0.02 mg/L and was 0.0005 mg/L for EtS. Calibration curves for EtG and EtS were constructed over the range of concentrations from 0.02 to 20 mg/L and 0.001 to 20 mg/L, respectively, which revealed a correlation of determination of R2 = 0.994 for EtG and R2 = 0.992 for EtS. Four controls for each analyte were examined for accuracy, precision, and recovery, and all were within the acceptable limits for method validation < ± 15%. Ion suppression values of 67% were observed of the target EtG concentration, which resulted in apparent low extraction recoveries less than 30%. The values for ion suppressions of 85% and ion enhancement of 115% were observed of the target EtS concentration and had a much better extraction recovery, exceeding by 80% that for EtS. Liu et al. [60] reported six cases with BAC 88–300 mg/dL which were investigated, and the results showed the range of EtG was 0.4–7.3 mg/L, while the EtS range was 0.3–1.5 mg/L. In these cases, the ratio value of the median of blood EtG/blood EtS was 1.9 (range 0.8–4.8), and a good correlation coefficient was observed between EtG and EtS with R2 = 0.582, y = 3.85x − 0.958. However, the correlation between ethanol biomarkers concentration and BAC was not good, with R2 = 0.319, y = 0.180x + 1.27 and R2 = 0.012, y = 0.1999x + 1.58, respectively, which was in agreement with previous studies [35, 54].
A recent study [1] investigated the EtG and EtS concentrations in blood samples using triple quadrupole LC–ESI/MS, operating with MRM and in negative ion mode. Fast elution of the analytes of interest in less than 2 min was obtained using a Raptor Biphenyl column (50 × 3.0 mm, 2.7 μm, Restek, USA) coupled with a Security Guard Cartridge (5 × 3.0 mm, 2.7 µm). The mobile phase consisted of 10 mM ammonium formate adjusted to pH 3 (A) and methanol (B) over a run time of 5 min at a flow rate of 0.3 mL/min. All calibration curves were linear, with coefficients of determination greater than 0.999. The LOD ranged from 0.05–0.06 mg/L for analytes of interest. The LOQs were 0.075 mg/L for both EtG and EtS. Within-run precision and between-run precision were acceptable with %CV less than 11% of the target concentration. Matrix effects for EtG and EtS observed an ion enhancement value ranging from 103 to 147%, and analytical recoveries ranged from 72 to 100%. This validated method was then used for routine postmortem forensic toxicology analyses in 592 postmortem cases to distinguish between antemortem ethanol consumption and postmortem ethanol formation by microbes. The median BAC in blood samples in the antemortem alcohol cases was 138 mg/dL (range 11–537 mg/dL), the median blood EtG concentration was 3 mg/L (range 1–156 mg/L), and the median blood EtS concentration was 2 mg/L (range 0.3–99 mg/L). The BAC, EtG and EtS were reported in cases that had some signs of putrefaction (5 cases), while only BAC was measured in heavily putrefied cases (7 cases). The use of EtG and EtS in parallel was found to provide accurate results for distinguishing between antemortem and postmortem BAC sources. Five samples were positive only for EtG and EtS and negative for ethanol, and 27 cases were positive for ethanol, EtG, and EtS. The ratio value of the median values for blood EtG/blood EtS, blood EtG/BAC (mg/mL), and blood EtS/BAC (mg/mL) were 3 (range 0.9–9), 3.5 (range 0.4–262), and 1.4 (range 0.2–231), respectively. A correlation coefficient between EtG and EtS was low with R2 = 0.378, y = 1.41x + 9.63. Similarly, the correlation between ethanol biomarkers and BAC was not good, with R2 = 0.003, y = − 0.0018x + 1.53 and R2 = 0.020, y = − 0.0114x + 1.57, which was in agreement with previous studies [35, 54].
Urine
Blood samples are not always available at autopsy due to trauma, decomposition, and exsanguinations that may occur with fatalities related to automobile accidents or other circumstances. Therefore, the ability to detect ethanol and metabolites in other bodily fluids and tissues is essential for toxicology analysis [61].
UAC is always tested in parallel with BAC, which is considered a reliable means for interpreting the source of alcohol at autopsy and thus whether it is due to antemortem alcohol consumption or postmortem synthesis by microorganisms. Urine is a useful specimen, especially for alcohol testing, because it is usually free of glucose in healthy individuals. In many postmortem forensic toxicology practices, negative UAC and positive BAC would be interpreted as an indication of postmortem synthesis of alcohol [4]. The role of urine alcohol testing in antemortem and postmortem analysis is discussed in detail by Jones [32]. A few studies have reported EtG alone in postmortem urine analysis [2, 16, 48, 53] and a single study reported urine EtS alone [62], while many studies have reported the simultaneous determination of EtG and EtS in routine postmortem urine analysis since 2006 [1, 11, 54, 59, 63]. The earliest studies found that urine had the highest EtG concentration compared with other bodily fluids and tissues [48, 51].
In 2006, LC–ESI-MS was used for the determination of EtG in different postmortem bodily fluids and tissues [48], and the LOD and LOQ were found to be low for analysis of urinary EtG at 0.02 and 0.06 mg/L, respectively. The details of the methods of analysis are discussed in the section about blood above. Most of the validation parameters were examined for blood samples [48]. The use of calibration curves derived from blood for calculating results from other postmortem specimens is acceptable according to SWGTOX guidelines; however, quality control samples prepared in matrices of interest should be examined which was not conducted in Schloegl et al. study [56], the focus of that study was to introduce new information on the stability of EtG in postmortem specimens rather than method validation of a new procedure.
LC coupled with single-mode MS was used to detect EtG in urine in two previous reports [16, 53]. In these reports, single-ion monitoring with negative ion mode at m/z 221 for EtG and m/z 226 for EtG-d5 were used to validate the analysis. Another two applications used MS/MS for autopsy urine specimens [2, 48]. In these applications, EtG and EtG-d5 were identified and quantified based on MRM negative ion mode using the following transitions: EtG m/z 221 → 75 and 221 → 85, and EtG-d5 m/z 226 → 75 and 226 → 85. In a study by Schloegl et al. [48], the median UAC/BAC ratio was 1.4 (range 1.2–1.8) and the median ratio of urine EtG concentration (mg/L)/UAC (mg/dL) was 0.3 (range 0.1–1). The correlation coefficient was poor between UAC and urine EtG, with R = 0.40.
In the paper by Hoiseth et al. [16] the analysis of urinary EtG testing had a LOQ of 0.1 mg/L, and the EtG calibration curve was found to be linear over the concentrations ranging up to 20 mg/L. Two controls were used to investigate the method precision (low: 0.15 mg/L and high: 6 mg/L), and relative standard deviations (RSD) in intraday and interday precision were < 10 and 2%, respectively. Keten et al. reported an LOD and an LOQ of 0.12 and 0.36 mg/L, respectively. The method enabled plotting a linear calibration line over a range from 0.05 to 50 mg/L, and the value of extraction recovery was almost 90% [53].
Santunione et al. [2] reported a fully validated LC–ESI/MS procedure for the analysis of 21 samples of various postmortem specimens including urine. This procedure was validated according to the SWGTOX guideline [56]. EtG was separated using an Atlantis T3 100 Å reversed-phase C18 column (150 × 2.1 mm; 3-μm particle size) with a guard column (50 × 2.1 mm) (Waters, Milford, MA), kept at 40 °C. The total run time was 20 min through operating a gradient elution system consisting of aqueous formic acid (0.1%) and acetonitrile. The LOQ was higher compared with previous reports [16, 53], and was reported to be 0.5 mg/L. Accuracy values were found to range from − 10 to + 10%, while intrabatch and interbatch precision values were lower and a %CV less than 10% was obtained. In 13 of the 21 cases, urine samples were available for EtG measurement. The median urine EtG concentration was 22 mg/L (range 2–373 mg/L), and the median ratio between urine EtG and blood EtG concentrations was 19.2 fold (range of 2–104) [2]. In another study by Schloegl et al. [48] urine EtG concentrations ranged from 15 to 509 mg/L, with a median concentration of 81 mg/L and the ratio of the concentrations of urine EtG/blood EtG of 15 fold (range 6–31). Similar ratios were also reported by Keten et al. [53], who showed a median ratio of 18.6 fold (range 3–142) and a higher urine EtG median concentration of 43 mg/L (range 0.3–632 mg/L).
Research on the usefulness of including EtS and EtG in postmortem alcohol testing was started at the same time. However, most of the investigations focused on EtG only, and methods were initially unavailable for measuring EtS with LC–MS and GC–MS. The first use of EtS measurement in postmortem urine analysis was published in 2006 [63], In that first study, LOD was set at 0.1 mg/L, matrix effects were observed for both EtG and EtS with ion suppression values up to 70% and ion enhancement values up to 115%. Two sets of calibration curves were constructed to accommodate the broad range of EtS concnentrations (0.1–15 mg/L and 5–750 mg/L), with three different controls for each set to examine the method accuracy and precision (0.1, 1.25, and 15 mg/L and 5, 62.5, and 750 mg/L, respectively). The RSDs for intraday and interday precision ranged from ± 0.6 to ± 4.7% and from ± 0.8 to ± 8.3%, respectively.
Bicker et al. [63] presented data for 52 autopsy urine samples (BAC, < 5–402 mg/dL; UAC, 24–542 mg/dL), with EtG and EtS being positive in 50, and 51 cases having median concentrations of 98.4 mg/L (range 0.1–1380 mg/L) and 28 mg/L (range 0.1–220 mg/L), respectively. In that study, good correlation was found between EtG and EtS, with median EtG concentration/EtS concentration ratios of 2 (0.35–5.6). However, UAC was poorly correlated with concentrations of both ethanol biomarkers.
Al-Asmari et al. [11] developed a method for urine analysis of EtG and EtS using a hydrophilic interaction liquid chromatography column for the first time in 2010, finding that the LC–ESI/ion trap MS/MS technique was efficient for direct determination of EtG and EtS in urine obtained from postmortem cases. They used selective reaction monitoring and negative ion mode. In that study, the following transitions were used in MRM mode: m/z 221 → 203 and m/z 221 → 113 for EtG; m/z 226 → 208 and m/z 226 → 118 for EtG-d5; m/z 125 → 97 and m/z 125 → 125 for EtS; and m/z 130 → 98.5 and m/z 130 → 130. for EtS-d5, and 10% of precursor ions remained as qualifier ions while the main product ions were used as quantifier ions. EtG and EtS were separated using a ZIC®-HILIC column (150 × 2.1 mm, 3.5 μm) protected by a guard column with identical packing material of a larger particle size (14 × 1.0 mm, 5 μm/200 Å) (Merck SeQuant, Umea, Sweden). The gradient mobile phase consisted of 5 mM ammonium acetate and acetonitrile at 0.2 mL/min, with a run time of 20 min. Low LOD values (0.001 mg/L) for EtG and EtS were obtained, with LOQ values of 0.13 mg/L and 0.2 mg/L for EtG and EtS, respectively. Linear calibration curves for EtG and EtS were established over the range of 0.05–10 mg/L. Three quality controls at low, medium, and high concentrations (0.05, 0.5 and 2.5 mg/L) were spiked into blank urine to investigate matrix effects, recoveries, accuracy, and precision. RSD values of < ± 8 and ± 11% were observed for intra-assay and interassay precision, respectively. The value for accuracies, matrix effect, and extraction recoveries ranged from − 3 to + 3%, from 99 to 105%, and from 96 to 101%, respectively. This method was used for routine urine postmortem analysis in 90 cases, and EtG concntrations ranged from 2 to 1500 mg/L and EtS from 0.5 to 400 mg/L, with mean (median) concentrations of 250 (120) mg/L for EtG and 33 (62) mg/L for EtS [11].
In 20 urine samples, the LOD of EtG was 0.09 mg/L and the range was 0.3–244 mg/L, based on analysis by LC–ESI/MS negative mode [59]. In addition, in 14 out of 19 cases that were analyzed to determine the levels of both EtG and EtS, the median concentrations of EtG and EtS in urine were 35.9 (range 1.0–182) and 8.5 mg/L (range 0.3–99), respectively. The median ratio between EtG and EtS concentrations in urine was 2.2 (range 0.03–7.8). Thierauf et al. conducted a similar study in 26 cases, and they found urine EtG concentrations ranged from not detected to 150 mg/L, with a UAC of 42–459 mg/L [54].
Al-Asmari and Al-Amoudi [1] have analyzed urine samples to determine EtG and EtS. The LOD of EtG and EtS was 0.05 mg/L and the LOQ was 0.075 mg/L. Moreover, the value for matrix effects and extraction recoveries were 107–147% and 72–89%, respectively. Linear calibration curves for EtG and EtS were established over the range of 0.05–4 mg/L. Three quality controls at low, medium, and high (0.1, 1.0 and 3 mg/L) were spiked into blank urine samples to investigate method accuracies (%E), within-run precision, and between-run precision, and the results ranged from; %E − 4 to + 1%, CV% 1–10%, and CV% 1–11%, for EtG and EtS, respectively. In this study [1], urine samples obtained from 32 autopsies were tested for ethanol biomarkers, in these cases, the median UAC was 125 mg/dL, and the mean UAC was 203 mg/dL (range 10–1180 mg/dL). All but eight cases were available for EtG and EtS testing, which had median concentrations of 52 mg/L (range 2–685 mg/L) and 22 mg/L (range 3–155 mg/L), respectively. Urine is typically not available in most cases involving alcohol synthesis, and it was available for only one-third of the cases reported in that study. The UAC/BAC ratios were calculated in many groups of samples to investigate their usefulness in distinguishing between antemortem alcohol consumption and postmortem alcohol synthesis. In fact, many UAC tests were positive despite the cases being classified as being due to alcohol postmortem synthesis. In contrast, ratios of UAC/BAC in putrefaction and nonputrefaction cases could not be distinguished, due to low BAC and UAC. Therefore, EtG and EtS are the indicators of choice for the identification of antemortem alcohol consumption. In the study Al-Asmari and Al-Amoudi, higher UAC/BAC concentrations ratios than expected (one third) were obtained for confirmed ethanol antemortem consumption cases for which the presence of antemortem and postmortem alcohol sources was proposed. EtG and EtS concentrations in cases in which alcohol was the only drug detected were higher than in cases in which alcohol was detected along with other drugs. The mean concentrations were 193 mg/L (18–649 mg/L) and 110 mg/L (2–475) for EtG and 43 mg/L (3–155 mg/L) and 29 mg/L (3–124 mg/L) for EtS, respectively. In addition, in cases that were positive for antemortem alcohol, EtG and EtS concentrations were higher than in cases that showed some putrefaction signs compared with cases without putrefaction. The median concentrations were 257 mg/L (34–445 mg/L) and 126 mg/L (2–649 mg/L) for EtG, while the EtS concentrations were 95 mg/L (3–156 mg/L) and 21 (3–124 mg/L), respectively. The UAC/BAC ratios in cases in which only ethanol was detected were almost within 1–2, with a mean and median of 1, compared with cases in which ethanol was detected with other drugs. In the latter cases, the ratios ranged from 1 to 10, with a mean and a median of 3 and 1, respectively. Al-Asmari and Al-Amoudi [1] stated that the correlation of BAC or UAC with both EtG and EtS was often poor, which was supported by previous reports [11, 63]. In a similar manner, UAC/BAC ratios that were higher than expected were found in 52 urine samples obtained from postmortem cases, with a mean and a median of 2 and 1 (0.5–22) [63]. No differences were observed in the urine EtG/EtS concentration ratios between groups of cases classified as alcohol only, alcohol with other drugs, nonputrefaction, and some putrefaction; the median ratio in all groups was 3. In previous studies, the median ratios of urine EtG/EtS concentrations were 3.5 [63], 4.8 [11], 1.8 [54] and 3.8 [59].
Vitreous humor
Blood, urine, and VH have been recommended as specimens for alcohol analysis. VH specimens are less prone to postmortem alcohol synthesis because they are free of glucose. Further, any microorganism contamination is limited in the early stages of the putrefaction process [4, 64], and in later postmortem stages, VH is hardly contaminated [54, 65]. These factors make VH a valuable specimen for ethanol testing in cases in which blood is not available or when postmortem ethanol synthesis is suspected [29]. VH has been extensively investigated in postmortem analyses for many drugs and metabolites and especially for alcohol; however, only five applications were reported for the analysis of EtG [29, 46, 47, 53, 54], with only two including EtS [47, 54].
In the first report of the process, VH samples for EtG testing were obtained from 25 autopsy cases [53]. The mean age of the deceased was 46 years old (range: 26–65 years). Information on the time interval between death and sample collection was available in 23 cases and it ranged from 12 to 36 h, with a median of 17 h. BACs were 33–358 mg/dL. All cases were positive for EtG and blood ethanol, except in one case that was negative for blood ethanol. EtG was measurable in blood in 21 out of 25 cases, in urine in 17 out of 19 cases, and in VH in 19 out of 25 cases. The negative VH could be explained by EtG pharmacokinetics, whereby alcohol starts to be detectable in VH 30 to 45 min after ingestion [66]. In Keten et al. [53] correlations between VH alcohol concentrations (VHAC) and BAC were not mentioned, but good correlation coefficients were found between VH EtG/blood EtG concentrations (R2 = 0.54) and VH EtG/urine EtG concentrations (R2 = 0.84). The median VH EtG/blood EtG ratios were calculated and found to be < 1 inall tested cases (range 0.02–0.8). The same study reported that VH EtG was present in measurable amounts in 19 cases. The VH EtG levels ranged from 0.05 to 1.90 mg/L and were below the LOD (0.03 mg/L) in six.
The study by Keten et al. [53] aimed to investigate VH EtG as a valuable postmortem tool to examine antemortem alcohol consumption. An LC–MS/MS method was developed to quantify EtG in blood, urine, and VH using a single-stage LC–ESI/MS instrument, and the EtG and EtG-d5 which were measured in negative mode with single ion monitoring at m/z 221 and m/z 226, respectively. Analytes of interest were separated using an isocratic mobile phase containing 0.1% formic acid and 5% acetonitrile over a run time of 30 min at a flow rate of 0.5 mL/min, with separation on Zorbax Bonus RP Rapid Resolution column (150 × 4.6 mm, particle size 3.5 µm). The LOD and LOQ for EtG were 0.083 and 0.249 mg/L, respectively, and the method produced a linear response over a range of 0.5–50 mg/L. Matrix effects on analyte ionization were examined using post-extraction blank blood, urine and VH samples and a recovery value of 80% of the target concentration was obtained. Although the method was well designed, some important validation parameters such as accuracy and precision were not conducted. Many of the positive cases had trace levels of the analyte of interest (between LOD and LOQ range). It is recommended to use LOQ concentrations as the lowest point on the calibration curve; however, in this procedure, the first point on the calibration curve was two times as high as the LOQ.
Thierauf et al. [54] reported the use of LC–MS/MS to analyze both EtG and EtS in VH obtained from autopsied corpses. Analytes of interest were separated using a polar-end capped phenylpropyl reverse-phase column (Synergi Polar-RP 250 × 2 mm, particle size 4 µm) with a guard column (4 × 2 mm) (Phenomenex, Aschaffenburg, Germany), using an isocratic elution (0.1% formic acid) with a flow rate of 0.2 mL/min and a run time of 20 min. The calibration curve extended from 0.05 to 50 mg/L for EtG and from 0.04 to 50 mg/L for EtS. The LOD was 0.05 mg/L for both EtG and EtS. In 26 cases in which EtG was analyzed in VH samples, the results showed that the range for EtG was 0.1–9.4 mg/L with a BAC level of 10–432 mg/dL, while the EtS range was 0.23–4.1 mg/L. The median BAC was 200 mg/dL (range 70–350 mg/dL), and the VHAC was 250 mg/dL (range 10–430 mg/dL), while the VHAC/BAC and blood EtG/VH EtG concentration ratios were 1.3 (range 1.1–1.8) and 2.2 (range 0.5–6.2), respectively. Good correlation coefficients were observed between BAC/VHAC and UAC/VHAC, with R2 = 895, y = 1.19x + 0.171 and R2 = 0.878, y = 0.798x + 0.313, respectively. However, poor correlation coefficients were obtained for EtG and EtS concentrations between matrices. Therefore, to distinguish between antemortem and postmortem alcohol sources, it is important to calculate ratios between alcohol concentrations in blood, urine, and VH, in addition to detecting EtG and EtS concentrations.
Many factors can influence the detectability in matrices, including the elimination rate and distribution based on the time of drinking and the time when analytes of interest reach various organs throughout the body. The authors [54] stressed that deaths may occur at any time point during ethanol elimination; for example, a UAC/BAC ratio of 1.25 or above indicates that ethanol was completely absorbed and distributed throughout the body. A higher urine concentration could be expected, however, and in some cases, a negative UAC and BAC with a low concentration in VH were encountered. Such cases can be interpreted in many ways, for example, either the VH is susceptible to alcohol synthesis post mortem or elimination was delayed. The authors excluded the first option due to high EtG and EtS concentrations being detected. In addition, the amount detected in VH could be a result of previous alcohol intake that has not yet been eliminated due to a slow elimination rate in VH.
Vezzoli et al. [29] reported a validated LC–MS/MS procedure for analyzing EtG in VH from a cadaver. This procedure achieved an LOD and an LOQ as low as 0.001 and 0.005 mg/L, respectively. Linear responses were obtained over a range from 0 and 1 mg/L, with a coefficient of determination better than 0.998. Matrix effects were examined using three controls at low, medium, and high concentrations (0.05, 0.1, and 0.2 mg/L) and the values ranged from 81 to 92%. Method accuracy and precision were investigated using similar controls examined for matrix effects. Acceptable %E and CV% values were obtained for accuracy and precision at ± 15% and 12%, respectively. However, matrix effects were not investigated at the LOQ level, and for the high control, neither precision nor accuracy was examined as recommended by SWGTOX guidelines [56]. In addition, the range of positive EtG values was considered low compared with most published methods, which considered 0.1 mg/L as the cutoff due to the probability of false-positive results. This is due to matrix effect and other ethanol-containing products such as mouthwash [57].
Vezzoli et al. [29], the VH EtG concentration was used as a marker of recent ethanol use in 63 cases, in which the median BAC was 30 mg/dL (range 10–230 mg/dL) and VHAC was 10 mg/dL (range 10–130 mg/dL). The VHAC/BAC and blood EtG/VH EtG concentration ratios were 0.6 (0.12–3.1) and 2 (0.1–18.2). Ethanol biomarkers and acetaldehyde and n-propanol were included to distinguish antemortem alcohol consumption and new formation of alcohol after death. Despite a good correlation between concentrations of EtG in blood and VH, with a correlation better than 90%, few cases with BAC > 6 mg/dL, were tested negative for acetaldehyde, and negative for EtG and n-propanol. This brought the use of putrefaction marker (n-propanol) and direct ethanol metabolites (acetaldehyde) into question regarding their usefulness in distinguishing between antemortem and postmortem ethanol production. In addition, three cases that were positive for BAC (49–125 mg/dL) had negative EtG results for blood and VH and positive acetaldehyde results. Four cases showed positive results for EtG in blood but were negative for the VH specimens. In contrast, one case showed a positive EtG in VH with negative EtG results for blood. Clearly, using VH as the only sample type for EtG would yield false-negative results. A negative VH EtG finding with positive blood EtG may be interpreted as indicative of a short amount of time elapsing between drinking and death (< 3 h). This conclusion is supported by work by Keten et al. [53] and a pharmacokinetic study of EtG reported by Hoiseth et al. [66]. Acetaldehyde is known to be a primary alcohol metabolite but it is not recommended as an antemortem biomarker of alcohol consumption because it is detected in low concentrations and there is a lack of proper analytical techniques for obtaining reproducible data [14, 15].
Wang et al. [47] developed a method for the analysis of EtG and EtS in blood and VH. In that procedure, MS/MS data were collected using MRM with a negative ion mode. Analytes of interest were separated on a Synergi Hydro-RP 100A column (2 × 100 mm, 2.5 μm) column oven kept at 40 °C during analysis, with gradient elution using 0.1% formic acid in water and 0.1% formic acid in acetonitrile with a flow rate of 0.25 mL/min and a 6-min run time. A linear calibration curve over a range from 0.005 to 10 mg/L for EtG and EtS was achieved. LOD and LOQ for analytes of interest were 0.002 and 0.005 mg/L, respectively. Accuracy values were ± 15%, and intraday and interday precision with %CV less than 10% of the target concentration was achieved in spiked controls for blood and VH. No obvious suppression effect was observed for EtG and EtS in both whole blood and VH, and extraction recovery values of analytes of interest ranged from 78 to 90% in blood and from 95 to 101% in VH. In 10 autopsy cases, both blood and VH were available for testing. In that study, blood EtG and EtS concentrations ranged from not detected to 22.3 mg/L and not detected to 2.1 mg/L, respectively. This method and previous investigations using real cases showed that ethanol biomarkers (EtG and EtS) in blood specimens were reliable and useful when interpreting the source of ethanol in postmortem samples.
Other biological fluids and tissues
Alternative specimens to blood and other traditional postmortem specimens such as urine and VH for ethanol biomarker testing are described in only three reports for EtG [2, 17, 48] and one report for EtS since 2006 [17]. Applications using hair were described in three reports [17, 49, 67].
Bone marrow, bones, and bile
Although the first application reported by Schloegl et al. [48] for EtG testing in multiple human specimens was promising, only EtG was examined and the stability of EtG under severe conditions had not been studied in 2006. In that study, multiple body fluids and tissues of 12 corpses were investigated, including blood, urine, bone marrow, liver, bones, bile, and gastric content. The LC–MS/MS method reported by Weinmann et al. [52] was adapted for that analysis. For 9 of 12 specimens, rib bone marrow was positive for EtG, with a median concentration of 1.5 mg/kg (range 0.5–9.4 mg/kg). Bile is another specimen that is commonly collected at autopsy, and in this study, bile from seven corpses was tested and the median concentration of EtG was 7 mg/L (range 1.1–42 mg/L). In addition, muscle and fat tissues were also available for testing, and the median EtG concentrations were 0.6 mg/kg (range 0.1–1.8 mg/kg) and 0.5 mg/kg (range 0.2–1.2 mg/kg), respectively. Correlation coefficients were good between blood and other tested matrices, except for muscle and fat tissues, which showed poor correlations: R2 = 0.401, y = 0.0557x + 0.50 and R2 = 0.058, y = 0.0132x + 0.5096, respectively. Other tissues showed a good correlation for EtG concentrations in bile, and bone marrow: R2 = 0.814, y = 2.17x + 2.48; and R2 = 0.662, y = 0.318x + 0.0047, respectively.
Liver, kidneys, and blood clots
Schloegl et al. [48] reported EtG analysis in liver obtained from putrefied corpses available in seven cases, for which the median concentration of EtG was 43 mg/kg (range 7–77 mg/kg). A good correlation for EtG concentrations between liver and blood was reported with R2 = 0.657, y = 3.2397x + 15.478. Liver and kidney tissues were the only types of matrix other than blood clots to be tested for EtG and EtS 27 years after death [17]. In this particular case, a 50-year-old man with a known history of alcoholism was found dead, with his body on a concrete floor. The cause of death was thought to be suicide by hanging, but 27 years later, the case was reopened as a possible homicide due to evidence that the man’s death was associated with a series of murders. The man’s body was exhumed and found to be naturally mummified upon autopsy. This allowed the analysis of blood clots and fragments of liver and kidneys for EtG and EtS in the biological material 27 years after death. EtG and EtS were measured in blood clot, liver, and kidney, and following PPE extraction, 20 µL of final extract was injected into LC–MS/MS instrument for analysis. EtG and EtS were identified and quantified using MRM and negative ion mode with the following transitions: m/z 221 → 75, m/z 221 → 85 for EtG, m/z 226 → 75 and m/z 226 → 85 for EtG-D5, m/z 125.1 → 96.9, m/z 125.1 → 79.9 for EtS, respectively. Analytes of interest were separated using the Chrompack Inertsil ODS-3 column (100 × 3 mm, 3-μm) equipped with a Chrompack (10 × 2 mm) RP guard column (Varian, Walnut Creek, CA, USA), and the flow rate and column temperature were kept at 0.2 mL/min and 25 °C, respectively. A gradient mobile phase containing 0.1% formic acid and acetonitrile was used for separation, within a run time of 30 min. The concentrations of EtG were 0.219, 0.141, and 0.250 mg/kg and those for EtS were 0.645 mg/L, 0.535 mg/kg, and 0.945 mg/kg in blood clots, liver, and kidney, respectively. Despite using an old validated method for their investigation [68], no validation parameters were detailed, given that the specimens differed from those used for validation. In contrast to the Schloegl et al. study [48], which showed that the liver had the highest EtG concentration, a study by Politi et al. [17] indicated that kidney tissues had almost double the concentration found in the liver. These studies clearly showed that EtG and EtS are stable over a long period of time under proper storage conditions. Although these studies provided important information, they lacked basic method validation with real case samples.
In the third application using liver, the tissue was tested for EtG in parallel with blood and urine samples [2]. Liver specimens were tested for 21 cases, and the median EtG concentration was 13 mg/kg (range 1.7–105 mg/kg). A good correlation was found between liver and peripheral blood, with R2 = 0.835, y = 0.167x − 0.190, while liver and central blood had an R2 = 0.643, y = 2.47x + 0.960. The median ratio between EtG concentrations in the liver and peripheral blood was 8 (range 2.1–37), while that for liver/central blood concentrations EtG was 2.4 (range 1.1–10), which indicated that the concentration of EtG was affected by postmortem redistribution. Therefore, the use of multiple specimens is recommended when EtG is being tested to avoid misinterpretation that could introduce error in the medical–legal investigation. It is seen that a higher concentration of EtG in the liver compared to the blood is due to the high level of the liver enzyme uridine-5'-diphospho-glucuronosyltransferase, which converts ethanol to EtG. In that study, urine EtG was found to have a higher concentration than liver EtG, which was in agreement with previous reports by Schloegl et al. [48].
Hair EtG testing
Hair specimens are preferable autopsy specimens for forensic investigations because they are distant from internal postmortem changes, especially in putrefied corpses. Therefore, hair testing has become a reliable tool for detecting drugs in forensic applications; however, ethanol itself cannot be directly tested in hair due to its volatile nature and because of possible contamination from outside sources. With advancing LC–MS technology and the discovery of EtG as a biomarker of recent alcohol use, the use of hair as a testing matrix became possible [67, 69]. The cutoff approved by the Society of Hair Testing (SoHT) to consider EtG in hair sample for repeated alcohol consumption was set at ≥ 7 pg/mg EtG in the 0–3 up to 0–6 cm proximal scalp hair segment, while chronic use cutoff was set at ≥ 30 pg/mg in 0–3 up to 0–6 cm proximal scalp hair [69, 70].
For investigating the suitability of using EtG in postmortem hair samples in cases of previous alcohol abuse, Bendroth et al. [67] studied 70 cases in which 100 mg of hair was treated with deionized water and incubated overnight in the sonicator, after which final extracts were injected into UPLC–ESI/MS. The EtG and EtG-d5 were identified in negative mode with selected ion monitoring at m/z 221 and m/z 226, respectively. Analytes of interest were separated using a high-strength silica (HSS) trifunctional C18 column (2.1 × 100 mm, 1.8 µm particle size). A gradient mobile phase consisting of 0.1% formic acid in water, pH 2.85, and 100% acetonitrile was used at a flow rate of 0.4 mL/min, with a run time of 2.6 min. The LOD for EtG was 0.9 pg/mg and the LOQ was 2.5 pg/mg. The method produced a linear response over a range of 5–15,000 pg/mg. Matrix effect values on EtG response were almost 70%, and the effect on quantification was compensated for by the use of the deuterated internal standard. This procedure achieved a %CV of < 10% for within-run and between-run precision.
In a study by Bendroth et al. [67], 70 autopsy hair samples for medical–legal investigations were examined. The average sample weight was 100 mg, and the mean age of decedents was 47 years (range 18–68 years); 17% of cases involved women. Despite EtG being detected in 65 cases, only 49 cases were considered positive, with the EtG concentration ranging from 8 to 10,400 pg/mg. Among the 49 positive cases, 39 were identified as indicative of chronic alcohol use based on EtG concentrations being ≥ 30 pg/mg according to SoHT guideline [69, 70], with the caveat that an EtG concentration ≤ 30 pg/mg in hair does not exclude alcohol abuse. In this application, testing EtG provided reliable information about the decedents’ history of alcohol abuse, given that EtG formation only occurs enzymatically when a person is alive. The use of liquid–liquid extraction provided results in most cases; however, the use of SPE could increase recoveries and provide much cleaner extracts.
SPE with LC–ESI/MS techniques was used to determine EtG in a hair sample from an autopsy completed 27 years after death [17]. The sample weight was 100 mg. The calibration curve was linear (y = 0.00894x + 0.00541, R2 = 1.0000), and quality controls were acceptable with an RSD of less than 11.4% for both accuracy and precision. Although the outcome was positive for EtG in hair, an absolute EtG concentration could not be determined due to ion suppression problems.
Other techniques for testing EtG and EtS in autopsy specimens
Electrophoresis technique
In one of the early applications using postmortem materials [62], EtS was identified and quantified using capillary electrophoresis paired with a photodiode array detector. Analysis was performed in a negative polarity mode with a background electrolyte composed of 15 mM maleic acid and an indirect UV detection at 220 nm (300 nm reference wavelength). Vinylsulfonic acid was used as the internal standard. Electrophoretic runs consisted of uncoated fused-silica capillaries that were 60 cm in length and 50 µm in internal diameter. EtS was separated using a mobile phase consisting of 1 M aqueous hydroxide solution for 5 min following by water for 2 min and background electrolytes for 5 min. A linear calibration curve was obtained over a range of 5–700 mg/L. Three quality control levels (low, medium, and high: 5, 60 and 700 mg/L) were used to examine method accuracy and precision. The RSDs for intraday, interday, and accuracy were all < ± 11%. The method was used for urine samples obtained from 16 autopsied cases. The EtS median concentration was 29 mg/L (range 6–200 mg/L), and the UAC for these cases was 260 mg/dL (range 39–444 mg/dL). Urine EtS concentration/UAC ratio was also calculated and found to be 11 (range 3.5–140). To examine the results of the capillary electrophoresis method, they were compared with the results obtained from the same samples based on LC–MS/MS. An excellent correlation was found between the two systems, with R2 = 0.975, y = 1.11x − 1.10. However, correlation between UAC and EtS concentrations in urine was very poor, which may have been due to postmortem ethanol production after deaths, degradation of ethanol due to putrefaction, or different elimination kinetics between ethanol and EtS.
Liquid chromatography coupled with electrochemical detector technique
A non-MS detector method for the analysis of EtG was reported by Kaushik et al. [71]. In that procedure, reverse-phase LC with pulsed electrochemical detection (PED) was used to measure EtG in urine samples from 29 postmortem cases. Methyl glucuronide was used as the internal standard in this application. Analytes of interest were separated using a mobile phase consisting of 1% acetic acid/acetonitrile (98/2, v/v). Post-column addition of NaOH allowed for the detection of all glucuronides using PED at a gold working electrode. LOD and LOQ values were 0.03 and 0.1 mg/L, respectively. Also, the percentage of recovery of compounds was 50%. Linearity was established over the range of the tested samples, which was within 3–700 mg/L using dilution in cases of higher concentration. The median concentration of EtG was 550 mg/L (range 47–1371 mg/L), BAC was 150 mg/dL (range 40–370 mg/dL), while UAC was 200 mg/dL (range 47–137 mg/dL) and the UAC/BAC ratio was 1.3 (range 0.6–4.3). A good coefficient of correlation between BAC and UAC was obtained at R2 = 0.6, y = 0.811x + 25.2; however, poor correlations were found for EtG concentration in urine with BAC and UAC, at R2 = 0.090, y = 0.733x + 1270 and at R2 = 0.138, y = 0.8284x + 1566.1, respectively. This outcome can be explained by ethanol concentration is not affected by increased water intake. Excretion of ethanol by the kidneys occurs by passive diffusion, compared with that of EtG, which is affected by interindividual variations and differences in the time lag within EtG excretion profiles. For example, EtG concentration in urine is found to be affected by many factors that are able to alter the production rate of urine in the kidneys. Dilution by drinking water or taking medications increases urine flow [72].
Gas chromatography–negative ion chemical ionization–mass spectrometry (GC–NIC–MS/MS)
In 2009, a GC-NIC-MS/MS method was reported [49] for the analysis of EtG in seven hair samples obtained at an autopsy. The average sample weight was 30 mg, and samples were extracted using mixed-mode extraction cartridges (Oasis MAX SPE) followed by derivation with perfluoropentanoic anhydride (Table 1). EtG was identified and quantified using the selected reaction monitoring mode with the transitions m/z 347 → 163 as a quantifier ion and m/z 347 → 119 as a qualifier ion; single-ion transition was used for EtG-d5 (m/z 352 → 163). Analytes of interest were separated on a fused silica capillary column (DB-5MS) with a 5% phenyl–95% methyl-polysiloxane stationary phase (15-m length × 0.25-mm I.D. × 0.25-µm film thickness). Helium was used as the carrier gas with a constant flow rate at 1 mL/min on splitless mode. The injection temperature was 250 °C, and the transfer line was maintained at 275 °C. The calibration curve was linear over a range of 8.4–260 pg/mg hair, with a coefficient of determination (R2) above 0.999. This method was sensitive, with LOD and LOQ values of 3 and 8.4 pg/mg, respectively. The RSD for method precision ranged from 1.6 to 13.2%, with method accuracy values of ± 30%. This method was applied to seven hair samples obtained at autopsy, and the concentration of EtG ranged from 60 to 820 pg/mg.
Immunoassays
An immunoassay is a bioanalytical method for determining the presence of analytes. It is highly sensitive and can measure molecules with low molecular weights in biological samples, using an antigen–antibody reaction procedure [73]. However, there are some limitations to immunoassay screenings. For EtG, the cutoff level is 100 ng/mL, and there is also the possibility of cross-reactivity with other substances, including drugs such as sevoflurane, or bacterially formed chemical compounds with a similar structure that could lead to a false-positive result for EtG [74]. Cuypers et al. [75] reported a urine sample that was screened positive for EtG via immunoassay testing, but the final result based on LC–MS/MS was negative. Therefore, immunoassay screening is not a selective, accurate, or reliable method for final confirmation, and any positive results must be confirmed by more advanced instrumentation with higher selectivity and specificity. Gas or liquid chromatography coupled with mass spectrometric instrumentation constitutes a more powerful technique than immunoassays, and it is characterized by both high selectivity and sensitivity [75]. Another drawback associated with the use of immunoassays is that a reagent is not yet available for EtS testing.
VH was available from 58 autopsied cases [46]. The immunoassay was based on the Microgenics DRI EtG enzyme immunoassay reagents used with an automated Abbott Architect c8000 clinical chemistry analyzer, while LC–MS/MS was used for confirmation as reported elsewhere by Helander et al. [76]. The EtG immunoassay showed an excellent correlation with the LC–MS/MS reference method (R2 = 0.94). The assay showed good linearity in the calibration range of 0.1–2 mg/L and excellent reproducibility of results (intra- and inter-assay coefficients of variation of 1.7 and 4.2%, respectively). In that study, VH EtG yielded a markedly higher sensitivity for ascertaining antemortem alcohol consumption than BAC testing (92 vs. 68%). A cutoff level of 0.3 mg/L for VH EtG was applied for routine immunoassays, which enabled identifying 34 of 37 (92%) cases with a history of excessive alcohol use. In comparison, a cutoff level of 10 mg/dL for BAC positively identified 25 (68%) of these 37 cases. A good coefficient of correlation between BAC and VH EtG concentration was found, with R2 = 0.62. In the 12 cases that were positive for VH EtG but negative for blood ethanol, the median VH EtG concentration was 1 mg/L (range 0.4–27.3 mg/L).
Stability of EtG and EtS in postmortem specimens
Studies on ethanol degradation and production after death are controversial issues in postmortem toxicology investigations. In contrast to postmortem alcohol synthesis, ethanol degradation due to putrefaction or storage conditions is rarely discussed in former studies [33, 77, 78]. Skopp et al. stated that ethanol postmortem synthesis or degradation after death should be taken into consideration when interpreting alcohol results in postmortem cases [42]. A similar conclusion reported by O’Neal et al. mentioned that ethanol can be lost or produced after death [8]. Weiler and Kloppel found that a BAC of zero was obtained 4 weeks after specimens in an open dish container were stored at 5 °C [79]. Jones and Ericsson found that even with refrigerating blood at 4 °C, BAC showed a gradual decrease, the ethanol degradation reached analytical significance values after 14–28 days of starting storage time [80]. In addition, Athanaselis et al. alleged that ethanol can be degraded due to evaporation during the putrefactive process [6]. Kugelberg and Jones [33] and others [65, 81] insisted that ethanol can be consumed by a wide variety of microorganisms as a source of carbon and energy after death which leads to lower or degraded antemortem alcohol concentrations, leading to incorrect decisions. After death, ethanol concentration could decrease through conversion to acetaldehyde [7]. Other studies have shown that antemortem blood ethanol was rapidly degraded postmortem, during the first 2 days at 30 °C [78, 82]. The amount of glucose, the substrate for the production of alcohol after death, is limited in the case of severe climatic conditions and after the putrefaction processes are completed, there will be a point where there is no substrate for the alcohol production at which time the microorganisms begin to use the alcohol available as a resource of energy and carbon. The possibility of evaporation of alcohol in cases of exposure to extreme heat may lead to the disappearance of all alcohol formed or consumed before death. In a previous study, blood samples were incubated with microorganisms at 37 °C for 24 h, maximum recorded BAC was followed by a gradual decline of BAC, this degradation was likely due to the reduction of the substrate availability and the subsequent ethanol hydrolyzed to acetate and finally to CO2 [83].
Sodium fluoride is the most commonly used preservative for postmortem specimens, and the mode of action is based on the fluoride ion preventing microbes from forming polysaccharides, which in turn prevents their growth [84]. In addition, studies have shown that sodium fluoride can prevent ethanol formation. One of these studies [85] showed that ethanol formation occurred in all specimens tested in the absence of sodium fluoride. That study also showed that the rate of ethanol formation decreased as the temperature became lower. However, over time, the ethanol concentration in the refrigerated specimens was similar to that of the specimens at room temperature. In fact, adding 1% sodium fluoride limited the formation of ethanol at both 4 and 25 °C. Not all investigations will be so straightforward though, and there could be circumstances in which a postmortem examination is performed days, weeks, or even longer after death. Nevertheless, what happens to the body from contamination simply parallels putrefaction, which is defined as “a type of decomposition due to the proliferation of endogenous bacterial microorganisms in the body” [86]. Putrefaction leads to discoloration and bloating of the body, a foul odor, and skin slippage. Microbial contamination of the blood may especially occur at a temperature over 5 °C, through skin wounds or punctures, or from intestinal bacteria invading the adjacent tissues and the vascular system [87].
The production of ethanol after death is dependent on the presence of particular microorganisms, most commonly yeasts (e.g., Candida parasitosis) and bacteria (e.g., Escherichia coli and Enterococcus faecalis); the availability of a substrate (e.g., glucose); storage temperature before and after autopsy; and the use of preservatives in the sample tubes or containers (e.g., sodium fluoride) that inhibit enzyme activities. The enzymes increase the conversion of glucose to ethanol in vivo or in vitro, and glucose is a good substrate for the anaerobic production of ethanol via glycolysis [87, 88]. Ethanol production is most likely to occur in a corpse if storage was not appropriate [86]. In addition, diluters, pipettes, and other laboratory factors can cause a considerable increase in BAC of the specimen if they are contaminated with microorganisms [88]. Therefore, the ethanol concentration in a postmortem sample cannot be viewed as a direct measure of alcohol consumption prior to death because there are several disturbance factors that can lead to ethanol postmortem synthesis and ethanol degradation as explained above. Accuracy is particularly questionable when the alcohol concentrations are low because they may reflect alcohol synthesis post mortem. Therefore, it is necessary to investigate additional biomarkers [1].
As mentioned previously, EtG and EtS are minor, nonoxidative ethanol metabolites that can be detected in several matrices, such as blood, hair, urine, and tissues, for longer periods of time than ethanol [20, 89]. In addition, these markers of antemortem ethanol consumption are considered highly sensitive and specific compared with other biochemical biomarkers [90]. They take a longer time to eliminate from the body than ethanol. EtG, for example, reaches its maximum concentration in blood after 4 h, with a median elimination half-life of 2.2 h [2]. However, the use of EtG biomarkers alone as evidence of alcohol intake may lead to false-negative results because it can be degraded in postmortem specimens due to severe putrefaction. For this reason, EtS is used in parallel with EtG because it is more stable and reliable. However, both biomarkers should be tested in cases of severe putrefaction [1, 11, 35].
In 2006, Schloegl et al. [85] examined nine urine samples and postmortem tissues (liver, skeletal muscle) by LC–MS/MS to determine the stability of EtG. EtG concentrations were constant when stored at 4 °C in airtight test tubes, but when they were stored in ventilated vials for 5 weeks at room temperature, the EtG concentrations changed within a range from a 30% decrease to an 80% increase. Postmortem tissue concentrations of EtG were reduced by 27.7% when samples were stored at room temperature for 4 weeks, but they were still detectable. Stored urine samples at 4 °C in airtight test tubes showed slight changes in EtG concentration ranging from − 12 to 32%. Higher variations were observed in EtG concentrations when samples were stored at room temperature (22–27 °C) in ventilated vials ranging from − 30 to + 83%.
In another study [16], blood samples were stored at 30 to 40 °C without preservatives. EtG was found to be extremely unstable in these conditions, but when the samples were stored at room temperature with potassium fluoride as a preservative, EtG was stable. No new formation of EtG occurred when samples were stored at 30 °C, even without preservatives, or at room temperature with potassium fluoride. In 15 routine cases in which blood EtG was negative and the detected ethanol was supposed to be endogenous, six cases were positive for EtG in urine. In these cases, ethanol was possibly ingested and the negative EtG result in the blood may have been a false-negative detection because of degradation during putrefaction.
In vitro experiments were carried out by Baranowski et al. [91] with bacterial colonies of E. coli, K. pneumoniae, and Clostridium sordellii that were isolated from autopsy specimens (liver, heart blood, urine, ascites, pericardial fluid, pleural fluid) and analyzed for β-glucuronidase activity. The three bacterial strains were added to a nutrient-deficient medium including EtG, EtS, or both and incubated at 36 ± 1 °C. Specimens were taken after different times up to 11 days, and EtS and EtG were measured by LC–ESI-MS/MS. Total degradation of EtG occurred with E. coli and C. sordellii in 3–4 days, but EtS was not affected during the entire 11 days of incubation. EtG is unstable in blood at temperatures around 30–40 °C due to bacterial degradation, yet it is stable at room temperature if potassium fluoride is added as a preservative.
The formation of EtS is catalyzed by sulfotransferases. EtS showed no indication of being decreased by E. coli in a study that showed bacterial degradation of EtG [78]. This finding indicates that the analysis of EtG alone could lead to false-negative results. Combining EtG analysis with EtS analysis is possible with LC–MS/MS. In one former report, time of alcohol consumption can be estimated to be within 1–2 h of blood samples collection if the ratio between BAC (mg/g) and EtG concentration (mg/l) is higher than 1.0, while if that ratio is less than 1.0, it indicates a longer time between ingestion and sample collection (> 2 h). This ratio dropped below 1.0 within 3 h for the ratio of BAC/EtG concentration and 5 h for BAC/EtS concentration. It takes the ethanol/EtS concentration ratio longer than the ethanol/EtG ratio to fall below 1.0. This indicates a slower elimination rate for EtS compared to that of EtG [92].
Therefore, a blood sample for alcohol concentration analysis should be stored under 4 °C with 1% sodium fluoride, which will keep the ethanol concentration for 29 weeks. The temperature is more influential than the presence or absence of sodium fluoride on the rate of ethanol concentration reduction [2, 16].
The stability of EtG and EtS in extracted postmortem samples was examined during analysis (autosampler stability) and found to be stable for up to 30 days at 4 to − 20 °C, as well as being stable in the autosampler for 7 days [90]. EtG and EtS were found to be stable in dried blood spots used as a storage technique, analytes of interest showed a high level of stability using this technique for 90 days regardless of the temperature [47, 93]. EtG and EtS spiked into postmortem specimens, or were positive in real postmortem cases, were found to be stable under different storage conditions; including long- and short-time stability, room temperature short-term stability for up to 48 h, autosampler up to 1 week, freeze–thaw for up to four cycles, refrigerated at 4 °C, and frozen at − 20 °C [3, 11, 47].
Conclusion
The measurement of EtG and EtS is needed to distinguish between antemortem and postmortem alcohol sources. Knowledge about the kinetics and the stability of postmortem EtG and EtS is required to interpret concentrations of these metabolites correctly. Sample types may be blood, urine, hair, and other types of specimens. The specimens can be analyzed directly or may need pretreatment. LC–MS/MS is a very accurate and highly sensitive technique compared with other methods used to investigate the ethanol biomarkers EtG and EtS in different biological samples. To ensure that the methods used for analysis are suitable, their sensitivity and selectivity should be tested before using them for forensic toxicology investigations. Methods should be validated to verify that a method’s performance parameters are suitable for analysis. In this review, the number of available investigations in the literature does not reflect the importance of ethanol post-mortem alcohol formation issues. In LC–MS reports, it is clear that the matrix effect can be a significant challenge for the analytical method performance in postmortem investigations. Matrix effects directly affect the ionization pattern of analytes, but due to high cutoff levels used to judge positive results for ethanol biomarkers and are often above 0.1–1 mg/L, which is away from the matrix effect zone. The use of deuterated internal standards can overcome matrix effects to a large extent since both analyte and internal standard should be affected to a similar extent by the matrix. In addition, improved chromatographic methods such as HILIC which move the EtG and EtS away from the column void volume can reduce matrix effects. Additional research on using tissue specimens is recommended for three main reasons; firstly, as most of the reported results were promising, secondly, as to be used as alternative samples in cases with highly decomposed bodies and no available bodily fluid specimens, and finally, as complementary specimens to blood samples.
References
Al-Asmari AI, Al-Amoudi DH (2020) The role of ethanol in fatalities in Jeddah, Saudi Arabia. Forensic Sci Int 316:110464. https://doi.org/10.1016/j.forsciint.2020.110464
Santunione AL, Verri P, Marchesi F, Rustichelli C, Palazzoli F, Vandelli D, Licata M, Silingardi E (2018) The role of ethyl glucuronide in supporting medico-legal investigations: analysis of this biomarker in different postmortem specimens from 21 selected autopsy cases. J Forensic Leg Med 53:25–30. https://doi.org/10.1016/j.jflm.2017.10.009
Krabseth H, Morland J, Hoiseth G (2014) Assistance of ethyl glucuronide and ethyl sulfate in the interpretation of postmortem ethanol findings. Int J Legal Med 128(5):765–770. https://doi.org/10.1007/s00414-014-1031-z
Levine B, Smith ML, Smialek JE, Caplan YH (1993) Interpretation of low postmortem concentrations of ethanol. J Forensic Sci 38(3):663–6675
De Lima IV, Midio AF (1999) Origin of blood ethanol in decomposed bodies. Forensic Sci Int 106(3):157–162. https://doi.org/10.1016/s0379-0738(99)00150-4
Athanaselis S, Stefanidou M, Koutselinis A (2005) Interpretation of postmortem alcohol concentrations. Forensic Sci Int 149(2–3):289–291. https://doi.org/10.1016/j.forsciint.2003.04.001
Ziavrou K, Boumba VA, Vougiouklakis TG (2005) Insights into the origin of postmortem ethanol. Int J Toxicol 24(2):69–77. https://doi.org/10.1080/10915810590936391
O’Neal CL, Poklis A (1996) Postmortem production of ethanol and factors that influence interpretation: a critical review. Am J Forensic Med Pathol 17(1):8–20. https://doi.org/10.1097/00000433-199603000-00002
Saady JJ, Poklis A, Dalton HP (1993) Production of urinary ethanol after sample collection. J Forensic Sci 38(6):1467–1471
Lewis RJ, Johnson RD, Angier MK, Vu NT (2004) Ethanol formation in unadulterated postmortem tissues. Forensic Sci Int 146(1):17–24. https://doi.org/10.1016/j.forsciint.2004.03.015
Al-Asmari AI, Anderson RA, Appelblad P (2010) Direct determination of ethyl glucuronide and ethyl sulfate in postmortem urine specimens using hydrophilic interaction liquid chromatography-electrospray ionization-tandem mass spectrometry. J Anal Toxicol 34(5):261–272. https://doi.org/10.1093/jat/34.5.261
Krabseth H, Mørland J, Høiseth G (2014) Assistance of ethyl glucuronide and ethyl sulfate in the interpretation of postmortem ethanol findings. Int J Legal Med 128(5):765–770. https://doi.org/10.1007/s00414-014-1031-z
Cowan DM, Maskrey JR, Fung ES, Woods TA, Stabryla LM, Scott PK, Finley BL (2016) Best-practices approach to determination of blood alcohol concentration (BAC) at specific time points: Combination of ante-mortem alcohol pharmacokinetic modeling and post-mortem alcohol generation and transport considerations. Regul Toxicol pharmacol 78:24–36. https://doi.org/10.1016/j.yrtph.2016.03.020
Tagliaro F, Lubli G, Ghielmi S, Franchi D, Marigo M (1992) Chromatographic methods for blood alcohol determination. J Chromatogr 580(1–2):161–190. https://doi.org/10.1016/0378-4347(92)80534-w
Musshof F (2002) Chromatographic methods for the determination of markers of chronic and acute alcohol consumption. Journal chromatogr B Analyt Technol Biomed Life Sci 781(1–2):457–480. https://doi.org/10.1016/s1570-0232(02)00691-8
Hoiseth G, Karinen R, Johnsen L, Normann PT, Christophersen AS, Morland J (2008) Disappearance of ethyl glucuronide during heavy putrefaction. Forensic Sci Int 176(2–3):147–151. https://doi.org/10.1016/j.forsciint.2007.08.002
Politi L, Morini L, Mari F, Groppi A, Bertol E (2008) Ethyl glucuronide and ethyl sulfate in autopsy samples 27 years after death. Int J Legal Med 122(6):507–509. https://doi.org/10.1007/s00414-008-0273-z
Schmitt G, Aderjan R, Keller T, Wu M (1995) Ethyl glucuronide: an unusual ethanol metabolite in humans. Synthesis, analytical data, and determination in serum and urine. J Anal Toxicol 19(2):91–94. https://doi.org/10.1093/jat/19.2.91
Palmer RB (2009) A review of the use of ethyl glucuronide as a marker for ethanol consumption in forensic and clinical medicine. Semin Diagn Pathol 26(1):18–27. https://doi.org/10.1053/j.semdp.2008.12.005
Biondi A, Freni F, Carelli C, Moretti M, Morini L (2019) Ethyl glucuronide hair testing: a review. Forensic Sci Int 300:106–119. https://doi.org/10.1016/j.forsciint.2019.05.004
Jatlow P, O’Malley SS (2010) Clinical (nonforensic) application of ethyl glucuronide measurement: are we ready? Alcohol Clin Exp Res 34(6):968–975. https://doi.org/10.1111/j.1530-0277.2010.01171.xx
Kummer N, Lambert WE, Samyn N, Stove CP (2016) Alternative sampling strategies for the assessment of alcohol intake of living persons. Clin Bioch 49(13–14):1078–1091. https://doi.org/10.1016/j.clinbiochem.2016.05.007
Lin Z, Wang H, Jones AW, Wang F, Zhang Y, Rao Y (2020) Evaluation and review of ways to differentiate sources of ethanol in postmortem blood. Int J Legal Med 134(6):2081–2093. https://doi.org/10.1007/s00414-020-02415-9
Rosman AS (1992) Utility and evaluation of biochemical markers of alcohol consumption. J Subst Abuse 4(3):277–297. https://doi.org/10.1016/0899-3289(92)90036-w
Sharpe PC (2001) Biochemical detection and monitoring of alcohol abuse and abstinence. Ann Clin Biochem 38(Pt 6):652–664. https://doi.org/10.1258/0004563011901064
Helander A, Beck O (2007) Chapter 17 Analytical markers of acute and chronic alcohol consumption. In: Bogusz M (ed) Handbook of analytical separations, vol 6. Elsevier Science B.V, pp 567–588
Jones AW (2006) Alcohol. In: Karch SB (ed) Drug abuse handbook, 2nd edn. CRC, Boca Raton, Fla., London, pp 313–427
Andresen-Streichert H, Müller A, Glahn A, Skopp G, Sterneck M (2018) Alcohol biomarkers in clinical and forensic contexts. Dtsch Arztebl Int 115(18):309–315. https://doi.org/10.3238/arztebl.2018.0309
Vezzoli S, Bernini M, De Ferrari F (2015) Ethyl glucuronide in vitreous humor and blood postmortem specimens: analysis by liquid chromatography-electrospray tandem mass spectrometry and interpreting results of neo-formation of ethanol. Ann Ist Super Sanita 51(1):19–27. https://doi.org/10.4415/ann_15_01_05
Pronko PS, Velichko MG, Moroz AR, Rubanovich NN (1997) Low-molecular-weight metabolites relevant to ethanol metabolism: correlation with alcohol withdrawal severity and utility for identification of alcoholics. Alcohol Alcohol 32(6):761–768. https://doi.org/10.1093/oxfordjournals.alcalc.a008327
Karch S (2007) Forensic Issues in Alcohol Testing. CRC Press, Boca Raton
Jones AW (2006) Urine as a biological specimen for forensic analysis of alcohol and variability in the urine-to-blood relationship. Toxicol Rev 25(1):15–35. https://doi.org/10.2165/00139709-200625010-00002
Kugelberg FC, Jones AW (2007) Interpreting results of ethanol analysis in postmortem specimens: a review of the literature. Forensic Sci Int1 65(1):10–29. https://doi.org/10.1016/j.forsciint.2006.05.004
Ismaiel OA, Jenkins RG, Karnes HT (2013) Investigation of endogenous blood lipids components that contribute to matrix effects in dried blood spot samples by liquid chromatography-tandem mass spectrometry. Drug Test Anal 5(8):710–715. https://doi.org/10.1002/dta.142
Hegstad S, Kristoffersen L, Liane VH, Spigset O (2017) EtG and EtS in autopsy blood samples with and without putrefaction using UPLC-MS-MS. J Anal Toxicol 41(2):107–113. https://doi.org/10.1093/jat/bkw123
Ferreiro-Vera C, Priego-Capote F, Luque de Castro MD (2012) Comparison of sample preparation approaches for phospholipids profiling in human serum by liquid chromatography–tandem mass spectrometry. J Chromatr A 1240:21–28. https://doi.org/10.1016/j.chroma.2012.03.074
Peters FT, Remane D (2012) Aspects of matrix effects in applications of liquid chromatography-mass spectrometry to forensic and clinical toxicology-a review. Anal Bioanal Chem 403(8):2155–2172. https://doi.org/10.1007/s00216-012-6035-2
Bylda C, Thiele R, Kobold U, Volmer DA (2014) Recent advances in sample preparation techniques to overcome difficulties encountered during quantitative analysis of small molecules from biofluids using LC-MS/MS. Analyst 139(10):2265–2276. https://doi.org/10.1039/c4an00094c
Chambers E, Wagrowski-Diehl DM, Lu Z, Mazzeo JR (2007) Systematic and comprehensive strategy for reducing matrix effects in LC/MS/MS analyses. J Chromatogr B Analyt Technol Biomed Life Sci 852(1–2):22–34
Sidgey D, Liane V, Kristoffersen L (2020) Quantitative determination of ethyl glucuronide and ethyl sulfate in postmortem and antemortem whole blood using phospholipid removal 96-well plate and UHPLC-MS-MS. J Anal Toxicol. https://doi.org/10.1093/jat/bkaa108
Zhang XY, Lin ZB, Li JL, Huang ZB, Rao YL, Liang H, Yan J, Zheng F (2017) Rapid determination of nine barbiturates in human whole blood by liquid chromatography-tandem mass spectrometry. Drug Test Anal 9(4):588–595. https://doi.org/10.1002/dta.2002/
Skopp G (2004) Preanalytic aspects in postmortem toxicology. Forensic Sci Int 142(2–3):75–100. https://doi.org/10.1016/j.forsciint.2004.02.012
Al-Asmari AI (2009) Applications of LC-MS/MS in Forensic Toxicology for the Analysis of Drugs and Their Metabolites. Ph.D. thesis, University of Glasgow, Glasgow, Scotland
Zhang X, Zheng F, Lin Z, Johansen SS, Yu T, Liu Y, Huang Z, Li J, Yan J, Rao Y (2017) Simultaneous determination of ethanol’s four types of non-oxidative metabolites in human whole blood by liquid chromatography tandem mass spectrometry. Anal Chim Acta 963:68–75. https://doi.org/10.1016/j.aca.2017.01.040
Kwon M, Choi HJ, Jo YH, Son MH, Min JS, Kim NY, Jung JE (2019) Analysis of ethyl glucuronide and ethyl sulfate in blood to determine the absorption or elimination phase of alcohol for Korean. Forensic Sci Int. https://doi.org/10.1016/j.forsciint.2019.06.015
Rainio J, Kultti J, Kangastupa P, Tuomi H, Ahola S, Karhunen PJ, Helander A, Niemelä O (2013) Immunoassay for ethyl glucuronide in vitreous humor: a new tool for postmortem diagnostics of alcohol use. Forensic Sci Int 226(1–3):261–265. https://doi.org/10.1016/j.forsciint.2013.01.040
Wang H, Li B, Wang F, Chang J, Zhang Y, Rao Y (2020) Determination of ethyl glucuronide and ethyl sulfate in human whole blood and vitreous humor by LC-MS/MS and applications to the interpretation of postmortem ethanol findings. J Anal Toxicol. https://doi.org/10.1093/jat/bkz082
Schloegl H, Rost T, Schmidt W, Wurst FM, Weinmann W (2006) Distribution of ethyl glucuronide in rib bone marrow, other tissues and body liquids as proof of alcohol consumption before death. Forensic Sci Int 156(2–3):213–218. https://doi.org/10.1016/j.forsciint.2005.03.024
Kharbouche H, Sporkert F, Troxler S, Augsburger M, Mangin P, Staub C (2009) Development and validation of a gas chromatography-negative chemical ionization tandem mass spectrometry method for the determination of ethyl glucuronide in hair and its application to forensic toxicology. J Chromatogr B Analyt Technol Biomed Life Sci 877(23):2337–2343. https://doi.org/10.1016/j.jchromb.2008.11.046
SOFT/AAFS (2006) Forensic toxicology laboratory guideline. http://www.soft-tox.org/files/Guidelines_2006_Final.pdf Accessed 17 Nov 2020
Hoiseth G, Karinen R, Christophersen AS, Olsen L, Normann PT, Morland J (2007) A study of ethyl glucuronide in post-mortem blood as a marker of ante-mortem ingestion of alcohol. Forensic Sci Int 165(1):41–45. https://doi.org/10.1016/j.forsciint.2006.02.045
Weinmann W, Schaefer P, Thierauf A, Schreiber A, Wurst FM (2004) Confirmatory analysis of ethylglucuronide in urine by liquid-chromatography/electrospray ionization/tandem mass spectrometry according to forensic guidelines. J Am Soc Mass Spectrom 15(2):188–193. https://doi.org/10.1016/j.jasms.2003.10.010
Keten A, Tumer AR, Balseven-Odabasi A (2009) Measurement of ethyl glucuronide in vitreous humor with liquid chromatography-mass spectrometry. Forensic Sci Int 193(1–3):101–105. https://doi.org/10.1016/j.forsciint.2009.09.018
Thierauf A, Kempf J, Perdekamp MG, Auwarter V, Gnann H, Wohlfarth A, Weinmann W (2011) Ethyl sulphate and ethyl glucuronide in vitreous humor as postmortem evidence marker for ethanol consumption prior to death. Forensic Sci Int 210(1–3):63–68. https://doi.org/10.1016/j.forsciint.2011.01.036
Høiseth G, Morini L, Polettini A, Christophersen A, Mørland J (2009) Blood kinetics of ethyl glucuronide and ethyl sulphate in heavy drinkers during alcohol detoxification. Forensic Sci Int 188(1–3):52–56. https://doi.org/10.1016/j.forsciint.2009.03.017
Scientific Working Group for Forensic Toxicology (SWGTOX) Standard Practices for Method Validation in Forensic Toxicology (2013). J Anal Toxicol 37(7):452–474. https://doi.org/10.1093/jat/bkt054
Beyer J, Gerostamoulos D, Drummer O, Costantino A (2007) Comments on "The effect of the use of mouthwash on ethylglucuronide concentration in urine". J Anal Toxicol 31(5):294–295; author reply 295–296. https://doi.org/10.1093/jat/31.5.294
Wang H, Li JL, Huang ZB, Wang FL, Zhang YF, Chang J, Rao YL (2019) Assessment of the role played by n-propanol in distinction of ethanol source in postmortem blood with the assistance of ethyl glucuronide and ethyl sulfate. Forensic Toxicol. https://doi.org/10.1007/s11419-019-00507-9
Hoiseth G, Karinen R, Christophersen A, Morland J (2010) Practical use of ethyl glucuronide and ethyl sulfate in postmortem cases as markers of antemortem alcohol ingestion. Int J Legal Med 124(2):143–148. https://doi.org/10.1007/s00414-009-0393-0
Liu YM, Zhang XY, Li JL, Huang ZB, Lin ZB, Wang JR, Zhang CQ, Rao YL (2018) Stability of ethyl glucuronide, ethyl sulfate, phosphatidylethanols and fatty acid ethyl esters in postmortem human blood. J Anal Toxicol 42(5):346–352. https://doi.org/10.1093/jat/bky010
Saenz SR, Lewis RJ, Angier MK, Wagner JR (2017) Postmortem fluid and tissue concentrations of THC, 11-OH-THC and THC-COOH. J Anal Toxicol 41(6):508–516. https://doi.org/10.1093/jat/bkx033
Esteve-Turrillas FA, Bicker W, Lammerhofer M, Keller T, Lindner W (2006) Determination of ethyl sulfate–a marker for recent ethanol consumption–in human urine by CE with indirect UV detection. Electrophoresis 27(23):4763–4771. https://doi.org/10.1002/elps.200600155
Bicker W, Lammerhofer M, Keller T, Schuhmacher R, Krska R, Lindner W (2006) Validated method for the determination of the ethanol consumption markers ethyl glucuronide, ethyl phosphate, and ethyl sulfate in human urine by reversed-phase/weak anion exchange liquid chromatography-tandem mass spectrometry. Anal Chem 78(16):5884–5892. https://doi.org/10.1021/ac060680+
Blackmore DJ (1968) The bacterial production of ethyl alcohol. J Forensic Sci Soc 8(2):73–78
Harper DR (1989) A comparative study of the microbiological contamination of postmortem blood and vitreous humour samples taken for ethanol determination. Forensic Sci Int 43(1):37–44. https://doi.org/10.1016/0379-0738(89)90120-5
Hoiseth G, Bernard JP, Karinen R, Johnsen L, Helander A, Christophersen AS, Morland J (2007) A pharmacokinetic study of ethyl glucuronide in blood and urine: applications to forensic toxicology. Forensic Sci Int 172(2–3):119–124. https://doi.org/10.1016/j.forsciint.2007.01.005
Bendroth P, Kronstrand R, Helander A, Greby J, Stephanson N, Krantz P (2008) Comparison of ethyl glucuronide in hair with phosphatidylethanol in whole blood as post-mortem markers of alcohol abuse. Forensic Sci Int 176(1):76–81. https://doi.org/10.1016/j.forsciint.2007.09.012
Morini L, Politi L, Zucchella A, Polettini A (2007) Ethyl glucuronide and ethyl sulphate determination in serum by liquid chromatography–electrospray tandem mass spectrometry. Clin Chim Acta 376(1):213–219. https://doi.org/10.1016/j.cca.2006.08.024
Kintz P (2015) 2014 consensus for the use of alcohol markers in hair for assessment of both abstinence and chronic excessive alcohol consumption. Forensic Sci Int 249:A1-2. https://doi.org/10.1016/j.forsciint.2014.11.001
Kintz P (2010) Consensus of the Society of Hair Testing on hair testing for chronic excessive alcohol consumption 2009. Forensic Sci Int 249:A1-2. https://doi.org/10.1016/j.forsciint.2014.11.001
Kaushik R, Levine B, LaCourse WR (2006) A brief review: HPLC methods to directly detect drug glucuronides in biological matrices (Part I). Anal Chim Acta 556(2):255–266. https://doi.org/10.1016/j.aca.2005.09.012
Dahl H, Stephanson N, Beck O, Helander A (2002) Comparison of urinary excretion characteristics of ethanol and ethyl glucuronide. J Anal Toxicol 26(4):201–204. https://doi.org/10.1093/jat/26.4.201
Salvador JP, Adrian J, Galve R, Pinacho DG, Kreuzer M, Sánchez-Baeza F, Marco MP (2007) Chapter 28 Application of bioassays/biosensors for the analysis of pharmaceuticals in environmental samples. In: Petrović M, Barceló D (eds) Comprehensive analytical chemistry, vol 50. Elsevier, pp 279–334
Böttcher M, Kühne D, Beck O (2018) Sevoflurane metabolite cross-react in the ethyl glucuronide DRI® immunoassay. Drug Test Anal 10(2):379–380. https://doi.org/10.1002/dta.2233
Cuypers E, Flanagan RJ (2018) The interpretation of hair analysis for drugs and drug metabolites. Clin Toxicol 56(2):90–100. https://doi.org/10.1080/15563650.2017.1379603
Helander A, Kenan N, Beck O (2010) Comparison of analytical approaches for liquid chromatography/mass spectrometry determination of the alcohol biomarker ethyl glucuronide in urine. Rapid commun in Mass Spectrom 24(12):1737–1743. https://doi.org/10.1002/rcm.4573
Ferrari LA, Triszcz JM, Giannuzzi L (2006) Kinetics of ethanol degradation in forensic blood samples. Forensic Sci Int 161(2–3):144–150. https://doi.org/10.1016/j.forsciint.2006.02.049
Thierauf A, Serr A, Halter CC, Al-Ahmad A, Rana S, Weinmann W (2008) Influence of preservatives on the stability of ethyl glucuronide and ethyl sulphate in urine. Forensic Sci Int 182(1–3):41–45. https://doi.org/10.1016/j.forsciint.2008.09.011
Weiler GaK A (1996) Alcohol evaporation from blood stored in unstoppered tube. ToxTalk 20(2):7
Jones AW, Ericsson E (2016) Decreases in blood ethanol concentrations during storage at 4 °C for 12 months were the same for specimens kept in glass or plastic tubes. Pract Lab Med 4:76–81. https://doi.org/10.1016/j.plabm.2016.02.002
Corry JE (1978) A review. Possible sources of ethanol ante- and post-mortem: its relationship to the biochemistry and microbiology of decomposition. J Appl Bacteriol 44(1):1–56. https://doi.org/10.1111/j.1365-2672.1978.tb00776.x
Takayasu T, Ohshima T, Tanaka N, Maeda H, Kondo T, Nishigami J, Nagano T (1995) Postmortem degradation of administered ethanol-d6 and production of endogenous ethanol: experimental studies using rats and rabbits. Forensic Sci Int 76(2):129–140. https://doi.org/10.1016/0379-0738(95)01807-7
Amick GD, Habben KH (1997) Inhibition of ethanol production by Saccharomyces cerevisiae in human blood by sodium fluoride. J Forensic Sci 42(4):690–692
Hoiseth G, Kristoffersen L, Larssen B, Arnestad M, Hermansen NO, Morland J (2008) In vitro formation of ethanol in autopsy samples containing fluoride ions. Int J Legal Med 122(1):63–66. https://doi.org/10.1007/s00414-007-0166-6
Schloegl H, Dresen S, Spaczynski K, Stoertzel M, Wurst FM, Weinmann W (2006) Stability of ethyl glucuronide in urine, post-mortem tissue and blood samples. Int J Legal Med 120(2):83–88. https://doi.org/10.1007/s00414-005-0012-7
Gill JR (2017) From death to death certificate: what do the dead say? J Med Toxicol 13(1):111–116. https://doi.org/10.1007/s13181-016-0551-y
Quintas MJ, Costa P, Melo P, Castro A, Franco JM, Teixeira HM (2017) Postmortem in vitro ethanol production-It could be more common than we think! Forensic Sci Int 274:113–116. https://doi.org/10.1016/j.forsciint.2016.12.040
Laurens JB, Sewell FJJ, Kock MM (2018) Pre-analytical factors related to the stability of ethanol concentration during storage of ante-mortem blood alcohol specimens. J Forensic Leg Med 58:155–163. https://doi.org/10.1016/j.jflm.2018.06.003
Ferraguti G, Ciolli P, Carito V, Battagliese G, Mancinelli R, Ciafrè S, Tirassa P, Ciccarelli R, Cipriani A, Messina MP, Fiore M, Ceccanti M (2017) Ethylglucuronide in the urine as a marker of alcohol consumption during pregnancy: comparison with four alcohol screening questionnaires. Toxicol lett 275:49–56. https://doi.org/10.1016/j.toxlet.2017.04.016
Casati S, Ravelli A, Angeli I, Durello R, Minoli M, Orioli M (2019) An automated sample preparation approach for routine liquid chromatography tandem-mass spectrometry measurement of the alcohol biomarkers phosphatidylethanol 16:0/18:1, 16:0/16:0 and 18:1/18:1. J Chromatogr A 1589:1–9. https://doi.org/10.1016/j.chroma.2018.12.048
Baranowski S, Serr A, Thierauf A, Weinmann W, Grobetae Perdekamp M, Wurst FM, Halter CC (2008) In vitro study of bacterial degradation of ethyl glucuronide and ethyl sulphate. Int J Legal Med 122(5):389–393. https://doi.org/10.1007/s00414-008-0229-3
Lundberg R (2018) Ethanol, ethyl glucuronide, and ethyl sulfate kinetics after multiple ethanol intakes: a study of ethanol consumption to better determine the latest intake of alcohol in hip flask defence cases. Student thesis
Nguyen VL, Paull P, Haber PS, Chitty K, Seth D (2018) Evaluation of a novel method for the analysis of alcohol biomarkers: ethyl glucuronide, ethyl sulfate and phosphatidylethanol. Alcohol 67:7–13. https://doi.org/10.1016/j.alcohol.2017.08.009
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Alsayed, S.N., Alharbi, A.G., Alhejaili, A.S. et al. Ethyl glucuronide and ethyl sulfate: a review of their roles in forensic toxicology analysis of alcohol postmortem. Forensic Toxicol 40, 19–48 (2022). https://doi.org/10.1007/s11419-021-00588-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11419-021-00588-5